
Abstract
Autophagy is a cellular bulk degradation system for long-lived proteins and organelles that operates during nutrient starvation and is thus a type of recycling system. In recent years, a series of mammalian orthologs of yeast autophagy-related (ATG) genes have been identified; however, the importance of the transcriptional regulation of ATG genes underlying autophagosome formation is poorly understood. In this study, we identified several ATG genes, including the genes ULK1, MAP1LC3B, GABARAPL1, ATG13, WIPI1, and WDR45/WIPI4, with elevated mRNA levels in thapsigargin-, C2-ceramide-, and rapamycin-treated as well as amino acid-depleted HeLa cells except for MAP1LC3B mRNA in rapamycin-treated HeLa cells. Rapamycin had a weaker effect on the expressions of ATG genes. The increase in WIPI1 and MAP1LC3B mRNA was induced prior to the accumulation of the autophagy marker protein MAP1LC3 in the thapsigargin- and C2-ceramide-treated A549 cells. By counting the puncta marked with MAP1LC3B in HeLa cells treated with different autophagy inducers, we revealed that the time-dependent mRNA elevation of a specific set of ATG genes was similar to that of autophagosome accumulation. The transcriptional attenuation of WIPI1 mRNA using RNA interference inhibited the puncta number in thapsigargin-treated HeLa cells. Remarkably, increases in the abundance of WIPI1 mRNA were also manifested in thapsigargin- and C2-ceramide-treated human fibroblasts (WI-38 and TIG-1), human cancer cells (U-2 OS, Saos-2, and MCF7), and rodent fibroblasts (Rat-1). Taken together, these results suggest that the detection of WIPI1 mRNA is likely to be a convenient method of monitoring autophagosome formation in a wide range of cell types.
Introduction
The process of autophagy has been conserved from yeast to mammals, and mammalian orthologous genes of budding yeast autophagy-related (ATG) genes have been identified and characterized in succession.Citation1 Mammalian autophagy has been shown to have fundamental roles in both physiology and pathology; thus, the molecular mechanism underlying autophagy regulation is considered to be an issue of primary importance in the field of life science.Citation2,Citation3 For example, a basal level of autophagy is essential for cellular homeostasis; thus, the inhibition of autophagy in nerve cells can play a causative role in degenerative diseases similar to the role that the death of nerve cells plays in Alzheimer disease. Moreover, autophagy is deeply involved in a variety of pathophysiological phenomena—from development and differentiation to lipid metabolism—and aberrations in autophagy can potentially lead to adult-onset diseases such as cancer, diabetes mellitus, and cardiac failure.Citation2,Citation3
Autophagy is regarded as a membrane-trafficking pathway and is actually a generic term that is used to refer to the following sequential process: First, a double membrane derived from the endoplasmic reticulum (ER) and/or other subcellular compartments becomes a precursor sequestering sac known as a phagophore; this membrane then transforms into an autophagosome and fuses with a lysosome to become an autolysosome.Citation4 Regarding the origin of the autophagosome membrane, multiple membrane sources have been advocated to serve in the formation of the phagophore.Citation5-Citation7 Evidence regarding the protein-protein interactions that occur during this process is accumulating.Citation8 At present, various means of indicating autophagy have been developed and are widely used, such as form shifts in the MAP1LC3 protein (mediated by cleavage and lipidation), consisting of the conversion of the cytoplasmic-liberated form of MAP1LC3 (LC3-I) to the autophagosome membrane-bound form of MAP1LC3 (LC3-II), and puncta observations of green fluorescent protein (GFP)-tagged MAP1LC3 (GFP-LC3) under fluorescent microscopy, as well as the detection of autophagosomes and autolysosomes using electron microscopy.Citation9,Citation10 However, interpretations of the form shift in MAP1LC3 protein as detected using western blotting frequently lead to false-positive predictions of autophagy; thus, caution is needed.Citation10-Citation13 Furthermore, GFP-LC3 tends to aggregate even in the absence of genuine autophagy.Citation11-Citation13 Therefore, technical innovations for monitoring autophagy, especially for monitoring autophagic flux, are needed.Citation10
Unlike our knowledge of protein levels during autophagy, the transcriptional regulation of basal and inducible levels of ATG genes remains poorly understood. Several lines of evidence indicate that the transcriptional regulation of ATG genes is fundamental to understanding autophagy from a pathophysiological perspective.Citation14 For instance, Becn1 (mouse ortholog of yeast VPS30/ATG6) heterozygous mice have an increased risk of developing cancer because of the haploinsufficiency of the Becn1 gene, which is to say that the Becn1 mRNA level is reduced by half.Citation15,Citation16 Details regarding recent imperative progress concerning transcriptional regulation in autophagy will be mentioned in the Discussion section.
In the present study, we aimed to develop and establish a more convenient and reliable method of detecting autophagosome formation. For this purpose, we quantitatively detected an increase or decrease in the mRNA abundance of ATG genes after exposure to different kinds of autophagy inducers and verified the usefulness of the expression levels of ATG genes as markers of autophagosome formation.
Results
Identification of WIPI1 mRNA as an indicator of chemical and physiological autophagy
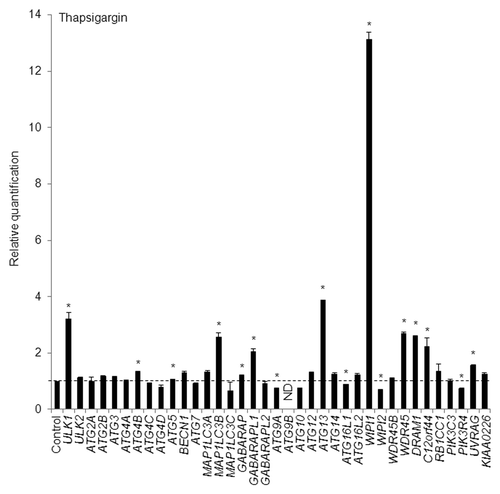
To identify the human ATG genes with accumulated mRNA levels after autophagy induction, we performed a quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis. The following 37 ATG genes were primarily examined (Table S1): ULK1, ULK2, ATG2A, ATG2B, ATG3, ATG4A, ATG4B, ATG4C, ATG4D, ATG5, BECN1 (ortholog of yeast VPS30/ATG6), ATG7, MAP1LC3A, MAP1LC3B (an ortholog of yeast ATG8), MAP1LC3C, GABARAP, GABARAPL1, GABARAPL2/GATE16, ATG9A, ATG9B, ATG10, ATG12, ATG13, ATG14, ATG16L1, ATG16L2, WIPI1 (ortholog of yeast ATG18), WIPI2, WDR45B/WIPI3/WDR45L, WDR45/WIPI4, DRAM1, C12orf44/ATG101, RB1CC1/FIP200, PIK3C3 (ortholog of yeast VPS34), PIK3R4, UVRAG, and KIAA0226/RUBICON. Among them, MAP1LC3A, MAP1LC3B, MAP1LC3C, GABARAP, GABARAPL1, and GABARAPL2 are yeast ATG8 orthologs, and WIPI (WD repeat protein interacting with phosphoinositides) family members including WIPI1, WIPI2, WDR45B, and WDR45 are yeast ATG18 orthologs. C12orf44 and RB1CC1 proteins are reportedly associated with the yeast Atg1 protein orthologs, ULK1 and ULK2, and the ATG13 protein complex.Citation17 First, we treated HeLa cells with 0.5 µM of thapsigargin, which induces cellular stress responses, such as ER stress and autophagy, by increasing the cellular calcium ion concentration,Citation18,Citation19 for 8 h. We selected 8 h as a time interval for thapsigargin treatment based on results obtained in time-course analyses of puncta formation and the WIPI mRNA level, as described later. As a result, we found that WIPI1 was the most significantly upregulated mRNA among the 37 ATG genes that were examined (). In addition to the 37 ATG genes, the mRNA levels of AMBRA1, COPZ1, RAB3GAP2, RPS6KA1, and WDR82 were not significantly changed (data not shown).
Figure 1. HeLa cells were exposed to 0.5 µM of thapsigargin for 8 h and the mRNA expression levels of the indicated ATG genes were quantitatively detected using TaqMan real-time RT-PCR. GAPDH was used as an internal standard. The means ± standard deviations (SDs) are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1 (horizontal broken line). *P < 0.05 (n = 2). ND indicates no signal was detected.
Similar to thapsigargin, tunicamycin, an inhibitor of N-glycosylation, has also been reported to induce ER stress and autophagy.Citation18,Citation19 ER stress has been shown to be capable of triggering autophagy.Citation18-Citation22 First, we observed that the mRNA levels for WIPI1 and the ER stress marker gene HSPA5/BiP were significantly elevated in tunicamycin (2 µg/mL, 8 h)-treated HeLa cells, compared with DMSO-treated HeLa cells (Fig. S1A and S1B). Next, the induction of autophagosome formation by tunicamycin was examined using HeLa cells stably expressing GFP-tagged MAP1LC3B (HeLa/GFP-LC3B) by counting the number of puncta marked by GFP-LC3B under a fluorescent microscope. We detected significantly larger numbers of puncta in the tunicamycin-treated HeLa/GFP-LC3B cells than in the DMSO-treated HeLa/GFP-LC3B cells after 24 h of tunicamycin treatment (Fig. S1C). These results suggest that WIPI1 mRNA could be a suitable and generally versatile indicator of cellular stress responses. We also detected the significant accumulation of mRNA for WIPI1 as well as HSPA5 in dithiothreitol (2 mM, 6 h)-treated HeLa cells, compared with mock-treated HeLa cells (unpublished data), but we have yet to determine the functional significance of WIPI1 mRNA accumulation during ER stress-induced or ER stress-associated autophagosome formation in HeLa cells.
