
Abstract
Defective autophagy has been implicated in mammary tumorigenesis, as the gene encoding the essential autophagy regulator BECN1 is deleted in human breast cancers and Becn1+/− mice develop mammary hyperplasias. In agreement with a recent study, which reports concurrent allelic BECN1 loss and ERBB2 amplification in a small number of human breast tumors, we found that low BECN1 mRNA correlates with ERBB2-overexpression in breast cancers, suggesting that BECN1 loss and ERBB2 overexpression may functionally interact in mammary tumorigenesis. We now report that ERBB2 overexpression suppressed autophagic response to stress in mouse mammary and human breast cancer cells. ERBB2-overexpressing Becn1+/+ and Becn1+/− immortalized mouse mammary epithelial cells (iMMECs) formed mammary tumors in nude mice with similar kinetics, and monoallelic Becn1 loss did not alter ERBB2- and PyMT-driven mammary tumorigenesis. In human breast cancer databases, ERBB2-expressing tumors exhibit a low autophagy gene signature, independent of BECN1 mRNA expression, and have similar gene expression profiles with non-ERBB2-expressing breast tumors with low BECN1 levels. We also found that ERBB2-expressing BT474 breast cancer cells, despite being partially autophagy-deficient under stress, can be sensitized to the anti-ERBB2 antibody trastuzumab (tzb) by further pharmacological or genetic autophagy inhibition. Our results indicate that ERBB2-driven mammary tumorigenesis is associated with functional autophagy suppression and ERBB2-positive breast cancers are partially autophagy-deficient even in a wild-type BECN1 background. Furthermore and extending earlier findings using tzb-resistant cells, exogenously imposed autophagy inhibition increases the anticancer effect of trastuzumab on tzb-sensitive ERBB2-expressing breast tumor cells, indicating that pharmacological autophagy suppression has a wider role in the treatment of ERBB2-positive breast cancer.
Introduction
Autophagy is a dynamic self-catabolic cellular process, whereby proteins and organelles are targeted to lysosomes for degradation. Autophagy is upregulated during periods of stress and maintains cell viability by enabling basic biomolecule and energy recycling. Under regular growth conditions, basal autophagy preserves cellular homeostasis by mediating degradation of misfolded proteins and aged or damaged organelles, thus mitigating cell damage.Citation1-Citation4
Defective autophagy has been implicated in tumorigenesis, as the essential autophagy gene, BECN1, is commonly deleted in breast, ovarian, and prostate cancers.Citation5 BECN1 is essential for autophagosome formation and, when ectopically expressed in partially autophagy-deficient human MCF7 breast cancer cells, it restores functional autophagy and suppresses tumorigenesis.Citation6 becn1−/− mice die early in embryogenesis, while aging Becn1+/− mice are tumor-prone, developing lymphomas and carcinomas of the lung and liver.Citation7,Citation8 Furthermore, mammary tissues from Becn1+/− mice display preneoplastic, hyperproliferative changes, but no spontaneous mammary carcinomas.Citation7 The seemingly paradoxical association between increased tumorigenesis and dysfunction and/or loss of a survival mechanism can be reconciled by the findings that autophagy defects render cells susceptible to metabolic stress and DNA damage, thus enhancing tumor necrosis, inflammation and genomic instability, which in turn accelerate tumorigenesis.Citation4,Citation9,Citation10 However, autophagy may also act as a tumor-promoting mechanism by supporting cancer cell survival, as it is readily induced in hypoxic tumor regions and in response to chemotherapy and radiation.Citation11-Citation13
Although allelic BECN1 loss has been implicated in the pathophysiology of breast cancer,Citation5 its specific role(s) in tumor initiation and progression have not been determined. A recent study reveals significant association between BECN1 deletion and ERBB2 amplification,Citation14 thus providing evidence for lower BECN1 expression in a particular breast cancer subtype.Citation15
ERBB2/HER2/neu (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2) is a member of the HER family of tyrosine kinases, along with EGFR (epidermal growth factor receptor), ERBB3, and ERBB4. In normal cells, a variety of extracellular ligands bind to HER receptor heterodimers, leading to activation of pathways that control growth, differentiation, motility, and adhesion.Citation16-Citation20 Deregulation of these signaling networks occurs frequently in cancer, as exemplified by ERBB2 gene amplification in breast cancer and by constitutive EGFR activation in lung and colon cancers.Citation21-Citation25 ERBB2 overexpression results in aberrant signaling of the PI3K-AKT1 and MAPK1/3 pathways, which in turn are associated with malignant transformation,Citation26 and ERBB2-positive breast malignancies are characterized by aggressive nature, poor clinical outcome, and chemotherapy resistance.Citation27 In addition to ERBB2 amplification, further genomic changes are commonly required for ERBB2-induced tumorigenesis, as abnormal ERBB2 signaling leads to apoptosis in cells carrying wild-type TP53.Citation28,Citation29
Interestingly, the human BECN1 and ERBB2 genes are both located on chromosome 17, specifically at the 17q12 and 17q21 locuses, respectively, which are characterized by frequent genomic instability events, such as ERBB2-TOP2A amplification and allelic loss events,Citation30-Citation32 in human tumors. In a small number of breast tumors examined by fluorescence in situ hybridization, genomic BECN1 loss correlates with ERBB2 amplification and this result has been confirmed in 2 independent, public copy number microarray data sets.Citation14 Furthermore, breast cancers with concurrent BECN1 deletion and ERBB2 amplification were also characterized by alterations in the TP53, PTEN, and PIK3CA genes.Citation14 However, despite the reported association between BECN1 loss and ERBB2-positive breast cancer, the role of BECN1 in ERBB2-induced mammary tumorigenesis has not yet been investigated. Quite intriguingly, ERBB2-positive tumors resistant to the humanized mouse monoclonal ERBB2 antibody trastuzumab (tzb) upregulate basal autophagy and are resensitized to treatment by autophagy inhibition,Citation33 thus implicating autophagy induction in development of treatment resistance and the high relapse rates observed in patients with metastatic ERBB2-positive breast cancer.
To determine the role of BECN1 deficiency in ERBB2-positive breast cancer pathogenesis and treatment, we investigated the impact of ERBB2 overexpression on the functional status of autophagy in immortalized mouse mammary epithelial cells and human breast cancer cell lines under metabolic stress. We also investigated the effect of monoallelic Becn1 loss on mammary tumorigenesis in the MMTV-Neu and MMTV-PyMT mouse tumor models. We now report that ERBB2 overexpression does not affect basal autophagy, but suppresses stress-induced autophagy in mammary tumor cells, even in a wild-type Becn1 background. Furthermore, monoallelic Becn1 deletion does not alter the tumorigenicity of ERBB2-expressing iMMECs in nude mice in vivo and does not impact spontaneous mammary tumorigenesis in the MMTV-Neu and MMTV-PyMT mouse models. We also found that low BECN1 expression correlates with the ERBB2-positive and basal-like human breast cancer subtypes and that ERBB2-positive breast tumors, independently of BECN1 mRNA levels, are likely functionally autophagy-deficient, as determined by gene expression profiling. Finally, both genetic and pharmacological autophagy inhibition enhance the response of tzb-sensitive, ERBB2-positive breast cancer cells to trastuzumab, indicating that autophagy modulation may improve the therapeutic efficacy of standard treatment in ERBB2-positive breast cancer.
Results
Low BECN1 expression correlates with ERBB2-positive and basal-like breast cancer subtypes
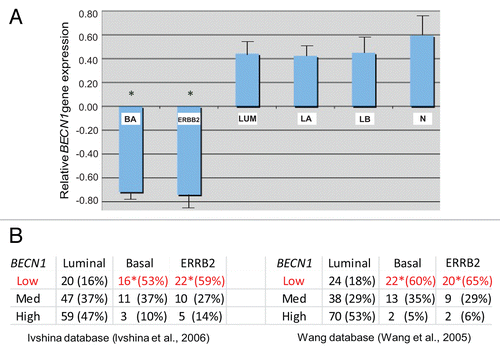
Given the recently reported association between genetic BECN1 loss and ERBB2 amplification in a small number of human breast tumors,Citation14 we investigated larger human breast cancer gene profiling databases to determine whether BECN1 expression correlates in any way with particular breast cancer subtypes. Using 3 independent DNA microarray databasesCitation34-Citation36 totaling 254 breast cancer specimens, we discovered that both ERBB2-positive and basal-like breast tumors commonly exhibit low BECN1 expression, whereas estrogen receptor (ER)-positive tumors are characterized by higher BECN1 mRNA levels (, P < 0.05).
