
Abstract
Autophagy maintains cellular homeostasis by sequestering unwanted material within autophagosomes and transferring these to lysosomes for degradation. Several signaling cascades activate or suppress autophagy in response to diverse environmental cues. However, whether autophagic structures per se regulate cell signaling was not known. The MAPK/ERK (mitogen-activated protein kinase) pathway controls several functions in the cell, and studies have identified the importance of scaffold proteins in modulating MAPK signaling through the spatial coordination of the RAF1-MAP2K/MEK-MAPK cascade. Growth factors increase the nuclear localization and activity of MAPK, and since the nucleus has been reported to contain LC3, an autophagy-related protein, we asked whether autophagic structures could serve as cytosolic and nuclear scaffolds for growth factor-induced MAPK phosphorylation.
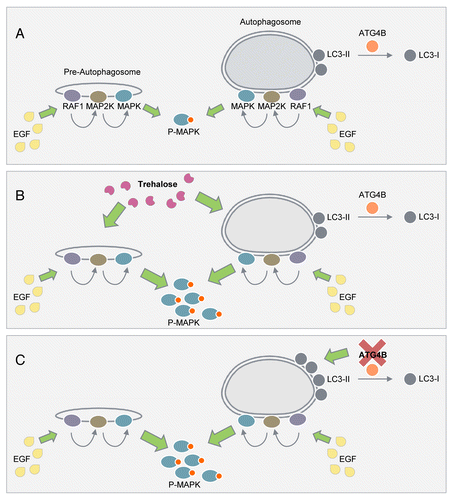
To determine the role of ATG (autophagy-related) proteins in regulating MAPK phosphorylation, we first investigated whether exposure of EGF (epidermal growth factor) increased association of the MAPK signaling components with ATGs. Much to our surprise, we observed that EGF stimulation increases the colocalization of phosphorylated (P)-RAF1, P-MAP2K, and P-MAPK with LC3-II in different cell types. EGF exposure also increases the colocalization of P-MAPK with phagophore markers ATG12–ATG5 and ATG16L1, with ATG8 family proteins LC3-II and GABARAP, and with the PtdIns3P effectors WIPI1/2, but not with upstream autophagy activators PIK3C3/VPS34 and P-ULK1. Associations between ATGs and MAPK signaling components could reflect 2 scenarios: i) MAPK cascade components are substrates degraded via autophagy, or ii) autophagic structures are scaffolds for MAPK signaling. Hence, we sought to distinguish whether RAF1, MAP2K, and MAPK were sequestered by autophagosomes as a means to terminate MAPK signaling, or whether MAPK localized at the external aspect of autophagosomes (). To determine the topology of MAPK’s association with autophagosomes, we used the LC3-II protease protection assay, which allows one to distinguish whether proteins are sequestered within autophagosomes or localized on the surface of autophagosomes. Exposing isolated autophagosomes to trypsin decreases autophagosome-associated P- and total MAP2K and MAPK while the levels of LC3-II are affected partially, indicating that MAP2K and MAPK are present on the extra-lumenal surface of autophagosomes. Furthermore, increased nuclear colocalization of P-MAPK with LC3-II is observed in EGF-treated cells, which is validated by the presence of P-MAPK and MAPK in LC3 affinity isolates from purified nuclear fractions. Intriguingly, immunofluorescence studies reveal increased colocalization of LC3-II with the MAPK scaffold complex protein KSR1 (kinase suppressor of ras 1) in response to EGF. Considering the localization of MAP2K and MAPK at the cytosolic face of autophagosomes and that LC3-II colocalizes with KSR1 raised the possibility that autophagic structures serve as transient cellular platforms for MAP2K-mediated MAPK phosphorylation. If this is true, then blocking autophagosome formation should blunt MAPK phosphorylation without affecting MAP2K phosphorylation. Indeed, liver-specific and brown adipose tissue-specific atg7−/− mice display reduced MAPK phosphorylation in cytosolic and nuclear fractions without modifying MAP2K phosphorylation. Nutrient-deprived atg5−/− mouse embryonic fibroblasts also exhibit decreased phosphorylation of MAPK, and MAPK’s downstream cytosolic and nuclear targets, RPS6KA1/p90RSK and ELK1, respectively. Loss of autophagy has no effect on STAT3, MAPK/JNK, MTOR, or ULK1 phosphorylation demonstrating the specificity of the role of ATGs in modulating MAPK phosphorylation. This result led to several new questions, for instance, was reduced MAPK phosphorylation in atg7−/− tissues secondary to possible developmental changes due to the early loss of autophagy, or was decreased MAPK phosphorylation a consequence of accumulation and/or hyperactivation of phosphatases? Indeed, transiently silencing key ATGs, ATG7, ATG5, or BECN1, or blocking autophagosome formation by expressing the lipidation-deficient LC3ΔG mutant each reduce MAPK phosphorylation. Furthermore, silencing MAPK phosphatases PPP2/PP2A and DUSP6/MKP-3 does not rescue MAPK phosphorylation in atg5−/− cells, demonstrating a direct role of ATGs in regulating MAPK phosphorylation. Conversely, increasing cellular LC3-II content by silencing ATG4B, which recycles LC3-II into cytosolic LC3-I, or exposing cells to the autophagy activator trehalose, increases MAPK phosphorylation. In fact, trehalose-induced MAPK phosphorylation is not detected in atg5−/− cells demonstrating the critical role of ATGs in regulating MAPK phosphorylation.