Contrary to previous reports, thapsigargin has been shown to inhibit autophagy by blocking the fusion of autophagosomes with the endocytic system.Citation23 More recently, thapsigargin has been reported to block an early stage of autophagic flux independently of ER stress.Citation24 These findings suggest that WIPI1 mRNA could be an indicator of autophagy dysregulation in addition to autophagy stimulation, since thapsigargin is likely to lead to autophagy stimulation in an attempt to compensate for the loss of autophagosome turnover. To determine whether WIPI1 mRNA accumulation can be seen after the blockage of the latter stage of autophagy, we treated HeLa cells with bafilomycin A1 or chloroquine. As well-known autophagy inhibitors, bafilomycin A1 inhibits vacuolar H+-ATPase while chloroquine increases the lysosomal pH.Citation25 Based on an autophagic flux assay, in which the presence of free GFP fragments (resulting from the degradation of GFP-LC3) within the autolysosome in HeLa cells was evaluated, unsaturating concentrations of bafilomycin A1 or chloroquine were shown to increase the level of free GFP fragments, indicating incomplete autophagy inhibition, while saturating concentrations of these compounds did not increase the level of free GFP fragments, indicating complete autophagy inhibition.Citation25 Therefore, unsaturating and saturating concentrations of bafilomycin A1 (2.5 nM and 25 nM, respectively) or chloroquine (10 µM and 50 µM, respectively) were used to treat HeLa cells for 6 h, and the resulting WIPI1 mRNA levels were then quantitatively measured. Saturating concentrations, but not unsaturating concentrations, of the autophagy inhibitors resulted in the accumulation of WIPI1 mRNA, but the extent of WIPI1 mRNA accumulation was considerably smaller than that obtained by thapsigargin treatment (Fig. S2; ).
Next, HeLa cells were treated with 100 µM of a well-known autophagy inducer, C2-ceramide (a kind of sphingolipid),Citation26 for 12 h; a quantitative real-time RT-PCR assay was then performed to detect any increases in the transcripts of the 37 ATG genes. We selected 12 h as a time interval for C2-ceramide treatment based on results obtained using time-course analyses of puncta formation and the WIPI mRNA level, as mentioned later. Remarkably, and in accordance with the results obtained using thapsigargin, C2-ceramide increased the mRNA abundance of WIPI1 to the greatest degree (). We also confirmed the WIPI mRNA level using 3 other TaqMan primers (Fig. S3A). In addition to WIPI1, the abundance of ULK1, MAP1LC3B, GABARAPL1, ATG13, WDR45, and DRAM1 mRNA was significantly increased by both thapsigargin and C2-ceramide in HeLa cells ( and ).
Figure 2. HeLa cells were exposed to 100 µM of C2-ceramide for 12 h and the mRNA expression levels of the indicated ATG genes were quantitatively detected using TaqMan real-time RT-PCR. GAPDH was used as an internal standard. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1 (horizontal broken line). *P < 0.05 (n = 2). ND indicates no signal was detected.
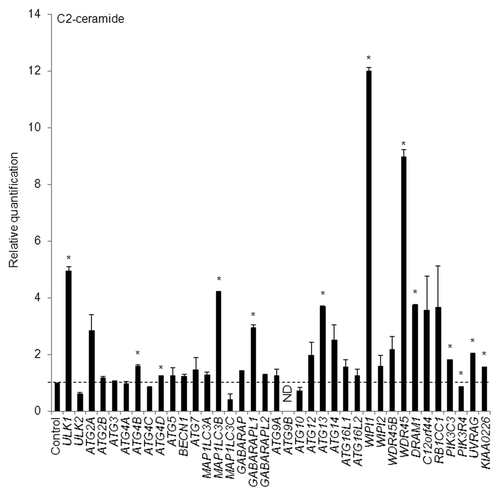
To further confirm these results, 100 µM of C2-ceramide was added to both the A549 cells as well as the HeLa cells for 24 h, and semi-quantitative RT-PCR was then performed to detect the mRNA expression levels of ULK1, MAP1LC3B, GABARAPL1, WIPI1, WDR45, and DRAM1. As a result, these mRNA levels were commonly upregulated in both cell types (Fig. S4). Collectively, we successfully demonstrated that the mRNA abundances for a unique set of ATG genes are commonly increased in 2 different cell lines.
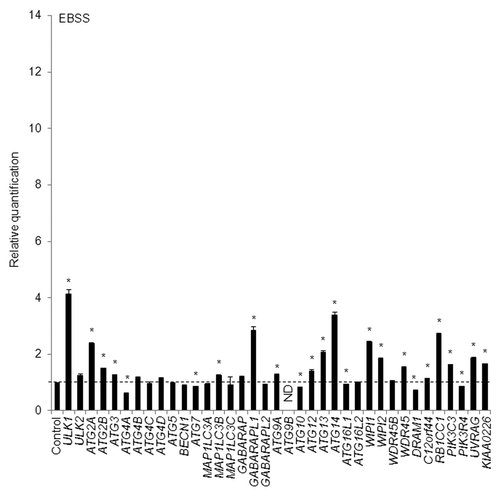
Furthermore, we checked different autophagy inducers to confirm the results obtained using thapsigargin and C2-ceramide using a quantitative real-time RT-PCR assay. For this purpose, we treated HeLa cells with amino acid-free Earle’s balanced salt solution (EBSS) medium, a physiological inducer of autophagy, for 4 h, and quantitative real-time RT-PCR was performed to detect any changes in the mRNA levels of 37 ATG genes. In accordance with the results obtained using thapsigargin- and C2-ceramide-treated HeLa cells, the mRNA levels for ULK1, MAP1LC3B, GABARAPL1, ATG13, WIPI1, and WDR45 were increased (). We also confirmed the WIPI mRNA level using 3 other TaqMan primers (Fig. S3B). Aside from these mRNAs, the mRNA levels of ATG2A, ATG14, and RB1CC1 more than doubled (). In addition to the HeLa cells, we also confirmed the accumulation of WIPI1 and MAP1LC3B mRNA in A549 cells cultured in EBSS medium for 3 h, rather than 30 min (Fig. S5A).
Figure 3. HeLa cells were exposed to EBSS for 4 h and the mRNA expression levels of the indicated ATG genes were quantitatively detected using TaqMan real-time RT-PCR. GAPDH was used as an internal standard. The means ± SDs are shown as the relative fold-induction when the values obtained in cells cultured in normal medium were set as 1 (horizontal broken line). *P < 0.05 (n = 2). ND indicates no signal was detected.
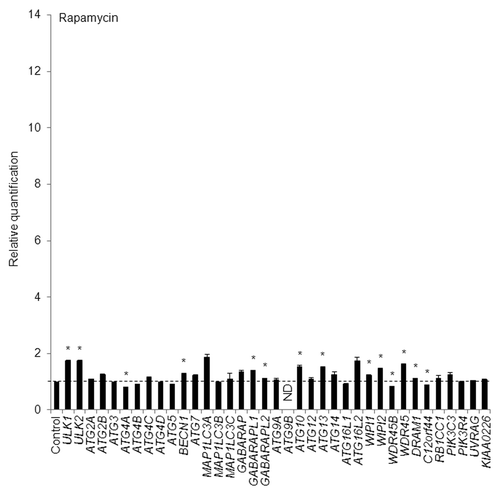
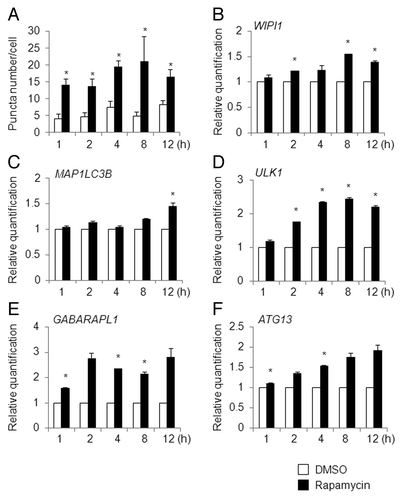
On the other hand, in HeLa cells treated with rapamycin, an inhibitor of mechanistic target of rapamycin (MTOR) and RPS6KB1/p70 S6 kinase,Citation27 at a concentration of 250 nM for 12 h, none of the ATG genes accumulated transcriptionally by more than 2-fold, though the mRNA levels of several ATG genes including ULK1, GABARAPL1, ATG13, WIPI1, and WDR45, but not MAP1LC3B, increased significantly (). The time interval for EBSS and rapamycin treatment was determined based on time-course analyses of puncta formation and the WIPI1 mRNA level, as described below. The reason why the transcription of ATG genes tended to remain low in the rapamycin-treated HeLa cells is discussed later. Finally, resveratrol, a famous natural polyphenol, exerts various biological activities including the induction of autophagy.Citation28 Remarkably, the mRNA levels for WIPI1 and MAP1LC3B were elevated in resveratrol-treated HeLa cells (Fig. S5B). The induction of autophagosome formation by resveratrol was examined using HeLa/GFP-LC3B cells by counting the number of puncta marked by GFP-LC3B under a fluorescent microscope. We detected significantly larger numbers of puncta in resveratrol-treated HeLa/GFP-LC3B cells, compared with DMSO-treated HeLa/GFP-LC3B cells (data not shown).