Figure 1. Low BECN1 mRNA levels correlate with the ERBB2-positive and triple negative breast cancer subtypes. (A) Relative expression of BECN1 mRNA in basal (BA), luminal (LUM), and ERBB2-amplified (ERBB2) breast cancer subclasses and normal breast tissue (N) from samples analyzed in Wang et al. and Richardson et al. The luminal class is shown further subdivided into luminal A (LA) and luminal B (LB) subclasses. The mean expression of BECN1 in the total sample set is shown normalized to 0. (B) The relative number of samples falling into the lower, middle, and high tertiles of BECN1 gene expression in BA, LUM and ERBB2 breast cancer subclasses in an independent set of samples from Ivshina et al. and Wang et al. is shown. Statistically significant subclasses (*) indicating a P value < 0.05 (Student t test).
ERBB2 overexpression in mammary tumor cells does not affect basal autophagy, but suppresses autophagy induction in response to metabolic stress
To examine the effect of ERBB2 overexpression on the functional status of autophagy, we used our previously described mouse mammary epithelial cell model,Citation37 and transfected Becn1+/+ and Becn1+/− iMMECs with a plasmid expressing wild-type human ERBB2. While the transfection efficiencies were similar for Becn1+/+ and Becn1+/− iMMECs as previously described,Citation4 we obtained several ERBB2-expressing Becn1+/+ iMMEC lines (7 out of 13 antibiotic-resistant colonies), but only one ERBB2-expressing Becn1+/− iMMEC line (1 out of 13 antibiotic-resistant colonies) (), possibly indicating that ERBB2 overexpression is not well tolerated in partially autophagy-deficient Becn1+/− iMMECs and is, thus, negatively selected for in vitro. Similar results were obtained when the experiment was repeated, at which time one more ERBB2-expressing Becn1+/− iMMEC line was recovered.
Figure 2. Stable ERBB2 overexpression inhibits stress-induced autophagy in mouse mammary tumor cells. (A) Autophagy-competent Becn1+/+ and partially autophagy-defective Becn1+/− iMMECs were stably transfected with a human wild-type ERBB2-expressing plasmid conferring resistance to G418. Seven out of 13 Becn1+/+ G418R-colonies and 1 out of 13 Becn1+/− G418R-colonies overexpressed ERBB2, as shown by ERBB2 and ACTB western blots of whole cell protein lysates. (B) GFP-fluorescence microscopy of BCL2- and EGFP-LC3B-expressing Becn1+/+ iMMECs, also stably expressing an ERBB2 or vector-control plasmid, under nutrient deprivation (Hanks medium) without or with bafilomycin A1 (BafA1, 25 nM). (C) Autophagy quantification of (B) based on number of GFP-fluorescent puncta per cell, as imaged by GFP-fluorescence microscopy. Each data point is an average of triplicate experiments ± SD after quantifying puncta in one hundred cells per experiment. **P value < 0.01. (D) GFP, SQSTM1 and ACTB western blots of whole cell protein lysates from apoptosis-deficient, EGFP-LC3B-expressing, and vector- or ERBB2-expressing Becn1+/+ iMMECs under nutrient deprivation without (-) and with (+) BafA1. ERBB2-expressing iMMECs exhibited lower GFP-LC3B expression than vector-expressing iMMECs after stable GFP-LC3B transfection, so protein lysates from ERBB2-expressing iMMECs were loaded at 2× the amount of vector-expressing lysates to better visualize lower GFP-LC3B levels in these cells. (E) Densitometric analysis of LC3B-II/LC3B-I ratio and SQSTM1 protein bands, as normalized to ACTB, using ImageJ. LC3B levels were corrected for 2× loading of ERBB2-expressing lysates. (F) Electron micrographs of Becn1+/+ iMMECs expressing BCL2 (top row), Becn1+/+ iMMECs expressing BCL2 and ERBB2 (middle row), and Becn1+/− iMMECs expressing BCL2 (bottom row) under metabolic stress (1% oxygen and no glucose). Images were taken at 3800×. White arrows indicate autophagic structures. Inlayed zoomed images show autophagosomes. (G) Autophagosome quantification of (F). Each data point is an average of the number of autophagosomes in 25 cells ± SD. *P value < 0.05.
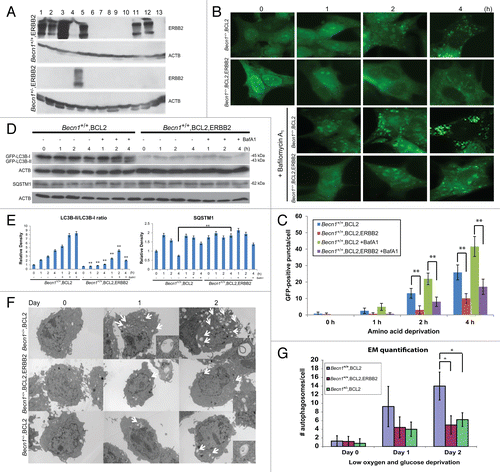
Given the recently documented upregulation of basal autophagy in mutant RAS-expressing mouse and human cancer cell lines and the resultant dependence of RAS-mutant tumors on autophagy for growth,Citation38 we hypothesized that the low recovery rate of ERBB2-expressing Becn1+/− cell lines might be secondary to a requirement for high functional autophagy in ERBB2-positive breast tumor cells. To investigate how ERBB2 overexpression impacts autophagy, we used stably and transiently ERBB2-expressing iMMEC and human breast cancer cell lines. First, BCL2-expressing, apoptosis-deficient Becn1+/+ iMMECs stably expressing EGFP-LC3B and either a human wild-type ERBB2 plasmid or vector control were subjected to nutrient deprivation (Hanks treatment) in the absence or presence of the autophagic flux inhibitor, bafilomycin A1 (BafA1). Autophagy induction was quantified by fluorescence microscopy, as previously described.Citation4,Citation39,Citation40 As shown in , ERBB2-overexpressing apoptosis-deficient Becn1+/+ iMMECs exhibited highly attenuated puncta formation in response to nutrient deprivation compared with vector-expressing Becn1+/+ iMMECs. This effect was observed even in the presence of BafA1 at a concentration (25 nM) that inhibits autophagic flux without affecting cell viability (Fig. S1), thus indicating that ERBB2 overexpression suppresses stress-induced autophagy in apoptosis-defective iMMECs (, P < 0.05). This result was verified by LC3B immunoblotting to follow the conversion of LC3B-I to LC3B-II.Citation41 In the absence of BafA1, LC3B-II increased over time in vector-expressing Becn1+/+ iMMECs under nutrient deprivation, indicating autophagy induction. In the presence of BafA1, LC3B-II was stabilized and displayed higher levels compared with non-BafA1 conditions, in agreement with LC3B-II accumulation in association with autophagic flux inhibition. In contrast, in ERBB2-expressing iMMECs, total LC3B protein levels and LC3B-I to LC3B-II conversion, normally observed in wild-type iMMECs under nutrient deprivation, were suppressed both in the absence and presence of BafA1 (), thus indicating that this result was not secondary to ERBB2-promoted acceleration of autophagic flux. The autophagy adaptor, SQSTM1/p62, commonly degraded during the process of autophagy exhibited higher protein levels in apoptosis-deficient ERBB2-expressing Becn1+/+ iMMECs compared with vector-expressing Becn1+/+ iMMECs, also indicating a suppression of the autophagic process ().Citation39 The same result was obtained using a different metabolic stressor and quantifying autophagy by electron microscopy (EM). In this case, apoptosis-deficient Becn1+/+ iMMECs stably expressing ERBB2 under low (1%) oxygen and glucose-deprivation conditions showed decreased number of autophagosomes compared with their non-ERBB2-expressing Becn1+/+ counterparts (). Interestingly, the level of autophagy induction in metabolically stressed ERBB2-expressing Becn1+/+ iMMECs was similar to that of vector-expressing Becn1+/− iMMECs (), confirming that ERBB2 overexpression renders mammary epithelial cells partially autophagy-deficient under stress.
To further investigate the impact of ERBB2 overexpression on stress-induced autophagy in an alternate system and in an apoptosis-competent background, we used a transient ERBB2 expression system.Citation40 To this intent, Becn1+/+ iMMECs stably overexpressing EGFP-LC3B were transiently transfected with a ERBB2-expressing or vector control plasmid and, after overnight recovery in regular culture medium, were incubated in Hanks medium for up to 3.5 h. Similar to the results described above (), transient ERBB2 overexpression did not affect basal autophagy, but suppressed autophagy induction in wild-type iMMECs in response to nutrient deprivation (, P < 0.01). This result was confirmed by decreased LC3B-I to LC3B-II conversion in iMMECs transiently overexpressing ERBB2 in both the absence and presence of BafA1 (). ERBB2 overexpression did not affect expression of the essential autophagy regulators BECN1 and ATG7 (Fig. S2), but resulted in decreased conversion of endogenous LC3B-I to LC3B-II (), indicating that ERBB2-promoted suppression of the autophagic response to stress was not associated with alterations in ATG expression.