Figure 1. An unconventional function of autophagic structures in the regulation of MAPK phosphorylation. (A) We propose that phagophores and autophagosomes serve as cellular signaling platforms that allow the RAF1-MAP2K-MAPK signaling cascade to dock on the surface to facilitate EGF-induced MAPK phosphorylation. (B) Acute exposure to trehalose that increases LC3-II content and autophagy without interfering with the MTOR pathway increases MAPK phosphorylation. (C) Increasing LC3-II availability by silencing ATG4B, which decreases LC3-II recycling, associates with increased MAPK phosphorylation.
MAPK1/ERK2 interacts with its substrates via its kinase-docking domains, which include the F-site recruitment sites (FRS) (Leu198, Tyr231/Leu232, Leu235 and Tyr261). Since mutations in these MAPK1 domains have been reported to decrease MAPK phosphorylation, we asked whether these mutants display reduced colocalization with ATGs, which in turn, leads to decreased MAPK phosphorylation. Indeed, L198A and L198A/L232A MAPK1 mutants display reduced colocalization with ATGs, while Y261A MAPK1 mutants show decreased colocalization with WIPI1, but increased colocalization with ATG12–ATG5 and WIPI2. Conversely, analyses of ATG12 and LC3B revealed the presence of D-domains that are found on MAPK substrates to interact with MAPK. Interestingly, ATG5 does not display D-domains, and thus it is likely that MAPK interacts with the ATG12–ATG5 conjugate because blocking ATG12–ATG5 conjugation blocks EGF-induced MAPK phosphorylation. Although it remains unclear why different MAPK1 mutations differentially modify MAPK1’s interaction with ATGs, these results suggest the possibility that different ATGs coordinately associate with MAP2K and MAPK to “fine-tune” MAPK phosphorylation during EGF stimulation. How blocking autophagy abrogates MAPK signaling is not known, and it needs to be determined whether blocking autophagosome formation disrupts MAP2K1/MEK1-MAPK interaction. It would require additional studies to identify the molecular signature of the cytosolic face of autophagosomes that facilitates MAPK recruitment. Furthermore, it needs to be determined whether phagophores and autophagosomes display different affinities to bind to MAPK, and whether changes in the molecular characteristics of the external aspect of autophagic structures during different stages in the autophagy process per se could differentially regulate MAPK signaling.
Our findings allow us to present a conceptual framework for considering a new unconventional function of phagophores and autophagosomes in the regulation of MAPK phosphorylation. We propose a simple model () wherein autophagic structures serve as cytosolic and nuclear signaling platforms for RAF1-MAP2K-MAPK to transiently dock to acquire the appropriate spatial coordination necessary for MAPK phosphorylation. Understanding how ATGs regulate MAPK signaling may help in the prevention of certain diseases, for instance, hepatocellular cancers that originate from inappropriate MAPK activity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Our laboratory is supported by NIH/NIA, NIH/NIDDK grants AG043517 and DK087776, and an Ellison Medical Foundation new scholar award (to RS). The authors have no competing financial interests to declare.