Figure 4. HeLa cells were exposed to 250 nM of rapamycin for 12 h and the mRNA expression levels of the indicated ATG genes were quantitatively detected using TaqMan real-time RT-PCR. GAPDH was used as an internal standard. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1 (horizontal broken line). *P < 0.05 (n = 2). ND indicates no signal was detected.
Collectively, these results suggest that among > 30 ATG genes, the detection of a specific set of ATG genes, especially WIPI1 mRNA, is likely to be a reliable means of monitoring the induction of autophagosome formation in HeLa and A549 cells in response to physiological as well as chemical inducers of autophagy.
Transcription of WIPI1 and MAP1LC3B genes prior to LC3-II accumulation in A549 cells
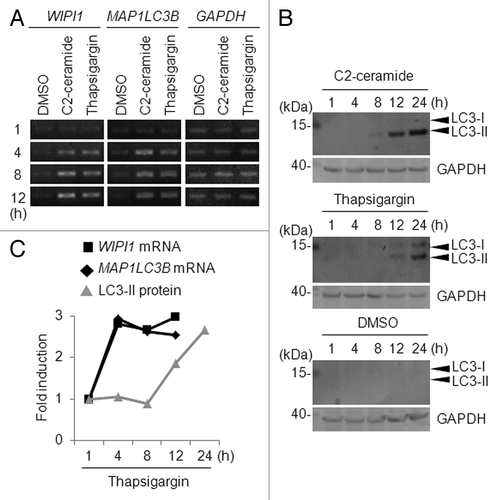
The MAP1LC3 protein exists in a soluble and phosphatidylethanolamine-conjugated form (membrane-bound form).Citation10 As a marker protein for the autophagosome membrane, MAP1LC3 has been found to shift from an 18-kDa cytoplasmic-liberated form (LC3-I) to a 16-kDa phagophore and autophagosome membrane-bound form (LC3-II) upon the induction of autophagy.Citation10 We detected the enhanced LC3-II form, rather than a shift in form from LC3-I to LC3-II, in C2-ceramide-treated HeLa and A549 cells, in a C2-ceramide concentration-dependent manner (Fig. S6). This observation suggests that the conversion from LC3-I to LC3-II might be rather quick and complete, depending on the stimuli and cell type, resulting in very little or undetectable amounts of LC3-I. These observations strengthened our hypothesis that the mRNA abundance of ATG genes might be an important indicator of the induction of autophagosome formation. Therefore, we decided to continue to analyze the importance of the regulation of ATG genes, particularly the time-course changes in the mRNA abundances of a unique set of ATG genes, upon autophagy induction.
The WIPI1 and MAP1LC3B mRNA levels tended to be higher after 12 h of treatment than after 24 h of treatment with 0.5 µM of thapsigargin in A549 cells, as shown using a quantitative real-time RT-PCR experiment (Fig. S7). To clarify the time-course changes in the WIPI1 and MAP1LC3B mRNA abundances during the early period of autophagy, we treated A549 cells with 100 µM of C2-ceramide or 0.5 µM of thapsigargin for 1, 4, 8, or 12 h. Consequently, we found that the mRNA expression levels of WIPI1 and MAP1LC3B were uniformly upregulated after 4 h of treatment with C2-ceramide or thapsigargin (). We also found that the amount of LC3-II increased markedly after 8 h of treatment with C2-ceramide or after 12 h of treatment with thapsigargin (). In contrast, no obvious conversion from LC3-I to LC3-II was observed in DMSO-treated cells (). In the thapsigargin-treated A549 cells, a slight accumulation of the LC3-I form was observed (). The time-course for mRNA abundance changes for the WIPI1 and MAP1LC3B genes and the accumulation of LC3-II, which was standardized using GAPDH as an internal control, clearly showed that the induction of WIPI1 and MAP1LC3B mRNA accumulation occurred prior to the accumulation of LC3-II (), suggesting that the accumulation of LC3-II occurred as a result of mRNA accumulation.
Figure 5. Time-course analysis of WIPI1 and MAP1LC3B expression. (A) A549 cells were treated with 100 µM of C2-ceramide, 0.5 µM of thapsigargin, or DMSO for the indicated time period (h), and a semiquantitative RT-PCR analysis was then performed. The primers used in this experiment have been previously reported.Citation29 The lengths of the PCR products for WIPI1 and MAP1LC3B were 420 and 480 base pairs, respectively. GAPDH was used as a loading control. (B) A549 cells were treated with 100 µM of C2-ceramide, 0.5 µM of thapsigargin, or DMSO for the indicated time period (h), and a western blot using the indicated antibody was performed (40 μg of lysate/lane). The arrowheads indicate the positions of LC3-I and LC3-II. The left side of the panel shows the position of the molecular weight marker (in kilodaltons). GAPDH was used as a loading control. (C) Relative fold-induction of WIPI1 mRNA (black line marked with box), MAP1LC3B mRNA (black line marked with rhombus), and LC3-II protein (gray line marked with triangle) in A549 cells treated with thapsigargin for the indicated time period (h). Quantification was performed using Quantity One (Bio-Rad Laboratories), and the GAPDH level was used as an internal control.
Relationship between puncta formation and the transcription of ATG genes in HeLa cells
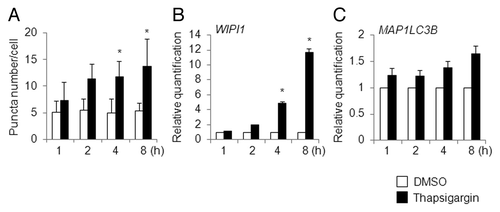
Next, we used HeLa/GFP-LC3B cells, instead of A549 cells, to count the number of puncta marked by GFP-LC3B under a fluorescent microscope during the time-course analysis, and the time-course changes in puncta numbers were compared with the time-course changes in the mRNA levels of select ATG genes, particularly that for WIPI1 mRNA. First of all, we checked the time-course changes in puncta number induced by thapsigargin. Based on the number of puncta per cell, 4 to 8 h of treatment with thapsigargin was sufficient to induce significant number of puncta, compared with DMSO-treated cells (). The puncta formation mediated by 8 h of treatment with thapsigargin was inhibited by cotreatment with 2 µg/mL of actinomycin D, an inhibitor of gene transcription (unpublished data). Similar to the results obtained in parental HeLa cells, where WIPI1 mRNA had significantly accumulated after 8 h treatment of thapsigargin (), WIPI1 mRNA had accumulated in the HeLa/GFP-LC3B cells at 4 to 8 h after the addition of thapsigargin (), and this time-dependent induction was well correlated with the results obtained using the puncta assay (). Contrary to the case observed for WIPI1 mRNA, the level of increased MAP1LC3B mRNA detected using quantitative real-time RT-PCR was minimal, probably because of the stable expression of MAP1LC3B in HeLa/GFP-LC3B cells (). Based on this observation, we used parental HeLa cells in place of HeLa/GFP-LC3B cells to detect mRNA level changes during the time-course of exposure to autophagy-inducible reagents other than thapsigargin, as described below.
Figure 6. Time-course analysis of puncta formation and WIPI1 and MAP1LC3B gene expression changes in thapsigargin-treated HeLa cells. (A) HeLa/GFP-LC3B cells were treated with DMSO (white bar) or 0.5 µM of thapsigargin (black bar) for the indicated time period (h) and the number of puncta per cell was counted. The bars indicate the mean ± SD of 128, 146, 246, 225, 200, 199, 121, and 126 cells, respectively (bars from left to right). HeLa/GFP-LC3B cells were treated with DMSO (white bar) or 0.5 µM of thapsigargin (black bar) for the indicated time period (h), and quantitative RT-PCR was performed to determine the WIPI1 (B) and MAP1LC3B (C) mRNA levels. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1. The GAPDH level was used as an internal standard. *P < 0.05 (5 areas for puncta formation assay and n = 2 for quantitative RT-PCR).
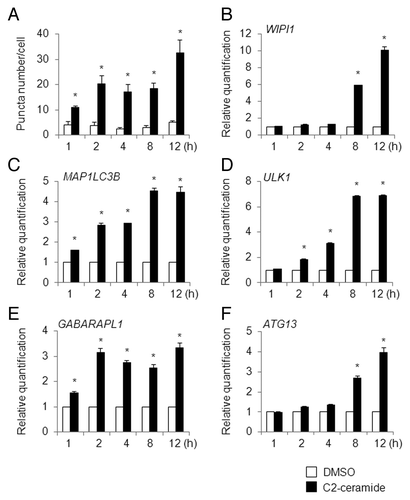
In C2-ceramide (100 µM)-treated HeLa/GFP-LC3B cells during a 12 h interval, the puncta number increased significantly after 1 h of treatment, and further increases in the puncta number were observed after 2 and 12 h of treatment, respectively (). In HeLa cells treated with C2-ceramide, time-dependent increments in the WIPI1, MAP1LC3B, ULK1, GABARAPL1, and ATG13 mRNA levels were clearly observed (); the WIPI1 and ATG13 mRNA levels increased rapidly after 8 h of treatment (), whereas the MAP1LC3B mRNA level increased in 3 stages after 1, 2, and 8 h of treatment (). The elevated mRNA levels for ULK1 and GABARAPL1 reached plateaus at 8 and 2 h, respectively (). The timing of WIPI1 mRNA accumulation in C2-ceramide-treated HeLa cells differed from that in A549 cells, where WIPI1 had accumulated after 4 h of treatment (), indicating that the timing of WIPI1 mRNA accumulation likely depends on the cell type.