Figure 3. Transient ERBB2 overexpression inhibits stress-induced autophagy in Becn1+/+ iMMECs to the level observed in partially autophagy-defective non-ERBB2-expressing Becn1+/− iMMECs. (A) GFP-fluorescence microscopy of EGFP-LC3B-expressing Becn1+/+ iMMECs transiently transfected with a ERBB2-expressing or vector control plasmid under nutrient deprivation conditions without or with bafilomycin A1 (BafA1, 25 nM). (B) Autophagy quantification of (A) based on number of GFP-fluorescent puncta per cell. Each data point is an average of triplicate experiments ± SD after quantifying puncta in 100 cells per experiment. *P value < 0.05; **P value < 0.01. (C) GFP and ACTB western blots of whole cell protein lysates from Becn1+/+ iMMECs transiently expressing ERBB2 under nutrient deprivation without and with BafA1. (D) Densitometric analysis of LC3B-II/LC3B-I ratio, as normalized to ACTB, using ImageJ. (E) EGFP-LC3B-expressing Becn1+/+ and Becn1+/− iMMECs transiently transfected with a ERBB2-expressing or vector control plasmid were subjected to nutrient deprivation, and autophagy was quantified by the number of GFP-fluorescent puncta per cell. Each data point is an average of triplicate experiments ± SD after quantifying puncta in 100 cells per experiment. *P value < 0.05; **P value < 0.01.
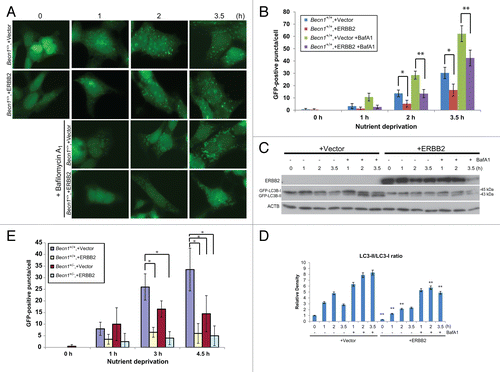
Finally, when EGFP-LC3B-expressing Becn1+/+ and Becn1+/− iMMECs were transiently transfected with a ERBB2-expressing or vector control plasmid and subjected to nutrient starvation, ERBB2 overexpression in either Becn1+/+ or Becn1+/− iMMECs induced similar number of autophagic puncta to those observed in vector-transfected Becn1+/− iMMECs (, P < 0.05), indicating that ERBB2 expression rendered mammary epithelial cells autophagy-defective, independent of allelic Becn1 status.
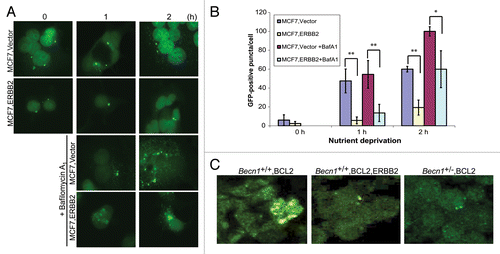
The effect of ERBB2 overexpression on the functional status of autophagy in human breast cancer cells was examined by transfection of stably EGFP-LC3B-expressing MCF7 cells with an ERBB2-expressing or vector control plasmid. Similar to the iMMEC results described above (; ), transient ERBB2 expression did not alter basal autophagy levels in MCF7 cells, but suppressed autophagy induction in response to nutrient starvation (). Inhibition of autophagic flux by bafilomycin A1 resulted in higher GFP puncta accumulation per cell in vector-compared with ERBB2-expressing MCF7 cells (, P < 0.05), again indicating that the ERBB2 signaling pathway decreases autophagy induction in response to stress and, thus, suggesting that ERBB2-positive breast tumors may be functionally autophagy-defective, independent of BECN1 expression.
Figure 4. Transient ERBB2 overexpression inhibits stress-induced autophagy in human breast cancer cells. (A) GFP-fluorescence microscopy of EGFP-LC3B-expressing MCF7 cells transiently transfected with a ERBB2-expressing or vector control plasmid under nutrient deprivation conditions for 0, 1, and 2 h without and with bafilomycin A1 (BafA1, 25 nM). (B) Autophagy quantification of (A) based on number of GFP-fluorescent puncta per cell. Each data point is an average of triplicate experiments ± SD after quantifying puncta in 100 cells per experiment. *P value < 0.05; **P value < 0.01. (C) GFP-fluorescence confocal microscopy of tumor cell plaques dissected 24 h post orthotopic implantation of BCL2-expressing Becn1+/+ (left panel), BCL2- and ERBB2-expressing Becn1+/+ (middle panel), and BCL2 expressing Becn1+/− (right panel) iMMECs in nude mice.
To examine the effect of ERBB2 overexpression on the functional status of autophagy in vivo, BCL2-, ERBB2- and EGFP-LC3B-expressing Becn1+/+ iMMECs, as well as BCL2-expressing Becn1+/+ and Becn1+/− iMMECs, were orthotopically implanted into the mammary fat pad of nude mice. Plaques were dissected 24 h post iMMEC implantation and LC3B translocation was qualitatively evaluated using fluorescence confocal microscopy. Similar to and , ERBB2-overexpressing Becn1+/+ mammary cells exhibited similar number and size of GFP-LC3B puncta to Becn1+/− cells in vivo, but fewer and smaller in size puncta than Becn1+/+ cells ().
Monoallelic Becn1 loss does not alter ERBB2- and PyMT-driven mammary tumorigenesis
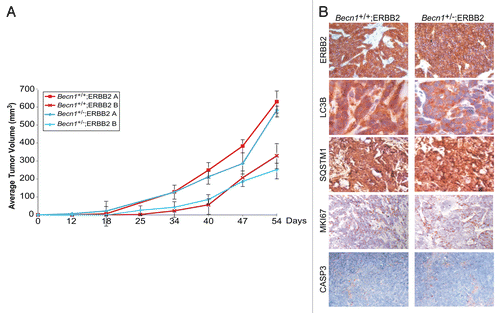
To investigate whether allelic Becn1 status impacts ERBB2-induced mammary tumorigenesis, ERBB2-overexpressing Becn1+/+ and Becn1+/− iMMECs were orthotopically implanted in NCR nude female mice. The kinetics of allograft mammary tumor formation were independent of Becn1 status (), indicating that, in our mouse mammary epithelial model,Citation37 monoallelic Becn1 loss does not alter ERBB2-induced mammary tumorigenesis. Mammary tumors generated by ERBB2-overexpressing Becn1+/+ and Becn1+/− iMMECs exhibited similar ERBB2, MKI67 (Ki67) and cleaved CASP3 levels, indicating that ERBB2-overexpressing Becn1+/+ and Becn1+/− iMMEC-generated tumors were similar in oncogene expression and in cell proliferation and death rates. However, given lower total LC3B expression, but similar SQSTM1 levels in Becn1+/− compared with Becn1+/+ tumors (), differences in the functional status of autophagy could not be reliably determined.
Figure 5. ERBB2-expressing Becn1+/+ and Becn1+/− iMMECs have similar tumor-forming capacities in nude mice. (A) Independent ERBB2-overexpressing Becn1+/+ and Becn1+/− iMMEC lines (e.g., A and B) were bilaterally implanted into the 3rd mammary fat pads of nude mice. Mice were monitored for tumor growth. Each data point represents the average volume of iMMEC-generated mammary tumors in 5 mice (2 tumors per mouse) per genotype ± SD (B) Representative images of ERBB2, LC3B, SQSTM1, MKI67, and cleaved CASP3 expression, as determined by IHC, in ERBB2-expressing Becn1+/+ (Becn1+/+;ERBB2) and ERBB2-expressing Becn1+/− (Becn1+/−;ERBB2) iMMEC-generated allograft mammary tumors from (A).
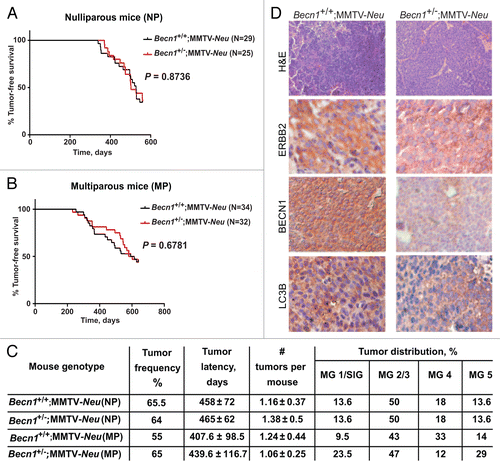
The functional interactions between the ERBB2 and autophagy pathways were further studied by crossing Becn1+/− mice to 2 well-characterized mouse mammary tumor models, namely the MMTV-NeuCitation42 and MMTV-PyMTCitation43 models, which show cosegregating tumor gene expression profiles when compared with other mouse mammary tumor models.Citation44 Similar to the iMMEC studies described above (), monoallelic Becn1 loss did not alter the incidence, MMTV-latency or multiplicity of ERBB2-induced mammary tumors, independent of parity status (). Similar ERBB2, but lower BECN1 and LC3B, levels were observed in Becn1+/−;Neu compared with Becn1+/+;MMTV-Neu mammary glands ().
Figure 6. Monoallelic Becn1 deletion does not affect ERBB2-driven mammary tumorigenesis. (A) Kaplan-Meier curve depicting percentage of tumor-free virgin (nulliparous-NP) mice over a period of 600 d post birth. (B) Kaplan-Meier curve depicting percentage of tumor-free retired breeder (multiparous-MP) mice over a period of 600 d post birth. (C) Table summarizing tumor frequency, latency, multiplicity, and anatomical distribution per genotype. MG, mammary gland; SlG, salivary gland. (D) Representative images of hematoxylin and eosin (H&E) and ERBB2, BECN1, and LC3B expression by IHC in mammary tumors from Becn1+/+;MMTV-Neu and Becn1+/−;MMTV-Neu virgin mice. Mice used: Becn1+/− (C57BL/6); MMTV-Neu (FVB/N).
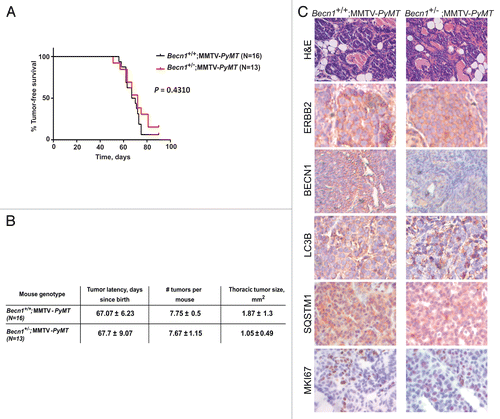
It is of interest to note that the cross between Becn1+/−Citation8 and MMTV-Neu mice resulted in a mixed C57BL/6:FVB (50:50) background and mammary tumors arose with greater latency and lower penetrance in Becn1+/+;MMTV-Neu mice than in the FVB/N MMTV-Neu model,Citation42 in agreement with earlier reports that the C57BL/6 background suppresses ERBB2-induced mammary tumor formation.Citation45,Citation46 To generate a Becn1+/− mouse model that is more readily amenable to mammary tumorigenesis studies, we changed the genetic background of the Becn1+/− mice from C57BL/6Citation8 to FVB/N. Rather than repeating the lengthier cross with MMTV-Neu mice, we instead crossed FVB Becn1+/− mice to the MMTV-PyMT mouse model, which develops mammary tumors in all mammary glands within 6 to 8 wkCitation43 and is frequently used as a surrogate model for ERBB2-driven mammary tumorigenesis, as NEU- and PyMT-induced mammary tumors exhibit cosegregating gene expression signatures and high ERBB2 expression.Citation47 Similar to the cross between Becn1+/− and MMTV-Neu mice (), Becn1 heterozygosity did not impact PyMT-induced mammary tumorigenesis (). Compared with Becn1+/+;MMTV-PyMT mammary tumors, Becn1+/−;MMTV-PyMT tumors exhibited lower BECN1 expression, but comparable ERBB2, LC3B, SQSTM1 and MKI67 levels (), indicating that cell proliferation and likely functional autophagy status in PyMT-driven mammary tumors were not affected by monoallelic Becn1 deletion.
Figure 7. Monoallelic Becn1 loss does not impact PyMT-driven mammary tumorigenesis. (A) Kaplan-Meier curve depicting percentage of tumor-free virgin mice over a period of 100 d post birth. (B) Table summarizing tumor latency, multiplicity, and size. (C) Representative images of H&E and ERBB2, BECN1, LC3B, SQSTM1, and MKI67 expression by IHC in mammary tumors from Becn1+/+;MMTV-PyMT and Becn1+/−;MMTV-PyMT mice. Mice used: Becn1+/− (FVB/N); MMTV-PyMT (FVB/N).
Autophagy inhibition enhances the response of tzb-responsive human breast cancer cells to trastuzumab
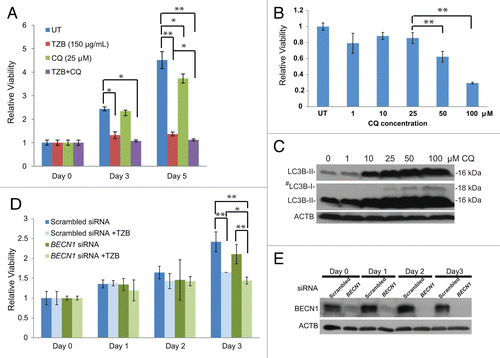
Our findings that ERBB2-positive breast tumors often exhibit low BECN1 expression (), ERBB2 overexpression suppresses stress-induced autophagy in mammary tumor cells in vitro and in vivo (–), and Becn1 heterozygosity does not impact ERBB2-induced mammary tumorigenesis in the mouse tumor models examined (–) suggest that, in contrast to mutant RAS-driven tumors,Citation38,Citation48-Citation50 ERBB2-overexpressing cancer cells do not depend on high functional autophagy levels for growth. It is possible, however, that the suppressed, but not absent, autophagic potential is still essential for ERBB2-positive cancer cell survival under stress and that further autophagy inhibition may promote tumor cell death. To investigate this clinically significant hypothesis, we examined whether pharmacological or genetic autophagy inhibition increased sensitivity of the ERBB2-positive human breast cancer cell line, BT474, to the humanized mouse monoclonal ERBB2 antibody trastuzumab.Citation51 Previous reports have shown that tzb-sensitive BT474 cancer cells exhibit low levels of basal autophagy and fail to upregulate autophagy in response to stress to the levels of other human breast cancer cell lines,Citation33,Citation52,Citation53 further supporting our finding that ERBB2 overexpression suppresses autophagy. As shown in , trastuzumab inhibited BT474 cell growth at 48 and 72 h of treatment (P < 0.05 and < 0.01, respectively), whereas the lysosomotropic agent and indirect autophagy inhibitor chloroquine (CQ), at a concentration that blocks autophagic flux (25 μM, ), had minimal effect on BT474 cell growth. The combination of trastuzumab and CQ showed statistically significant enhanced antitumor effect relative to the single agent trastuzumab (), indicating that pharmacological autophagy inhibition with CQ augments the therapeutic efficacy of trastuzumab on tzb-sensitive BT474 breast cancer cells (P < 0.05). Near-complete BECN1 knockdown with siRNA () did not affect BT474 cell growth, but increased the antitumor effect of trastuzumab at 72 h (, P < 0.05), indicating that targeted suppression of BECN1 expression also impacts ERBB2-positive breast cancer cell responsiveness to trastuzumab.
Figure 8. Autophagy inhibition sensitizes tzb-responsive ERBB2-positive breast cancer cells to trastuzumab. (A) Viability assays of BT474 cells treated with trastuzumab (TZB, 150 µg/ml), chloroquine (CQ, 25 µM), or combination of both for 0, 3, and 5 d. (B) Viability of BT474 after 3 d of treatment with increasing CQ concentrations. (C) LC3B western blot of BT474 cells treated with increasing CQ concentrations. #middle panel is higher exposure of top panel (D) Viability assays of BT474 cells treated with TZB (150 µg/ml) for 0 to 3 d, starting at 24 h after transfection with BECN1 or scrambled siRNA. (E) BECN1 immunoblot confirms target knockdown by siRNA during 0 to 3 d of treatment. P values were calculated using paired Student t test. Each data point is an average of triplicate experiments ± SD. *P value < 0.05; **P value < 0.01.