Figure 7. Time-course analysis of puncta formation and ATG gene expression changes in C2-ceramide-treated HeLa cells. (A) HeLa/GFP-LC3B cells were treated with DMSO (white bar) or 100 µM of C2-ceramide (black bar) for the indicated time period (h) and the number of puncta per cell was counted. The bars indicate the mean ± SD of 129, 159, 127, 135, 128, 145, 148, 152, 167, and 109 cells, respectively (bars from left to right). HeLa cells were treated with DMSO (white bar) or 100 µM of C2-ceramide (black bar) for the indicated time period (h), and quantitative RT-PCR was performed to determine the WIPI1 (B), MAP1LC3B (C), ULK1 (D), GABARAPL1 (E), and ATG13 (F) mRNA levels. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1. The GAPDH level was used as an internal standard. *P < 0.05 (5 areas for puncta formation assay and n = 2 for quantitative RT-PCR).
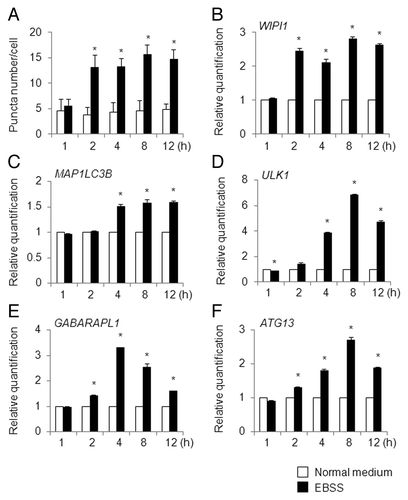
In EBSS-cultured HeLa/GFP-LC3B cells, the puncta number increased significantly after 2 h of treatment, and the increased puncta number was maintained until 12 h after treatment (; Table S2). In accordance with the time-dependent regulation of puncta formation, the WIPI1 and MAP1LC3B mRNA levels were elevated in HeLa cells after 2 and 4 h of EBSS culture, respectively, and the levels reached plateaus that persisted until the end of the time-course (). In contrast, the ULK1, GABARAPL1, and ATG13 mRNA levels increased in a time-dependent manner, but the mRNA levels decreased after 8 to 12 h of EBSS culture ().
Figure 8. Time-course analysis of puncta formation and ATG gene expression changes in EBSS-treated HeLa cells. (A) HeLa/GFP-LC3B cells were cultured in normal medium (white bar) or EBSS (black bar) for the indicated time period (h) and the number of puncta per cell was counted. The bars indicate the mean ± SD of 82, 105, 88, 99, 90, 99, 104, 111, 99, and 93 cells, respectively (bars from left to right). HeLa cells were cultured in normal medium (white bar) or EBSS (black bar) for the indicated time period (h), and quantitative RT-PCR was performed to determine the WIPI1 (B), MAP1LC3B (C), ULK1 (D), GABARAPL1 (E), and ATG13 (F) mRNA levels. The means ± SDs are shown as the relative fold-induction when the values obtained in cells cultured in normal medium were set as 1. The GAPDH level was used as an internal standard. *P < 0.05 (5 areas for puncta formation assay and n = 2 for quantitative RT-PCR).
Finally, rapamycin (250 nM) promptly increased the puncta formation in HeLa/GFP-LC3B cells after 1 h of treatment, and this increment reached a maximum at 4 to 8 h after treatment (). WIPI1 mRNA had modestly accumulated after 8 to 12 h of treatment with rapamycin (). In contrast to the results shown in , MAP1LC3B mRNA was weakly increased after 12 h of treatment with rapamycin (). Based on these results, we considered that WIPI1 mRNA, rather than MAP1LC3B mRNA, is more reliable for monitoring the induction of autophagy. Similar to the results for WIPI1, the time-course changes in the mRNA levels of ULK1, GABARAPL1, and ATG13 were reasonably enhanced in rapamycin-treated HeLa cells ().
Figure 9. Time-course analysis of puncta formation and ATG gene expression changes in rapamycin-treated HeLa cells. (A) HeLa/GFP-LC3B cells were treated with DMSO (white bar) or 250 nM of rapamycin (black bar) for the indicated time period (h) and the number of puncta per cell was counted. The bars indicate the mean ± SD of 100, 112, 101, 123, 95, 101, 102, 98, 100, and 110 cells, respectively (bars from left to right). HeLa cells were treated with 250 nM of DMSO (white bar) or rapamycin (black bar) for the indicated time period (h), and quantitative RT-PCR was performed to determine the WIPI1 (B), MAP1LC3B (C), ULK1 (D), GABARAPL1 (E), and ATG13 (F) mRNA levels. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO-treated cells were set as 1. The GAPDH level was used as an internal standard. *P < 0.05 (5 areas for puncta formation assay and n = 2 for quantitative RT-PCR).
Taken together, among the ATG genes that commonly accumulated in autophagy-induced cells, we decided to focus on WIPI1 mRNA because the extent of elevation in response to different autophagy inducers was the greatest.
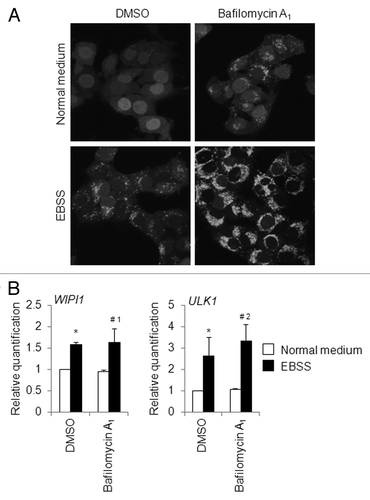
To further determine whether there are any changes in the WIPI1 mRNA level during autophagy flux, we induced autophagosome formation by culturing HeLa cells with EBSS for 4 h, while simultaneously cotreating with 25 nM of bafilomycin A1. We selected 4 h as the time interval for EBSS treatment based on results obtained using time-course analyses of puncta formation and the WIPI and ULK1 mRNA levels, as mentioned above. The puncta assay showed that bafilomycin A1 clearly inhibited the latter stage of autophagy, since the number of puncta per cell was significantly increased in the EBSS-plus-bafilomycin A1-treated HeLa/GFP-LC3B cells when compared with that obtained in the EBSS-treated HeLa/GFP-LC3B cells (). A quantitative real-time RT-PCR assay was then performed to detect any changes in the WIPI1 and ULK1 mRNA levels. We demonstrated that the WIPI1 mRNA level was almost unchanged in EBSS-treated HeLa cells with or without bafilomycin A1 treatment (, left panel). The ULK1 mRNA level was increased to some degree in HeLa cells cultured with EBSS and bafilomycin A1, compared with the level observed in HeLa cells cultured with EBSS and DMSO (, right panel). Collectively, these results suggest that the WIPI1 mRNA level seems to be a reliable indicator of autophagosome formation, meaning that the WIPI1 mRNA level is unlikely to represent an ostensible increase in the number of autophagosome. As we will discuss later, the accumulation of WIPI1 mRNA is thought to be part of the preparation for new protein synthesis as a backup, since WIPI1 protein is eventually degraded in autolysosomes. On the other hand, WIPI1 protein is marshaled to increase the number of autophagosomes upon bafilomycin A1 treatment, but the degradation cycle of WIPI1 protein is not permitted under such conditions.
Figure 10. Changes in WIPI1 mRNA levels during autophagy flux in HeLa cells. (A) HeLa/GFP-LC3B cells were cultured in normal medium or amino acid-free EBSS with DMSO or 25 nM of bafilomycin A1 for 4 h, and the puncta appearance was observed under a fluorescence microscope. (B) HeLa cells were cultured in normal medium or EBSS with or without 25 nM of bafilomycin A1 for 4 h, and quantitative real-time RT-PCR was performed to determine the WIPI1 and ULK1 mRNA levels. The means ± SDs are shown as the relative fold-induction when the values obtained in cells cultured in DMSO-treated normal medium were set as 1. The GAPDH level was used as an internal standard. *P < 0.05 (vs. DMSO-treated normal medium), #1no significant difference (vs. DMSO-treated EBSS), and #2P < 0.05 (vs. DMSO-treated EBSS), according to a 2-tailed Student t test (n = 6).
Reduction of WIPI1 mRNA level leads to the inhibition of puncta accumulation in HeLa cells
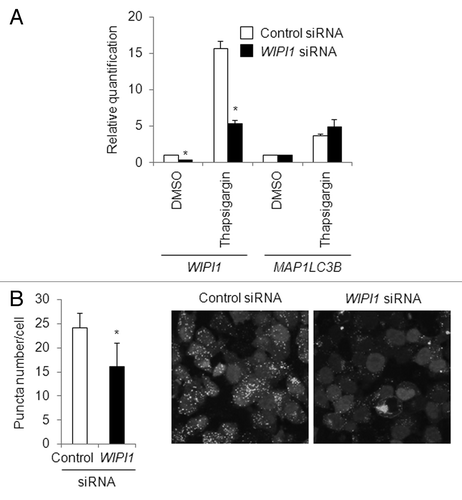
To further verify the importance of the mRNA abundance of WIPI1 in puncta formation, a WIPI1-specific short interference RNA (siRNA) was used to reduce the WIPI1 mRNA level. A quantitative real-time RT-PCR experiment demonstrated that WIPI1 siRNA clearly reduced the thapsigargin-induced mRNA abundance of WIPI1 as well as the endogenous mRNA expression level of WIPI1, whereas MAP1LC3B mRNA was unaffected by the WIPI1 siRNA in HeLa cells (). Next, we performed a puncta assay in control siRNA- or WIPI1 siRNA-introduced HeLa/GFP-LC3B cells. As expected, thapsigargin-induced puncta accumulation, as determined based on the increase in the puncta number, was significantly suppressed by the WIPI1 siRNA, compared with the control siRNA (). Collectively, these results indicated that the suppression of WIPI1 inhibited puncta accumulation.