ERBB2-positive human breast tumors exhibit a low autophagy gene signature independent of BECN1 mRNA status
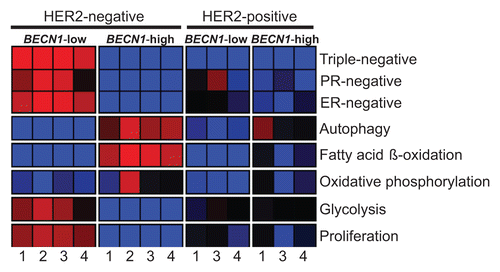
Given our initial observation that low BECN1 expression in human breast tumors correlates with the ERBB2 and basal-like subtypes in independent, but rather small, gene profiling databases (), we examined whether this finding holds true in much larger and unrelated breast cancer cohorts.Citation54-Citation57 Tumors were clustered into 4 subgroups based on BECN1 expression levels [high (BECN1+) vs. low (BECN1−) for BECN1 levels above or below the mean across samples, respectively] and reported ERBB2 status [positive (ERBB2+) vs. negative (ERBB2−)] and their expression profiles were compared regarding hormone receptor status, autophagy-related gene expression, and gene signatures of metabolic pathways (Fig. S3) previously reported to be affected by functional autophagy status.Citation38,Citation48,Citation49,Citation58-Citation60 This analysis confirmed that ERBB2-positive and triple negative breast tumors commonly express low levels of BECN1 mRNA (, Fig. S4, P = 5.70E-18). We also discovered that ERBB2-positive tumors, independent of BECN1 expression and very similar to non-ERBB2-expressing BECN1-low tumors, exhibited low expression of autophagy-regulated genes, possibly indicating functional autophagy suppression in ERBB2-positive breast cancers even when BECN1 is highly expressed (, Fig. S4, P = 2.60E-03). It is of great interest and worthy of further investigation that, similar to non-ERBB2-expressing BECN1-low tumors and in contrast to non-ERBB2-expressing BECN1-high tumors, ERBB2-positive breast tumors showed decreased fatty acid β-oxidation and oxidative phosphorylation gene signatures, independent of BECN1 expression. Intriguingly, ERBB2-positive breast cancers seem to have glycolysis and cell proliferation gene signatures between the significantly upregulated and significantly downregulated patterns observed in non-ERBB2-expressing BECN1-low (i.e., mostly triple negative) and non-ERBB2-expressing BECN1-high (i.e., mostly hormone receptor-positive) tumors, respectively (, Fig. S4, P = 1.60E-05 and P = 3.10E-06).
Figure 9. ERBB2-positive breast cancers have, independent of BECN1 expression, gene expression signatures similar to those of non-ERBB2-expressing breast cancers with low BECN1 mRNA levels. The heatmap reports gene set enrichment analysis (GSEA) for selected gene signatures (rows) in breast cancer cohorts defined by ERBB2 and BECN1 status. The color indicates the enrichment (Fischer exact test) of samples with gene signature upregulation (red, P+, enrichment < 0.05), downregulation (blue, P−, enrichment < 0.05), or no difference (black, P+, enrichment ≥ 0.05 and P−, enrichment ≥ 0.05) within a cohort subgroup relative to the remaining samples in the cohort. Gene signature lists and statistical analysis are provided in Figures S3 and S4, respectively. Databases: 1) Decremoux et al. 2011, 2) Hatzis et al. 2011 (ERBB2-negative breast cancers only), 3) Servant et al. 2012, and 4) Sabatier et al. 2011.
Discussion
Role of autophagy in mammary tumorigenesis
The human epidermal growth factor receptor and tyrosine kinase ERBB2 has been widely studied because of its strong transforming potential, its role in the pathogenesis of breast cancer, and its use as a therapeutic target in patients with ERBB2-positive breast tumors. ERBB2 amplification is likely an early genetic event in mammary tumorigenesis, as it is commonly observed in ductal carcinoma in situ, in the absence of invasive disease.Citation61 ERBB2 status remains constant as disease progresses to invasive and then metastatic stages.Citation62-Citation65 Becn1 has been identified as a haplo-insufficient tumor suppressor, as Becn1+/− mice develop mammary hyperplasias, lymphomas, and lung and liver carcinomas, which retain a wild-type Becn1 allele.Citation4,Citation7 Epithelial cells with autophagy defects, including Becn1 heterozygosity, exhibit susceptibility to metabolic stress, which is accompanied by DNA damage and increased genomic instability, in turn likely driving cancer progression.Citation4 In our study, and in agreement with earlier work,Citation14 ERBB2 overexpression and low BECN1 mRNA levels are positively correlated in human breast cancers (), indicating that many ERBB2-positive breast malignancies may be functionally autophagy-deficient. In support of this hypothesis, we found that ERBB2 axis activation suppresses stress-induced autophagy (–), suggesting that ERBB2-positive premalignant and malignant breast lesions may exhibit decreased autophagic potential, even if BECN1 is genomically intact. Given the contribution of allelic Becn1 loss to DNA damage and genomic instability,Citation4 it is conceivable that, by functionally suppressing autophagy, early ERBB2 activation may lead to further ERBB2 amplification and, thus, a positive feedback loop maintaining and increasing the protumorigenic function of the ERBB2 axis. In this case, defective autophagy, but not necessarily allelic BECN1 deletion, may indeed play a role in ERBB2-positive breast cancer, particularly during tumor initiation. Recent studies support the concept that autophagy suppression is important for aberrant tyrosine kinase-induced tumorigenesis, such as those mediated by AKT1 and EGFR,Citation66,Citation67 which inhibit autophagy by phosphorylation of BECN1. Upon expression of a BECN1 mutant resistant to phosphorylation in cancer cell lines, autophagy could not be inhibited and tyrosine kinase-mediated xenograft tumor formation in nude mice was suppressed.Citation66,Citation67 It would be very informative to further investigate the role of autophagy in ERBB2-induced tumorigenesis by generating a transgenic mouse model that combines ERBB2 overexpression and a constitutively activated or non-suppressible autophagy status. As it is evident from our in vitro and in vivo allograft and spontaneous tumorigenesis studies, unless autophagy is ectopically induced or engineered to be in a “nonsuppressible state,” activation of the ERBB2 axis suppresses the autophagic response to stress and renders ERBB2-positive breast cancer cells autophagy-deficient, thus providing an explanation for why partial Becn1 deficiency does not impact ERBB2-driven mammary tumor formation (–).
Our bi-transgenic mouse models combining monoallelic Becn1 loss and ERBB2 or PyMT activation under the MMTV-promoter join the efforts to study the role of defective autophagy in mammary tumorigenesis using mammary tumor-prone mouse models.Citation68,Citation69 In the first such publication, mammary gland-targeted deletion of the positive autophagy regulator Rb1cc1/Fip200 suppresses mammary tumor initiation and progression in the MMTV-PyMT model, in association with defective autophagy in tumor cells, as indicated by accumulation of ubiquitinated protein aggregates and SQSTM1, deficient LC3B conversion, and increased number of abnormal-appearing mitochondria.Citation68 In a more recently published study, monoallelic Becn1 loss suppresses mammary tumor formation driven by Palb2 deletion in mammary epithelial cells (MECs) in a wild-type Trp53 background, but fails to impact tumorigenesis induced by combined MEC-specific Palb2 and Trp53 loss,Citation69 suggesting that the role of allelic Becn1 status in mammary tumorigenesis is greatly influenced by other oncogenic events. The results mentioned above, including our studies, once more indicate that the role of autophagy in breast cancer is complex and warrants further investigation.
Autophagy modulation for ERBB2-positive breast cancer treatment
Our treatment studies () demonstrate that pharmacological inhibition of autophagy increases sensitivity of tzb-responsive breast cancer cells to trastuzumab, indicating that the functionally reduced autophagy status in ERBB2-overexpressing breast cancer cells still preserves cell viability and provides protection against chemotherapy. This finding is in agreement with previously published studies, which implicated autophagy in resistance to breast cancer treatment, as autophagy inhibition by CQ or silencing of Atg genes resensitized tzb-resistant SKBR3 cells and hormone-resistant MCF7 cells to trastuzumab and tamoxifen, respectively.Citation33,Citation70
Tumor cell addiction to autophagy
The suppressive effect of ERBB2 overexpression on the functional status of autophagy is in sharp contrast to the upregulation of basal autophagy and the strong dependence of RAS-mutant tumors on autophagy for growth.Citation38,Citation48-Citation50,Citation58 RAS-mediated adhesion-independent transformation is dependent on autophagy, as autophagy inhibition reduced glycolytic capacity and attenuated cell proliferation and transformation.Citation48 Furthermore, RAS-expressing cells have high basal autophagy to maintain a functional mitochondrial pool and meet energy demands imposed by oncogenic RAS, as autophagy suppression decreased tumor cell survival under starvation and abrogated tumorigenesis in nude mice, in association with depletion of oxidative phosphorylation and tricarboxylic acid cycle intermediates.Citation38,Citation49 Whereas the aforementioned workCitation38,Citation48-Citation50,Citation58 suggests an oncogene-induced requirement for autophagy induction during tumorigenesis, our present studies indicate that “autophagy addiction” is not a generalized phenomenon in cancer pathophysiology, but its activation is instead specific to particular oncogenic events.