Figure 11. Effect of WIPI1 transcriptional inhibition on puncta accumulation. (A) HeLa cells were transfected with control siRNA (white bar) or WIPI1 siRNA (black bar) and 24 h later, the cells were treated with DMSO or 0.5 µM of thapsigargin for 8 h. A quantitative real-time RT-PCR analysis was then performed to determine the WIPI1 and MAP1LC3B mRNA expression levels. GAPDH was used as an internal control. The means ± SDs are shown as the relative fold-induction when the values obtained in DMSO (control siRNA)-treated cells were set as 1. *P < 0.05 (n = 2). (B) Puncta formation assay in WIPI1 siRNA-introduced HeLa/GFP-LC3B cells treated with thapsigargin. HeLa/GFP-LC3B cells were transfected with control siRNA (white bar) or WIPI1 siRNA (black bar) and 24 h later, the cells were treated with 0.5 µM of thapsigargin for 12 h. A puncta assay was then performed. The bars indicate the mean ± SD of 417 and 491, respectively. *P < 0.05 (10 areas; two independent experiments). A representative microscopic view of the GFP-LC3B fluorescence observed is shown.
Changes in WIPI1 mRNA level upon the suppression of autophagosome formation
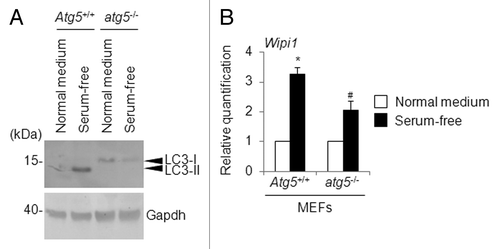
To check whether autophagy inhibition is directly associated with a lack of WIPI1 mRNA accumulation, we compared atg5-deficient mouse embryonic fibroblasts (atg5−/− MEFs) and their wild-type counterpart (Atg5+/+ MEFs).Citation30 We observed the accumulation of LC3-II in Atg5+/+ MEFs, but not in atg5−/− MEFs, after 4 h of serum starvation (). For the “serum starvation,” the MEFs were cultured in MEM without fetal bovine serum and nonessential amino acids. Using quantitative real-time RT-PCR, we also examined the Wipi1 mRNA levels in both Atg5+/+ MEFs and atg5−/− MEFs cultured under the above-mentioned conditions. Consequently, we detected an increase in the Wipi1 mRNA level in both serum-starved Atg5+/+ MEFs and atg5−/− MEFs, when compared with the levels obtained in MEFs cultured in a normal medium. Notably, the extent of the Wipi1 mRNA level was significantly suppressed in the serum-starved atg5−/− MEFs compared with that obtained in the serum-starved Atg5+/+ MEFs (). The reason why Wipi1 mRNA was still accumulated in the atg5−/− MEFs may be due to the existence of an Atg5-independent alternative autophagosome formation pathway.Citation31 Taken together, these results support the notion that WIPI1 mRNA could be useful as a functional indicator of autophagosome formation.
Figure 12. Changes in the Wipi1 mRNA level in atg5-deficient MEFs. (A) Atg5+/+ MEFs and atg5−/− MEFs were cultured in normal medium or serum-free medium for 4 h, and a western blot using the indicated antibody was performed (40 μg of lysates/lane). The arrowheads indicate the position of LC3-I and LC3-II. The left side of the panel indicates the position of the molecular weight marker (in kilodaltons). As a loading control, Gapdh was detected. (B) Atg5+/+ MEFs and atg5−/− MEFs were cultured in normal medium or serum-free medium for 4 h, and quantitative RT-PCR was performed to detect the Wipi1 mRNA level. The means ± SDs are shown as the relative fold-induction when the values obtained in cells cultured in normal medium were set as 1. The Gapdh level was used as an internal standard. *P < 0.05 (vs. Atg5+/+ MEFs cultured with normal medium) and #P < 0.05 (vs. atg5−/− MEFs cultured with normal medium and Atg5+/+ MEFs cultured with serum-free medium), according to a 2-tailed Student t test (n = 4).
Next we aimed to determine whether the accumulation of WIPI1 mRNA is perturbed upon the suppression of autophagosome formation in HeLa cells. Phosphoinositide 3-kinase (PI3K) and phosphatidylinositol 3-kinase (PtdIns3K) inhibitors, such as 3-methyladenine (3-MA) and wortmannin, have been widely used as autophagy inhibitors based on their inhibitory effect on class III PtdIns3K activity, which is known to be essential for the induction of autophagy. Although 3-MA has been widely used as an autophagy inhibitor in published literature, 3-MA has been shown to exert a dual role in autophagy, which means that the autophagy promotion activity of 3-MA arises from its differential temporal effects on class I PI3K and class III PtdIns3K; 3-MA blocks class I PI3K persistently, whereas its suppressive effect on class III PtdIns3K is transient.Citation32 Therefore, we selected wortmannin to inhibit autophagosome formation. First, HeLa/GFP-LC3B cells were cultured in normal medium or amino acid-free EBSS with DMSO or 50 nM of wortmannin for 4 h, and the number of puncta per cell was counted. As a result, the number of puncta induced by EBSS was significantly diminished by cotreatment with wortmannin (Fig. S8A). Next, we determined the WIPI1 and ULK1 mRNA level using quantitative real-time RT-PCR and revealed that the WIPI1 and ULK1 mRNA levels were almost unchanged between the EBSS-plus-DMSO-treated HeLa cells and the EBSS-plus-wortmannin-treated HeLa cells (Fig. S8B). Precisely, a statistically significant difference in the WIPI1 mRNA level was observed in EBSS-treated HeLa cells treated with DMSO or with wortmannin; however, the difference was minimal (Fig. S8B). Collectively, these results do not suggest any correlation between the WIPI1 mRNA level and the inhibition of autophagosome formation. We have no explanation for the discrepancy between the results obtained from serum-starved atg5−/− MEFs and EBSS-plus-wortmannin-treated HeLa cells.
Accumulation of WIPI1 and MAP1LC3B mRNAs in cultured cells other than HeLa and A549 cells
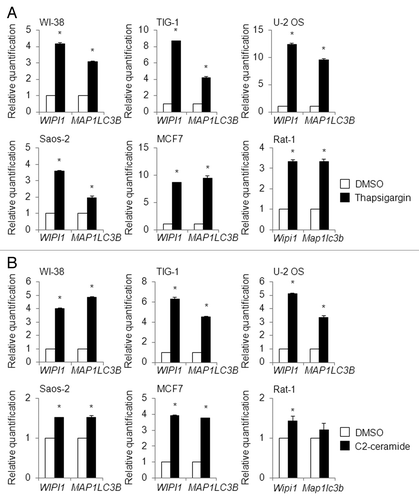
To expand our findings regarding the usefulness of WIPI1 mRNA as a beneficial biomarker for monitoring autophagosome formation, we also tested human cell lines other than HeLa and A549. For this purpose, we used 0.5 µM of thapsigargin or 100 µM of C2-ceramide to treat the human diploid fibroblasts WI-38 and TIG-1, the human osteosarcoma cells U-2 OS and Saos-2, and the human breast adenocarcinoma cell line MCF7 for 12 h; we then performed a quantitative real-time RT-PCR analysis to determine the MAP1LC3B as well as the WIPI1 mRNA expression level. Remarkably, the WIPI1 and MAP1LC3B mRNA expression levels were clearly upregulated by both thapsigargin and C2-ceramide in all the human cells that were examined (). In addition, we also tested whether the mRNA accumulation of WIPI1 and MAP1LC3B is a common phenomenon in other species-derived cell lines. For this purpose, rat fibroblasts (Rat-1) were subjected to thapsigargin or C2-ceramide, as used for the human cells. Notably, the rat Wipi1 mRNA level was also apparently elevated by both thapsigargin and C2-ceramide, whereas the rat Map1lc3b mRNA level was not significantly accumulated by C2-ceramide, compared with DMSO treatment (). We also treated 0.5 µM of thapsigargin with MEFs for 12 h and performed a quantitative RT-PCR experiment using TaqMan primers for Wipi1 (Mm00461219_m1, Mm00461220_m1, and Mm00461228_g1) and Map1lc3b (Mm00782868_sH). The mRNA levels of the 2 genes increased significantly, but the increase was within 2-fold (data not shown).
Figure 13.WIPI1 and MAP1LC3B mRNA levels in various cell lines. WI-38, TIG-1, U-2 OS, Saos-2, MCF7, and Rat-1 cells were treated with 0.5 µM of thapsigargin (A) (black bar), 100 µM of C2-ceramide (B) (black bar), or DMSO (white bar) for 12 h, and a quantitative RT-PCR analysis was performed to determine the WIPI1 and MAP1LC3B mRNA levels. The bars indicate the means ± SDs. *P < 0.05 (n = 2 and n = 4 for human cell lines and Rat-1 cells, respectively).
Next, we performed western blotting for LC3-I and/or LC3-II in thapsigargin- or C2-ceramide-treated cells. Unlike the phenomena observed for HeLa and A549 cells, LC3-II accumulation was not seen, but almost equal levels of LC3-I and/or LC3-II were detected in both the DMSO- and the thapsigargin-treated cells, while the accumulation of LC3-I was observed in the thapsigargin-treated MCF7 cells (Fig. S9A). On the other hand, the accumulation of LC3-II, but not a form shift from LC3-I to LC3-II, were apparently observed in C2-ceramide-treated WI-38, U-2 OS, and MCF7 cells, and modest accumulation was observed in Rat-1 cells (Fig. S9B). Overall, the detection of form shifts before and after thapsigargin or C2-ceramide treatment for the monitoring of autophagy seems to be difficult, though LC3-II accumulation is likely to be suitable for the confirmation of autophagy induction in some limited cases. Regarding LC3-I and/or LC3-II detection as an autophagy marker using western blotting, the basal level of LC3-I and/or LC3-II is not necessarily constant, even in the same cells (compare the DMSO-treated cells shown in Fig. S9A and S9B).
Taken together, these results suggest that the mRNA level of WIPI1 and also that of MAP1LC3B might be useful for detecting autophagosome formation in a wide variety of cells, including both normal and tumor cells, irrespective of the LC3-I and/or LC3-II status.
Discussion
In this study, we demonstrated that among ATG genes, the WIPI1 mRNA level could be used as a beneficial biomarker of autophagosome formation in certain cell types, including the widely used cell lines HeLa and A549, regardless of the chemical or physiological induction of autophagy. Generally, autophagy is considered to be tightly regulated at the post-translational level; however, as we demonstrated in this study, WIPI1 tends to accumulate at the mRNA level upon autophagosome formation to prepare for new protein synthesis, since WIPI1 protein is eventually degraded in autolysosomes. As a human orthologous gene of yeast Atg18, WIPI1 has been shown to tether to the autophagosome membrane in a manner similar to that of MAP1LC3.Citation33 Therefore, it is very intriguing that WIPI1 and MAP1LC3B, which are essential genes for autophagy, concomitantly accumulate at the transcriptional level during autophagosome formation, though as an exception, the MAP1LC3B mRNA level was unchanged in rapamycin-induced autophagy.
Besides WIPI1 and MAP1LC3B, we demonstrated that the time-course changes in ULK1, GABARAPL1, and ATG13 mRNA can be used to monitor autophagosome formation. To date, several ATG genes have been reported to accumulate transcriptionally or to be regulated, the details of which will be mentioned below. Indeed, BECN1 mRNA accumulates in response to C2-ceramide treatment (75 µM, 1 h) in MCF7 cells and HT-29 cells.Citation34 Importantly, the restoration of the autophagic capacity in MCF7 cells is exemplified by the gene transfer of BECN1.Citation35 Along with results obtained using Becn1 heterozygous mice,Citation15,Citation16 not only post-translational modifications but also transcriptional changes in ATG genes could be a rate-controlling step for certain cellular phenotypes such as autophagy. In this study, the BECN1 mRNA level was unchanged in the initial screening; however, it is plausible that a specific set of ATG genes could accumulate uniquely in response to different autophagy inducers and cell types. Among > 30 ATG genes, we demonstrated that WIPI1 mRNA is highly likely to be a common indicator of autophagy.
With regard to the mRNA accumulation of both WIPI1 and MAP1LC3B induced by resveratrol, it is interesting to consider how the concomitant accumulation of WIPI1 and MAP1LC3B mRNAs is responsible for the noncanonical autophagic degradation pathway. Under nutrient-free conditions, resveratrol has been shown to induce noncanonical autophagy and to inhibit the localization of WIPI1 protein to the phagophore membrane but to require the accumulation of LC3-II, which is derived from an LC3-II pool that is distinct from the membranes of canonical autophagic phagophores.Citation36
The spatiotemporal post-translational regulation of MAP1LC3 renders the shift in form from LC3-I to LC3-II enigmatic, since LC3-II is subjected to degradation when the autophagosome fuses with a lysosome. Of concern, the detection of the shift in form from LC3-I to LC3-II using western blotting seems to be inconsistent, since the ratio of autophagosome formation to autolysosome formation differs in each cell and/or under different conditions.Citation10 Also, the differential appearance in the shift in form could be affected by the endogenous levels of MAP1LC3. Taken together, our results suggest that the monitoring of the mRNA levels of WIPI1 and/or MAP1LC3B instead of or together with the LC3-I to LC3-II ratio might be a straightforward approach to determining whether autophagosome formation actually occurs in some cell lines.
The depletion of amino acids and glucose from HeLa cells by incubating them in Hank’s balanced salt solution (HBSS) demonstrates that LC3-II accumulation had occurred at 24 h.Citation37 In the present study, EBSS culture increased the puncta formation and elevated the WIPI and MAP1LC3B mRNA levels within 4 h in HeLa cells. Collectively, these results suggest that similar to the effects of thapsigargin and C2-ceramide, the physiological induction of autophagosome formation is accompanied by the accumulation of WIPI and MAP1LC3B mRNA. On the other hand, rapamycin (100 nM) has been shown to induce the accumulation of LC3-II after 6 h of treatment, and this accumulation reached a maximum after 15 h of treatment in HeLa cells.Citation37 Recently, low-dose rapamycin (500 nM), but not high-dose rapamycin (30 mM), has been shown to induce autophagic flux after 24 h treatment in HeLa cells.Citation38 In the present study, we demonstrated that autophagosome formation occurred after 1 h of treatment with rapamycin (250 nM) but that the extent of the mRNA accumulation of WIPI1 and MAP1LC3B was limited, compared with that caused by other autophagy inducers. Taken together, these results indicate that in contrast to the other autophagy inducers used in this study, rapamycin seems to induce autophagy through a different mechanism or be a poor inducer of autophagy. This point is discussed in further detail below.
Some reports describing the transcriptional regulation of ATG genes during autophagy have been published, but in many cases, the evidence originates from global expression analyses, such as DNA microarrays.Citation39 It is important to consider the kinds of signal cascades or transcription factors that are involved in the regulation of WIPI1 and MAP1LC3B. Recently, Ca2+-dependent signaling, including that induced by calmodulin-dependent kinase (CaMK) I, has been shown to promote WIPI1-positive puncta formation.Citation40 In one important report, FoxO (Forkhead box transcription factor class O) is identified as the first transcription factor to be involved in the regulation of autophagy in the Drosophila larval fat body.Citation41 FOXO3 has also been shown to induce autophagy in mouse skeletal muscle cells, along with the transcriptional induction of the selected ATG genes.Citation42,Citation43 Based on these findings, 2 mechanisms, transcriptional and nontranscriptional mechanisms, for the regulation of autophagy have been modeled.Citation43 Briefly, upon a reduction in the activity of PI3K-AKT signaling, autophagy is rapidly induced through MTOR at a nontranscriptional level and is slowly induced by FOXO3 at a transcriptional level.Citation43 Indeed, MTOR is a kinase downstream of AKT that can inhibit autophagy by enhancing protein synthesis.Citation44 In line with this scenario, rapamycin had a minimal effect on the transcriptional regulation of ATG genes, compared with the observed effects for thapsigargin, C2-ceramide, and EBSS, in the present study. Of note, the transcriptional mechanism regulated by FOXO3 for autophagy was shown to be more predominant than nontranscriptional mechanisms regulated by MTOR in skeletal muscle, since rapamycin failed to induce autophagy.Citation45 Moreover, the importance of FOXO transcription factors in the regulation of autophagy has been well documented.Citation46,Citation47 For instance, the regulation of Atg14 by FOXO transcription factors has been shown in mouse hepatocytes.Citation48
Based on the present findings and others, the transcriptional regulation of a specific set of ATG genes seems to provide a unique platform for elucidating basal and inducible autophagy in a variety of cell fate determinations, especially for the regulation of cell-type-specific autophagy. Indeed, besides FOXO transcription factors, several transcription factors, including E2F1, TP53/p53, transcription factor EB (TFEB), and GATA1, have been shown to be involved in the regulation of selected ATG genes.Citation49-Citation54 On the other hand, E2F1 lacking a transcriptional activity domain is sufficient to induce autophagosome formation.Citation55 In addition, JUNB/Junb has been shown to suppress starvation-induced autophagy in an MTOR-independent manner.Citation56 The rapidly growing number of transcription factors that are involved in the regulation of autophagy strongly supports the concept that the transcriptional regulation of specific ATG genes may be a fundamental tool for detecting autophagy induction that could probably be applied to the monitoring of autophagy flux, though additional studies are needed. Interestingly, CEBPB/C/EBPβ regulates the rhythmic expression of specific mouse Atg genes, including Ulk1, Map1lc3b, and Gabarapl1, in mouse liver.Citation57 Recently, spermidine, a kind of polyamine, has been shown to extend the life spans of a series of model organisms via the induction of autophagy.Citation58 Uniquely, many yeast ATG genes, including Atg8 (human MAP1LC3B ortholog) and Atg18 (human WIPI1 ortholog), can be transcriptionally upregulated, potentially via the modification of the histone acetylation status, during chronological aging upon treatment with spermidine, although the extent of the differential mRNA expression level was minimal.Citation58 Overall, among the selected ATG genes, the transcriptional regulation of yeast Atg8 orthologs, such as MAP1LC3B, seems to be key, suggesting that WIPI1 as well as MAP1LC3B mRNAs per se could be rate-limiting factors for the transcriptional regulation of autophagy, thus providing unique indicators for autophagosome formation. In the course of the review process, WIPI1 and MAP1LC3B have been shown to be epigenetically repressed by the histone methyltransferase G9a (EHMT2), and these 2 genes were transcriptionally activated in autophagy-induced naïve T cells under physiological conditions.Citation59 Finally, the transcriptional repressor ZKSCAN3 has been recently reported to suppress a large set of genes, particularly MAP1LC3B, involved in various steps of the autophagy process in several cell lines, including HeLa cells.Citation60 Aside from MAP1LC3B, WIPI2, ULK1, and GABARAPL2 were also identified as direct targets of ZKSCAN3.Citation60
The relationship between the transcriptional and nontranscriptional regulation of autophagy should be elucidated, particularly for the transcriptional regulation of WIPI1 and MAP1LC3B mRNAs. Considering that orchestrating transcription factors are likely involved in the regulation of autophagy in a variety of cells, the nontranscriptional regulation of autophagy, such as MTOR-based regulation, is likely to be integrated into the transcriptional regulation of autophagy in a feedback manner. Nevertheless, special attention is needed when discriminating the autophagy route as transcriptional or nontranscriptional. We investigated the possibility of the transcriptional and nontranscriptional regulation of autophagy by assessing the phosphorylation level at Thr389 in RPS6KB, which is a well-characterized target of MTOR.Citation61 Actually, rapamycin and EBSS, which showed a relatively weaker effect on the expression of ATG genes, suppressed the phosphorylation level at Thr389 in RPS6KB, whereas thapsigargin and C2-ceramide, which were considered to be potent inducers of the expression of ATG genes based on our present results, had little effect on the phosphorylation level at Thr389 in RPS6KB, compared with that obtained in mock-treated HeLa cells (Fig. S10).