Despite the differences in autophagy functional status in RAS-mutant and ERBB2-positive tumors and the potentially discrete roles of defective autophagy in RAS- and ERBB2-driven tumor initiation and maintenance, our studies reveal a common role for autophagy in resistance to cancer therapy. Similar to the sensitization of different tumor types to standard anticancer agents,Citation13,Citation71-Citation73 tzb-responsive breast cancer cells were rendered more sensitive to trastuzumab by pharmacological and genetic autophagy suppression, thus further supporting use of autophagy inhibitors in combination with conventional cancer therapies.
Autophagy functional status in ERBB2-positive breast tumors
Our finding that ERBB2 activation suppresses stress-induced autophagy in breast cancer cells in vitro and in vivo (–) is in agreement with our analysis of human breast tumor DNA microarray data showing that ERBB2-expressing breast cancers exhibit lower expression of autophagy-related genes (Figs. S3 and S4), independent of BECN1 expression levels (). It is, thus, likely that ERBB2-positive breast tumors are functionally autophagy-defective and, similar to Becn1+/− iMMECs,Citation74 sensitive to oxidative and endoplasmic reticulum (ER) stress-inducing agents; this hypothesis will be investigated in subsequent studies.
An interesting and thought-provoking finding from our gene expression analysis is the striking downregulation of glycolysis and proliferation gene signatures in non-ERBB2-expressing BECN1-high breast cancers, which are highly enriched in hormone receptor-positive tumors and also exhibit high and relative upregulation of fatty acid β-oxidation and oxidative phosphorylation gene signatures, respectively. ERBB2-positive breast cancers, independent of BECN1 expression, are characterized by a relative upregulation of glycolysis and proliferation gene signatures compared with non-ERBB2-expressing BECN1-high (mostly hormone receptor-positive) tumors, but not to levels observed in non-ERBB2-expressing BECN1-low (mostly triple negative breast) tumors. Upregulation of glycolysis in association with ERBB2 activation has been reported before.Citation75-Citation77 However, the high expression of glycolysis-related genes in conjunction with a low autophagic gene signature is surprising considering that, in RAS-mediated transformation, defective autophagy reduces glycolytic capacity.Citation48 It is possible that breast tumors with high functional autophagy do not rely on glycolysis for meeting their metabolic demands, as fatty acid β-oxidation and oxidative phosphorylation can be sustained at high levels in autophagy-maintained healthy mitochondria. In contrast, low BECN1 expression, and likely defective autophagy and deregulation of mitochondrial homeostasis, correlates with significant suppression of fatty acid β-oxidation and oxidative phosphorylation, as previously reported,Citation38,Citation60 thus forcing the cancer cell metabolic machinery toward glycolysis. The relationship between autophagy regulation and metabolic reprogramming is obviously quite complex,Citation58 and further studies are needed to explore the metabolic profiles of the different breast cancer subtypes and incorporate the knowledge acquired in the design of more effective therapeutic regimens.
Materials and Methods
Cell line generation and culture conditions
Primary mouse mammary epithelial cells (pMMECs) from Becn1+/+ and Becn1+/− miceCitation8 were immortalized to generate iMMEC cell lines, which were then engineered to stably express BCL2, EGFP-LC3B or wild-type human ERBB2, as previously described.Citation4 The BT474 (HTB-20) cell line was obtained from American Type Culture Collection. Hank’s balanced salt solution (Life Technologies, 14025-092) was used for nutrient-deprivation studies. Bafilomycin A1 (BafA1; Sigma-Aldrich, B1793) was used at a concentration of 25 nM.
Fluorescence and electron microscopy
Autophagy was quantified by quantification of EGFP-LC3B puncta per cell using fluorescence microscopy, using an Olympus IX51 fluorescent microscopy system at 60× magnification. One hundred cells per cell line were evaluated for number of EGFP-LC3B puncta per cell at each time point. Three independent experiments were performed, and the average number of GFP-fluorescent puncta per cell with standard deviation for each cell line at each time point is presented. For EM, cells were fixed in a 2.5% glutaraldehyde, 4% paraformaldehyde, 8 µM calcium chloride, 0.1 M cacodylate, pH 7.4 fixative buffer. Electron microscopy was performed with a JEOL 1200EX electron microscope at 3800× magnification. Statistical analysis (2-tailed Student t test) was performed by Excel’s Data Analysis ToolPak (Microsoft, www.microsoft.com).
Western blotting and immunohistochemistry
Western blotting using whole-cell protein extracts and immunohistochemistry (IHC) were performed as previously described.Citation78 Antibodies used were against ERBB2 (Cell Signaling, 2165); Ki67 (Leica Microsystems, NCL-L-Ki67-MM1); LC3B (Novus Biologicals, NB100-2331); BECN1/Beclin1 (Santa Cruz Biotechnology, sc-11427); SQSTM1 (p62, Enzo Life Sciences, BML-PW9860); ATG7 (A2856), ACTB/β-Actin (Sigma-Aldrich, A4527). Cleaved CASP3 IHC was performed by Rutgers Cancer Institute of New Jersey Tissue Analytical Services. Densitometry analysis was performed by ImageJ.Citation79
Tumorigenicity assays
Orthotopic mammary gland implantation of iMMECsCitation4 and trangenic mouse tumorigenicity studies were performed according to Institutional Animal Care and Use Committee-approved protocols. C57BL/6 Becn1+/− mice were crossed to MMTV-Neu mice [FVB/N-Tg(MMTVneu) 202 Mul/J] (The Jackson Laboratory, 002376). To circumvent suppression of ERBB2-induced mammary tumorigenesis by the C57BL/6 background,Citation45 C57BL/6 Becn1+/− mice were backcrossed into the FVB/N background for 10 generations. FVB Becn1+/− mice were subsequently crossed to MMTV-PyMT mice.Citation80 Progeny cohorts of all resultant genotypes were observed for spontaneous mammary tumor formation by weekly palpation. Kaplan-Meier survival curves and subsequent P values (2-tailed logrank test) were generated using GraphPad Prism version 5.0 for Windows (GraphPad Software, www.graphpad.com).
Cell viability assays
Cells were plated in 6-well plates at medium density, so as to ensure nonconfluency after 5 d of vehicle treatment. Media and drugs were changed after 3 d. Cell viability was assessed using the trypan blue exclusion method automated by a Vi-Cell (Beckman Coulter). Trastuzumab (Herceptin) was supplied as an aqueous solution at a concentration of 25 mg/mL and it was a generous gift from the Rutgers Cancer Institute of New Jersey clinical pharmacy. Chloroquine diphosphate salt was purchased from Sigma-Aldrich (C6628). Statistical analysis (2-tailed Student t test) was performed by Excel’s Data Analysis ToolPak.
Gene expression analysis
For the data shown in , gene expression array data from early stage breast cancers published by Wang et al. and by Richardson et al. (which included some normal breast samples) were combined and analyzed.Citation35,Citation36 The data set of Richardson et al., obtained on U133-Plus Affymetrix arrays, was made compatible with that of Wang et al., obtained on Affymetrix U133A arrays, by restricting it to the probe sets of the U133A chip and processing it with the mas5 software available at http://www.bioconductor.org.Citation81 The distance-weighted discrimination method was used for systematic source and batch bias adjustment in the 2 data sets.Citation82 Breast cancers were classified into basal-like cancers (BA), ERBB2-positive, Luminal (LUM), Luminal A (LA) and Luminal B (LB) by robust consensus clustering.Citation83 The average expression of each gene across all samples was normalized to 0. The mean relative expression of probes corresponding to gene of interest in each subtype was calculated and graphed. Statistical analysis (Student t test) was performed by Excel’s Data Analysis ToolPak (Microsoft, www.microsoft.com).
For the data shown in , 4 reported breast cancer data sets were downloaded from Gene Expression Omnibus (GEO): de Cremoux et al.Citation54 (226 breast tumors, Affymetrix U133 Plus 2.0 Array, GEO series GSE26639), Hatzis et al.Citation56 (508 ERBB2+ breast tumors, Affymetrix U133A Array, GEO series GSE25066), Servant et al.Citation57 (343 breast tumors, Illumina HumanWG-6 v3.0 expression bead chip Array, GEO series GSE30682) and Sabatier et al.Citation55 (266 breast tumors, Affymetrix U133 Plus 2.0 Arrays, GEO series GSE21653). All the Affymetrix data sets were processed using the justRMA function in R Bioconductor, obtaining a log2 expression values. For the Servant et al. data set assayed with an Illumina array, log2 of the reported variance stabilized expression values were used.Citation57 The samples were classified into ERBB2-positive (ERBB2+) or ERBB2-negative (ERBB2−) based on the reported ERBB2 amplification status. The samples were classified into BECN1-high (Beclin+) or BECN1-low (Beclin−) depending on whether BECN1 expression (probe 208945_s_at) was above or below the mean across samples. Gene expression signatures were analyzed using Gene Set Enrichment Analysis,Citation84 obtaining a quantification of the statistical significance for upregulation (P+) or downregulation (P−) for each signature and sample pair. A sample was said to have a signature significantly upregulated if P+ < 0.05, significantly downregulated if P− < 0.05, and no significant change otherwise. The complete lists of gene signatures are listed in Figure S3. Statistical analysis is listed in Figure S4.
| Abbreviations: | ||
| AKT1 | = | v-akt murine thymoma viral oncogene homolog 1 |
| ATG | = | autophagy-related |
| BA | = | basal-like |
| BafA1 | = | bafilomycin A1 |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BECN1 | = | Beclin 1 |
| CASP3 | = | caspase 3 |
| CQ | = | chloroquine |
| EGFP | = | enhanced green fluorescent protein |
| EM | = | electron microscopy |
| ER | = | estrogen receptor |
| RB1CC1/FIP200 | = | RB1-inducible coiled-coil 1 |
| ERBB2 | = | v-erb-b2 avian erythoblastic leukemia viral oncogene homolog 2 |
| ERBB3 | = | v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 3 |
| ERBB4 | = | v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4 |
| IHC | = | immunohistochemistry |
| iMMECs | = | immortalized mouse mammary epithelial cells |
| MAP1LC3B (LC3B) | = | microtubule-associated protein 1 light chain 3 beta |
| MAPK1 | = | mitogen-activated protein kinase 1 |
| MAPK3 | = | mitogen-activated protein kinase 3 |
| MG | = | mammary gland |
| MKI67 | = | marker of proliferation Ki-67 |
| MMTV | = | mouse mammary tumor virus |
| PALB2 | = | partner and localizer of BRCA2 |
| PI3K | = | phosphoinositide 3-kinase |
| PIK3CA | = | phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha |
| pMMECs | = | primary mouse mammary epithelial cells |
| PTEN | = | phosphatase and tensin homolog |
| PyMT | = | polyoma middle T |
| RAS (KRAS and HRAS) | = | Kirsten and Harvey rat sarcoma viral oncogene homolog |
| SlG | = | salivary gland |
| SQSTM1/p62 | = | sequestosome 1 |
| TOP2A | = | topoisomerase (DNA) II alpha 170 kDa |
| TP53 | = | tumor protein 53 |
| TZB and tzb | = | trastuzumab |
| UT | = | untreated |
Additional material
Download Zip (1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Z Yue and S Jin (Mount Sinai School of Medicine and Rutgers Robert Wood Johnson Medical School, respectively; Becn1+/− mice), E Lattime (Rutgers Cancer Institute of New Jersey; human wild-type ERBB2-expressing plasmid), R Patel (Rutgers Robert Wood Johnson Medical School) for assistance with electron microscopy, N Goldsmith-Kane (Rutgers University) for assistance with confocal microscopy, and the Rutgers Cancer Institute of New Jersey Tissue Analytical Services. We are grateful to the New Jersey Commission on Cancer Research (Predoctoral Fellowship to FL), NIH-NCI (R00 grant to VK) and Damon Runyon Cancer Research Foundation (Clinical Investigator Award to VK) for their generous financial support.
References
- Dunn WA Jr.. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol 1994; 4:139 - 43; http://dx.doi.org/10.1016/0962-8924(94)90069-8; PMID: 14731737
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999; 59:59 - 65; http://dx.doi.org/10.1006/geno.1999.5851; PMID: 10395800
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res 2007; 13:7271 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-07-1595; PMID: 18094407
- Mathew R, White E. Why sick cells produce tumors: the protective role of autophagy. Autophagy 2007; 3:502 - 5; PMID: 17611387
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther 2011; 11:157 - 68; http://dx.doi.org/10.4161/cbt.11.2.14622; PMID: 21228626
- Negri T, Tarantino E, Orsenigo M, Reid JF, Gariboldi M, Zambetti M, Pierotti MA, Pilotti S. Chromosome band 17q21 in breast cancer: significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosomes Cancer 2010; 49:901 - 9; http://dx.doi.org/10.1002/gcc.20798; PMID: 20589936
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747 - 52; http://dx.doi.org/10.1038/35021093; PMID: 10963602
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995; 378:390 - 4; http://dx.doi.org/10.1038/378390a0; PMID: 7477376
- Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J 1998; 17:719 - 31; http://dx.doi.org/10.1093/emboj/17.3.719; PMID: 9450997
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995; 378:394 - 8; http://dx.doi.org/10.1038/378394a0; PMID: 7477377
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature 1995; 378:386 - 90; http://dx.doi.org/10.1038/378386a0; PMID: 7477375
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 1995; 269:230 - 4; http://dx.doi.org/10.1126/science.7618084; PMID: 7618084
- Pierce JH, Arnstein P, DiMarco E, Artrip J, Kraus MH, Lonardo F, Di Fiore PP, Aaronson SA. Oncogenic potential of erbB-2 in human mammary epithelial cells. Oncogene 1991; 6:1189 - 94; PMID: 1713661
- Liu Y, el-Ashry D, Chen D, Ding IY, Kern FG. MCF-7 breast cancer cells overexpressing transfected c-erbB-2 have an in vitro growth advantage in estrogen-depleted conditions and reduced estrogen-dependence and tamoxifen-sensitivity in vivo. Breast Cancer Res Treat 1995; 34:97 - 117; http://dx.doi.org/10.1007/BF00665783; PMID: 7647336
- Franklin WA, Veve R, Hirsch FR, Helfrich BA, Bunn PA Jr.. Epidermal growth factor receptor family in lung cancer and premalignancy. Semin Oncol 2002; 29:Suppl 4 3 - 14; http://dx.doi.org/10.1053/sonc.2002.31520; PMID: 11894009
- Hirsch FR, Franklin WA, Veve R, Varella-Garcia M, Bunn PA Jr.. HER2/neu expression in malignant lung tumors. Semin Oncol 2002; 29:Suppl 4 51 - 8; http://dx.doi.org/10.1053/sonc.2002.31523; PMID: 11894014
- Yarom N, Jonker DJ. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov Med 2011; 11:95 - 105; PMID: 21356164
- Ménard S, Pupa SM, Campiglio M, Tagliabue E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003; 22:6570 - 8; http://dx.doi.org/10.1038/sj.onc.1206779; PMID: 14528282
- Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release 2010; 146:264 - 75; http://dx.doi.org/10.1016/j.jconrel.2010.04.009; PMID: 20385184
- Daly JM, Jannot CB, Beerli RR, Graus-Porta D, Maurer FG, Hynes NE. Neu differentiation factor induces ErbB2 down-regulation and apoptosis of ErbB2-overexpressing breast tumor cells. Cancer Res 1997; 57:3804 - 11; PMID: 9288791
- Giani C, Casalini P, Pupa SM, De Vecchi R, Ardini E, Colnaghi MI, Giordano A, Ménard S. Increased expression of c-erbB-2 in hormone-dependent breast cancer cells inhibits cell growth and induces differentiation. Oncogene 1998; 17:425 - 32; http://dx.doi.org/10.1038/sj.onc.1201954; PMID: 9696035
- Ellsworth RE, Ellsworth DL, Patney HL, Deyarmin B, Love B, Hooke JA, Shriver CD. Amplification of HER2 is a marker for global genomic instability. BMC Cancer 2008; 8:297; http://dx.doi.org/10.1186/1471-2407-8-297; PMID: 18854030
- Mano MS, Rosa DD, De Azambuja E, Ismael GF, Durbecq V. The 17q12-q21 amplicon: Her2 and topoisomerase-IIalpha and their importance to the biology of solid tumours. Cancer Treat Rev 2007; 33:64 - 77; http://dx.doi.org/10.1016/j.ctrv.2006.10.001; PMID: 17113234
- Futreal PA, Söderkvist P, Marks JR, Iglehart JD, Cochran C, Barrett JC, Wiseman RW. Detection of frequent allelic loss on proximal chromosome 17q in sporadic breast carcinoma using microsatellite length polymorphisms. Cancer Res 1992; 52:2624 - 7; PMID: 1568230
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 2009; 4:e6251; http://dx.doi.org/10.1371/journal.pone.0006251; PMID: 19606230
- Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res 2006; 66:10292 - 301; http://dx.doi.org/10.1158/0008-5472.CAN-05-4414; PMID: 17079448