In contradiction to these 2 route mechanisms, the MTOR complex 1 has been recently shown to control a transcriptional program for autophagy by directly regulating the subcellular localization and thus the activity of TFEB.Citation62-Citation64 Out of the 51 ATG genes that were analyzed, several ATG genes, including WIPI (not specifically identified as WIPI1 or one of 3 other WIPIs) and MAP1LC3B, were transcriptionally upregulated in both HeLa cells that had been starved and TFEB-overexpressing HeLa cells, but these identified ATG genes were transcriptionally unchanged in HeLa cells that had been treated with rapamycin (0.25 mg/mL, 4 h).Citation53 The extent of the differential mRNA expression levels in the HeLa cells induced by starvation or TFEB overexpression was modest and was comparable to the results obtained using EBSS treatment in this study.Citation53
Recently, transcription factors involved in the regulation of the ER stress response have been shown to be required for the direct control of MAP1LC3B and ATG5 gene promoters, and this regulation was thought to be responsible for the protective effect on cancer cells subjected to hypoxic stress.Citation65 Interestingly, the induction of MAP1LC3B mRNA is reportedly required for the replenishment of MAP1LC3B protein, since MAP1LC3B protein undergoes rapid turnover during hypoxia-induced autophagy. Moreover, regarding the relationship between ER stress and autophagy, MAP1LC3B gene regulation mediated by ATF4 seems to be a key factor. For instance, Bortezomib, an inhibitor of the 26S proteasome, induces an unfolded protein response (UPR) to cope with ER stress in addition to inducing autophagy in MCF7 cells; this process resulted from MAP1LC3B mRNA induced by stabilized ATF4 protein that had escaped from degradation in the proteasome, consequently leading to the bypass of apoptosis.Citation66 Moreover, ATF4 regulates the MAP1LC3B gene promoter directly.Citation67 As mentioned above, in some human cancer cells, hypoxia induced MAP1LC3B and ATG5 mRNAs, and this phenomenon was found to be regulated by the transcription factors ATF4 and DDIT3/CHOP, the functions of which are governed by the primary UPR sensor EIF2AK3/PERK.Citation65 Recently, a subset of ATG genes has been shown to be transcriptionally upregulated by leucine starvation in MEFs, and UPR genes, such as ATF4 and DDIT3, have been shown to be involved in the transcriptional regulation of a subset of ATG genes.Citation68 ATF4 also upregulates the transcription of the ULK1 gene.Citation69 Because UPR genes form a gene regulatory network,Citation70 an investigation of how these UPR genes govern autophagy as well as ER stress, especially with regard to the regulation of ATG genes, would likely be interesting.
In conclusion, this report describes the importance of the transcriptional regulation of several ATG genes during autophagosome formation, and changes in the WIPI1 mRNA level could potentially be used as a key biomarker for monitoring autophagosome formation in some cell types. The molecular mechanisms underlying complex cellular systems, such as autophagy, ER stress, and apoptosis, can likely be revealed by further investigation of the transcriptional regulation of ATG genes.
Materials and Methods
Cells
Human cervical carcinoma (HeLa) cells (BioResource Center, RCB0007, RIKEN Tsukuba Institute), human lung carcinoma A549 cells (Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer, TKG0184, Tohoku University), human breast adenocarcinoma MCF7 cells (BioResource Center, RCB1904, RIKEN Tsukuba Institute), and the human normal fibroblast cell lines WI-38 (Health Science Research Resources Bank, IFO50075) and TIG-1 (Health Science Research Resources Bank, PDL 20, JCRB0501), and Atg5+/+ MEFs and atg5−/− MEFs (BioResource Center, RCB2710 and RCB2711, respectively, RIKEN Tsukuba Institute), which were obtained with the prior approval from Dr Noboru Mizushima (University of Tokyo/Tokyo Medical and Dental University), were cultured in minimum essential medium (MEM; Life Technologies, 11095), while the human osteosarcoma cell lines U-2 OS (DS Pharma Biomedical, EC92022711) and Saos-2 (Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer, TKG0469, Tohoku University) were cultured in McCoy’s 5A medium (Life Technologies, 16600) and Rat-1 fibroblasts (BioResource Center, RCB1830, RIKEN Tsukuba Institute) were cultured in Dulbecco’s modified Eagle medium (Life Technologies, 11995). These media were supplemented with 10% fetal bovine serum, 1% nonessential amino acids (Life Technologies, 11140), and antibiotic-antimycotics (Life Technologies, 15240) in 5% CO2 at 37 °C. HeLa cells stably expressing GFP-tagged MAP1LC3B protein (HeLa/GFP-LC3B) were kindly provided by Dr Noboru Mizushima.
Reagents
Thapsigargin (Sigma-Aldrich, T9033), C2-ceramide (N-acetyl-D-erythro-sphingosine; Merck KGaA, 110145), rapamycin (Merck KGaA, 553210), tunicamycin (Sigma-Aldrich, T7765), trans-resveratrol (Cayman Chemical, 70675), bafilomycin A1 (Sigma-Aldrich, B1793), and wortmannin (Merck KGaA, 681675) were dissolved in dimethyl sulfoxide (Wako Pure Chemical Industries, DMSO) and stored at −30 °C before use. Chloroquine (Sigma-Aldrich, C6628) was dissolved in distilled water. As an amino acid-free medium, Earle’s balanced salt solution (EBSS; Sigma-Aldrich, E2888) was used.
RT-PCR
The total RNA (500 ng) was treated with deoxyribonuclease I (Life Technologies, 18068-015) and was reverse transcribed using High-Capacity cDNA Reverse Transcription kit (Life Technologies), in accordance with the manufacturer’s instructions. The PCR mixtures included the TaqMan Gene Expression Assay primer (Table S1), TaqMan Universal PCR Master Mix, and synthesized cDNA (1/20) in a total reaction volume of 20 μL. The reactions were performed using the 7500 standard program on a 7500 Fast Real-Time PCR System (Life Technologies). The cycling parameters were 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and annealing/extension at 60 °C for 1 min. The cycle threshold (Ct) values, corresponding to the PCR cycle number at which the fluorescence emission reaches a threshold above the baseline emission, were determined, and the mRNA expression values were calculated using GAPDH as an endogenous control according to the comparative Ct (ΔΔCt) method. Basically, all the reactions were performed at least in duplicate (n = 2).
Puncta formation assay
Cells were fixed using a 4% paraformaldehyde-phosphate buffer solution (Wako Pure Chemicals) and were encapsulated using SlowFade Gold Antifade Reagent (Life Technologies). Fluorescence was detected using confocal scanning laser microscopy (IX80; Olympus) equipped with a FV1000 system and a UPlanSApo (×60) oil immersion objective lens (Olympus). The images were obtained using a CCD camera (Olympus) and were analyzed using an FV10-ASW 3.1 viewer software. Basically, the puncta were counted in five areas per slide (n = 5), and the average number of puncta per cell was calculated. The puncta counts were confirmed by 2 readers.
Western blotting
The cells were harvested and lysed in modified RIPA lysis buffer (50 mM TRIS-HCl [pH7.5], 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 1 mM EDTA) containing a protease inhibitor cocktail (Nacalai Tesque, 25955-11) for 20 min on ice. The cell lysates were centrifuged, and the protein concentrations were determined using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories). Before performing sodium dodecyl sulfate PAGE, the reaction was stopped by the addition of Laemmli sample buffer containing 100 mM of dithiothreitol. Equal amounts of cellular protein were electrophoresed on NuPAGE 4–12% Bis-Tris gel using MES running buffer (Life Technologies) and were then transferred to a Hybond-PVDF membrane (GE Healthcare, RPN303N). The membrane was first blocked using phosphate-buffered saline containing 0.1% Tween 20 and 5% non-fat dried milk and was then incubated with antibodies for LC3-I and LC3-II (Medical & Biological Laboratories, M115-3) and GAPDH (Life Technologies, AM4300). Alkaline phosphatase-labeled secondary antibody (Promega, S3721) and a Western Blue-stabilized substrate (Promega, S3841) were used to detect the signals, according to the manufacturer’s protocol.