- Wang Y, Klijn J, Zhang Y, Atkins D, Foekens J. Gene expression profiles and prognostic markers for primary breast cancer. Methods Mol Biol 2007; 377:131 - 8; http://dx.doi.org/10.1007/978-1-59745-390-5_7; PMID: 17634613
- Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006; 9:121 - 32; http://dx.doi.org/10.1016/j.ccr.2006.01.013; PMID: 16473279
- Karantza-Wadsworth V, White E. A mouse mammary epithelial cell model to identify molecular mechanisms regulating breast cancer progression. Methods Enzymol 2008; 446:61 - 76; http://dx.doi.org/10.1016/S0076-6879(08)01604-2; PMID: 18603116
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Lee HK, Myers RA, Marzella L. Stimulation of autophagic protein degradation by nutrient deprivation in a differentiated murine teratocarcinoma (F9 12-1a) cell line. Exp Mol Pathol 1989; 50:139 - 46; http://dx.doi.org/10.1016/0014-4800(89)90063-4; PMID: 2646143
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491 - 502; http://dx.doi.org/10.1016/j.biocel.2004.02.005; PMID: 15325587
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A 1992; 89:10578 - 82; http://dx.doi.org/10.1073/pnas.89.22.10578; PMID: 1359541
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 1992; 12:954 - 61; PMID: 1312220
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 2007; 8:R76; http://dx.doi.org/10.1186/gb-2007-8-5-r76; PMID: 17493263
- Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, Hunter KW. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer 1998; 77:640 - 4; http://dx.doi.org/10.1002/(SICI)1097-0215(19980812)77:4<640::AID-IJC26>3.0.CO;2-8; PMID: 9679770
- Rowse GJ, Ritland SR, Gendler SJ. Genetic modulation of neu proto-oncogene-induced mammary tumorigenesis. Cancer Res 1998; 58:2675 - 9; PMID: 9635596
- Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI, Dickson R, Furth P, Hunter K, Kucherlapati R, et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci U S A 2002; 99:6967 - 72; http://dx.doi.org/10.1073/pnas.102172399; PMID: 12011455
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 78; http://dx.doi.org/10.1091/mbc.E10-06-0500; PMID: 21119005
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 2011; 286:12924 - 32; http://dx.doi.org/10.1074/jbc.M110.138958; PMID: 21300795
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 2012; 9:16 - 32; http://dx.doi.org/10.1038/nrclinonc.2011.177; PMID: 22124364
- Xiao D, Bommareddy A, Kim SH, Sehrawat A, Hahm ER, Singh SV. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS One 2012; 7:e32597; http://dx.doi.org/10.1371/journal.pone.0032597; PMID: 22457718
- Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013; 32:2261 - 72, 1-11; http://dx.doi.org/10.1038/onc.2012.252; PMID: 22733132
- de Cremoux P, Valet F, Gentien D, Lehmann-Che J, Scott V, Tran-Perennou C, Barbaroux C, Servant N, Vacher S, Sigal-Zafrani B, et al. Importance of pre-analytical steps for transcriptome and RT-qPCR analyses in the context of the phase II randomised multicentre trial REMAGUS02 of neoadjuvant chemotherapy in breast cancer patients. BMC Cancer 2011; 11:215; http://dx.doi.org/10.1186/1471-2407-11-215; PMID: 21631949
- Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra JM, Jacquemier J, et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat 2011; 126:407 - 20; http://dx.doi.org/10.1007/s10549-010-0897-9; PMID: 20490655
- Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, Vidaurre T, Holmes F, Souchon E, Wang H, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011; 305:1873 - 81; http://dx.doi.org/10.1001/jama.2011.593; PMID: 21558518
- Servant N, Bollet MA, Halfwerk H, Bleakley K, Kreike B, Jacob L, Sie D, Kerkhoven RM, Hupé P, Hadhri R, et al. Search for a gene expression signature of breast cancer local recurrence in young women. Clin Cancer Res 2012; 18:1704 - 15; http://dx.doi.org/10.1158/1078-0432.CCR-11-1954; PMID: 22271875
- Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol 2012; 23:395 - 401; http://dx.doi.org/10.1016/j.semcdb.2012.01.005; PMID: 22281437
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Vasko R, Goligorsky MS. Dysfunctional lysosomal autophagy leads to peroxisomal oxidative burnout and damage during endotoxin-induced stress. Autophagy 2013; 9:442 - 4; http://dx.doi.org/10.4161/auto.23344; PMID: 23328407
- Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007; 26:6469 - 87; http://dx.doi.org/10.1038/sj.onc.1210477; PMID: 17471238
- Tsuda H, Akiyama F, Terasaki H, Hasegawa T, Kurosumi M, Shimadzu M, Yamamori S, Sakamoto G. Detection of HER-2/neu (c-erb B-2) DNA amplification in primary breast carcinoma. Interobserver reproducibility and correlation with immunohistochemical HER-2 overexpression. Cancer 2001; 92:2965 - 74; http://dx.doi.org/10.1002/1097-0142(20011215)92:12<2965::AID-CNCR10156>3.0.CO;2-A; PMID: 11753973
- Latta EK, Tjan S, Parkes RK, O’Malley FP. The role of HER2/neu overexpression/amplification in the progression of ductal carcinoma in situ to invasive carcinoma of the breast. Mod Pathol 2002; 15:1318 - 25; http://dx.doi.org/10.1097/01.MP.0000038462.62634.B1; PMID: 12481013
- Carlsson J, Nordgren H, Sjöström J, Wester K, Villman K, Bengtsson NO, Ostenstad B, Lundqvist H, Blomqvist C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer 2004; 90:2344 - 8; PMID: 15150568
- Park K, Han S, Kim HJ, Kim J, Shin E. HER2 status in pure ductal carcinoma in situ and in the intraductal and invasive components of invasive ductal carcinoma determined by fluorescence in situ hybridization and immunohistochemistry. Histopathology 2006; 48:702 - 7; http://dx.doi.org/10.1111/j.1365-2559.2006.02403.x; PMID: 16681686
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956 - 9; http://dx.doi.org/10.1126/science.1225967; PMID: 23112296
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269 - 84; http://dx.doi.org/10.1016/j.cell.2013.08.015; PMID: 24034250
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510 - 27; http://dx.doi.org/10.1101/gad.2051011; PMID: 21764854
- Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, Barnard N, Ganesan S, Karantza V, White E, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov 2013; 3:894 - 907; http://dx.doi.org/10.1158/2159-8290.CD-13-0011; PMID: 23650262
- Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, Gorski SM. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat 2008; 112:389 - 403; http://dx.doi.org/10.1007/s10549-007-9873-4; PMID: 18172760
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528 - 39; http://dx.doi.org/10.1038/nrclinonc.2011.71; PMID: 21587219
- Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis 2011; 32:955 - 63; http://dx.doi.org/10.1093/carcin/bgr031; PMID: 21317301
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533 - 41; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047; PMID: 21878654
- Kongara S, Kravchuk O, Teplova I, Lozy F, Schulte J, Moore D, Barnard N, Neumann CA, White E, Karantza V. Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol Cancer Res 2010; 8:873 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-09-0494; PMID: 20530580
- Suárez E, Bach D, Cadefau J, Palacin M, Zorzano A, Gumá A. A novel role of neuregulin in skeletal muscle. Neuregulin stimulates glucose uptake, glucose transporter translocation, and transporter expression in muscle cells. J Biol Chem 2001; 276:18257 - 64; http://dx.doi.org/10.1074/jbc.M008100200; PMID: 11278386
- Zhang D, Tai LK, Wong LL, Chiu LL, Sethi SK, Koay ES. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol Cell Proteomics 2005; 4:1686 - 96; http://dx.doi.org/10.1074/mcp.M400221-MCP200; PMID: 16048908
- Walsh A, Cook RS, Rexer B, Arteaga CL, Skala MC. Optical imaging of metabolism in HER2 overexpressing breast cancer cells. Biomed Opt Express 2012; 3:75 - 85; http://dx.doi.org/10.1364/BOE.3.000075; PMID: 22254170
- Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev 2004; 18:2095 - 107; http://dx.doi.org/10.1101/gad.1204904; PMID: 15314031
- Rasband W. ImageJ. Bethesda, MD: U.S. National Institute of Health, 1997–2012
- Guy CT, Muthuswamy SK, Cardiff RD, Soriano P, Muller WJ. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes Dev 1994; 8:23 - 32; http://dx.doi.org/10.1101/gad.8.1.23; PMID: 7507074
- Bioconductor: http://www.bioconductor.org
- Benito M, Parker J, Du Q, Wu J, Xiang D, Perou CM, Marron JS. Adjustment of systematic microarray data biases. Bioinformatics 2004; 20:105 - 14; http://dx.doi.org/10.1093/bioinformatics/btg385; PMID: 14693816
- Alexe G, Dalgin GS, Scanfeld D, Tamayo P, Mesirov JP, DeLisi C, Harris L, Barnard N, Martel M, Levine AJ, et al. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res 2007; 67:10669 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-07-0539; PMID: 18006808
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545 - 50; http://dx.doi.org/10.1073/pnas.0506580102; PMID: 16199517