RNA interference
Silencer select predesign siRNA for WIPI1 (Life Technologies, ID: s30083) and negative control siRNA with a high GC (Life Technologies) were transfected into the cells using Lipofectamine 2000 (Life Technologies, 11668), according to the manufacturer’s protocol. Briefly, 125 pmol of RNA and 2.5 μL of transfection reagent were incubated in 0.5 mL of Opti-MEM I reduced serum medium (Life Technologies, 31985) for 15 min to facilitate complex formation at room temperature. The resulting mixture was added to the cells cultured in 1.5 mL of MEM in a 6-well plate. After 24 h, the RNA/transfection reagent mixture was readded.
Statistical Analysis
Statistical difference was determined by 2-tailed Student t test. P < 0.05 was considered as a statistical difference.
| Abbreviations: | ||
| ATG | = | autophagy-related |
| EBSS | = | Earle’s balanced salt solution |
| ER | = | endoplasmic reticulum |
| GFP | = | green fluorescent protein |
| LC3-I | = | cytoplasmic-liberated form of MAP1LC3 |
| LC3-II | = | phagophore and autophagosome membrane-bound form of MAP1LC3 |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| RT-PCR | = | reverse transcription-polymerase chain reaction |
| siRNA | = | small interfering RNA |
| UPR | = | unfolded protein response |
| WIPI | = | WD repeat protein domain, phosphoinositide interacting |
Additional material
Download Zip (880.3 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed. This work was supported in part by a Research Project Grant (2009–2011) from the Institute of Science and Technology, Meiji University, and by a grant (2010–2012) from the Organization for the Strategic Coordination of Research and Intellectual Property, Meiji University.
Acknowledgments
This work was supported in part by a Research Project Grant (2009–2011) from the Institute of Science and Technology, Meiji University, and by a grant (2010–2012) from the Organization for the Strategic Coordination of Research and Intellectual Property, Meiji University.
References
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Yoshimori T, Noda T. Toward unraveling membrane biogenesis in mammalian autophagy. Curr Opin Cell Biol 2008; 20:401 - 7; http://dx.doi.org/10.1016/j.ceb.2008.03.010; PMID: 18472412
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Hamasaki M, Yoshimori T. Where do they come from? Insights into autophagosome formation. FEBS Lett 2010; 584:1296 - 301; http://dx.doi.org/10.1016/j.febslet.2010.02.061; PMID: 20188731
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol 2012; 22:R29 - 34; http://dx.doi.org/10.1016/j.cub.2011.11.034; PMID: 22240478
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117 - 24; http://dx.doi.org/10.1002/path.2694; PMID: 20225337
- Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 2013; 23:310 - 22; http://dx.doi.org/10.1016/j.semcancer.2013.05.008; PMID: 23726895
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973 - 9; http://dx.doi.org/10.4161/auto.5.7.9296; PMID: 19597335
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220 - 31; http://dx.doi.org/10.1128/MCB.01453-06; PMID: 17030611
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem 2008; 283:15370 - 80; http://dx.doi.org/10.1074/jbc.M710209200; PMID: 18356160
- Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14:1576 - 82; http://dx.doi.org/10.1038/sj.cdd.4402200; PMID: 17612585
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282:4702 - 10; http://dx.doi.org/10.1074/jbc.M609267200; PMID: 17135238
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 9; http://dx.doi.org/10.1038/sj.cdd.4401984; PMID: 16794605
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731 - 43; http://dx.doi.org/10.1016/j.molcel.2011.04.024; PMID: 21700220
- Engedal N, Torgersen ML, Guldvik IJ, Barfeld SJ, Bakula D, Sætre F, Hagen LK, Patterson JB, Proikas-Cezanne T, Seglen PO, et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013; 9:1475 - 90; http://dx.doi.org/10.4161/auto.25900; PMID: 23970164
- Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7:188 - 204; http://dx.doi.org/10.4161/auto.7.2.14181; PMID: 21107021
- Jiang W, Ogretmen B. Autophagy paradox and ceramide. [Epub ahead of print] Biochim Biophys Acta 2013; http://dx.doi.org/10.1016/j.bbalip.2013.09.005; PMID: 24055889
- Gibbons JJ, Abraham RT, Yu K. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol 2009; 36:Suppl 3 S3 - 17; http://dx.doi.org/10.1053/j.seminoncol.2009.10.011; PMID: 19963098
- Delmas D, Solary E, Latruffe N. Resveratrol, a phytochemical inducer of multiple cell death pathways: apoptosis, autophagy and mitotic catastrophe. Curr Med Chem 2011; 18:1100 - 21; http://dx.doi.org/10.2174/092986711795029708; PMID: 21291372
- Kusama Y, Sato K, Kimura N, Mitamura J, Ohdaira H, Yoshida K. Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 2009; 14:1165 - 75; http://dx.doi.org/10.1007/s10495-009-0390-2; PMID: 19657746
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850 - 61; http://dx.doi.org/10.1074/jbc.M109.080796; PMID: 20123989
- Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett 2007; 581:3396 - 404; http://dx.doi.org/10.1016/j.febslet.2007.06.040; PMID: 17618624
- Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 2004; 279:18384 - 91; http://dx.doi.org/10.1074/jbc.M313561200; PMID: 14970205
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, Proikas-Cezanne T. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy 2011; 7:1448 - 61; http://dx.doi.org/10.4161/auto.7.12.17802; PMID: 22082875
- Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010; 6:1125 - 38; http://dx.doi.org/10.4161/auto.6.8.13547; PMID: 20930550
- Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, Chen Y, Deng X, Xia B, Lin J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8:1215 - 26; http://dx.doi.org/10.4161/auto.20284; PMID: 22647982
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol Pharmacol 2011; 80:1066 - 75; http://dx.doi.org/10.1124/mol.111.071761; PMID: 21896713
- Juhász G, Puskás LG, Komonyi O, Erdi B, Maróy P, Neufeld TP, Sass M. Gene expression profiling identifies FKBP39 as an inhibitor of autophagy in larval Drosophila fat body. Cell Death Differ 2007; 14:1181 - 90; http://dx.doi.org/10.1038/sj.cdd.4402123; PMID: 17363962
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458 - 71; http://dx.doi.org/10.1016/j.cmet.2007.11.001; PMID: 18054315
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472 - 83; http://dx.doi.org/10.1016/j.cmet.2007.11.004; PMID: 18054316
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 2008; 4:524 - 6; PMID: 18367868
- Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008; 20:126 - 36; http://dx.doi.org/10.1016/j.ceb.2008.02.005; PMID: 18394876
- Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011; 25:310 - 22; http://dx.doi.org/10.1101/gad.1984311; PMID: 21325132
- Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem 2012; 287:39107 - 14; http://dx.doi.org/10.1074/jbc.M112.412569; PMID: 22992773
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860 - 4; http://dx.doi.org/10.1038/onc.2008.117; PMID: 18408756
- Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27:6229 - 42; http://dx.doi.org/10.1128/MCB.02246-06; PMID: 17576813
- Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009; 8:1571 - 6; http://dx.doi.org/10.4161/cc.8.10.8498; PMID: 19377293
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181 - 5; http://dx.doi.org/10.1016/j.ceb.2009.12.001; PMID: 20044243
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429 - 33; http://dx.doi.org/10.1126/science.1204592; PMID: 21617040
- Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, Farnham PJ, Keles S, Bresnick EH. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 2012; 32:226 - 39; http://dx.doi.org/10.1128/MCB.06166-11; PMID: 22025678
- Garcia-Garcia A, Rodriguez-Rocha H, Tseng MT, Montes de Oca-Luna R, Zhou HS, McMasters KM, Gomez-Gutierrez JG. E2F-1 lacking the transcriptional activity domain induces autophagy. Cancer Biol Ther 2012; 13:1091 - 101; http://dx.doi.org/10.4161/cbt.21143; PMID: 22825328
- Yogev O, Goldberg R, Anzi S, Yogev O, Shaulian E. Jun proteins are starvation-regulated inhibitors of autophagy. Cancer Res 2010; 70:2318 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-09-3408; PMID: 20197466
- Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J 2011; 30:4642 - 51; http://dx.doi.org/10.1038/emboj.2011.322; PMID: 21897364
- Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11:1305 - 14; http://dx.doi.org/10.1038/ncb1975; PMID: 19801973
- Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, Herreros-Villanueva M, Gress TM, Ellenrieder V, Bujanda L, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 2013; 33:3983 - 93; http://dx.doi.org/10.1128/MCB.00813-13; PMID: 23918802
- Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, Boyd DD. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell 2013; 50:16 - 28; http://dx.doi.org/10.1016/j.molcel.2013.01.024; PMID: 23434374
- Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 2012; 441:1 - 21; http://dx.doi.org/10.1042/BJ20110892; PMID: 22168436
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903 - 14; http://dx.doi.org/10.4161/auto.19653; PMID: 22576015
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; http://dx.doi.org/10.1126/scisignal.2002790; PMID: 22692423
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095 - 108; http://dx.doi.org/10.1038/emboj.2012.32; PMID: 22343943
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127 - 41; http://dx.doi.org/10.1172/JCI40027; PMID: 20038797
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-08-2839; PMID: 19417138
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29:4424 - 35; http://dx.doi.org/10.1038/onc.2010.191; PMID: 20514020
- B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683 - 99; http://dx.doi.org/10.1093/nar/gkt563; PMID: 23804767
- Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, Li JL, Simon AK, Murray JT, Harris AL. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J 2013; 449:389 - 400; http://dx.doi.org/10.1042/BJ20120972; PMID: 23078367
- Takayanagi S, Fukuda R, Takeuchi Y, Tsukada S, Yoshida K. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones 2013; 18:11 - 23; http://dx.doi.org/10.1007/s12192-012-0351-5; PMID: 22802018