
Abstract
The islet in type 2 diabetes mellitus (T2DM) is characterized by a deficit in β-cells and increased β-cell apoptosis attributable at least in part to intracellular toxic oligomers of IAPP (islet amyloid polypeptide). β-cells of individuals with T2DM are also characterized by accumulation of polyubiquitinated proteins and deficiency in the deubiquitinating enzyme UCHL1 (ubiquitin carboxyl-terminal esterase L1 [ubiquitin thiolesterase]), accounting for a dysfunctional ubiquitin/proteasome system. In the present study, we used mouse genetics to elucidate in vivo whether a partial deficit in UCHL1 enhances the vulnerability of β-cells to human-IAPP (hIAPP) toxicity, and thus accelerates diabetes onset. We further investigated whether a genetically induced deficit in UCHL1 function in β-cells exacerbates hIAPP-induced alteration of the autophagy pathway in vivo. We report that a deficit in UCHL1 accelerated the onset of diabetes in hIAPP transgenic mice, due to a decrease in β-cell mass caused by increased β-cell apoptosis. We report that UCHL1 dysfunction aggravated the hIAPP-induced defect in the autophagy/lysosomal pathway, illustrated by the marked accumulation of autophagosomes and cytoplasmic inclusions positive for SQSTM1/p62 and polyubiquitinated proteins with lysine 63-specific ubiquitin chains. Collectively, this study shows that defective UCHL1 function may be an early contributor to vulnerability of pancreatic β-cells for protein misfolding and proteotoxicity, hallmark defects in islets of T2DM. Also, given that deficiency in UCHL1 exacerbated the defective autophagy/lysosomal degradation characteristic of hIAPP proteotoxicity, we demonstrate a previously unrecognized role of UCHL1 in the function of the autophagy/lysosomal pathway in β-cells.
Introduction
Type 2 diabetes is characterized by insufficient insulin secretion in the setting of insulin resistance resulting in hyperglycemia. Impaired insulin secretion in T2DM is associated with a deficit in β-cell mass and increased β-cell apoptosis.Citation1 The islet in T2DM is also characterized by extracellular islet amyloid deposits derived from islet amyloid polypeptide,Citation2 a 37-amino acid protein co-expressed with insulin by pancreatic β-cells. Moreover, membrane-permeant intracellular IAPP oligomers are also present to a greater extent in β-cells in T2DM.Citation3 Transgenic overexpression of human-IAPP (hIAPP) in β-cells of rodents may lead to diabetes development with an islet pathology that is comparable to that in humans, both being characterized by endoplasmic reticulum (ER) stress-induced β-cell apoptosis and formation of toxic intracellular IAPP oligomers.Citation3-Citation5
A structure of the toxic form of amyloidogenic proteins including IAPP has recently been elucidated, being composed of an out-of-register hexamer in the form of a cylinder (cylindrin).Citation6 Since intracellular formation of toxic IAPP oligomers is likely to contribute to β-cell dysfunction and apoptosis in T2DM, the questions arise, why do they form and why are they not removed? By implication, the cellular mechanisms present in long-lived secretory cells, such as β-cells, to prevent the formation of intracellular IAPP oligomers must be overcome in β-cells in T2DM.
The pathways of protein clearance, the ubiquitin/proteasome system and macroautophagy (hereafter referred to as autophagy), have been recently shown to play a crucial role in β-cell function and survival.Citation7-Citation15 Both of these protein degradation pathways are compromised in β-cells in T2DM.Citation9,Citation15 We recently reported that polyubiquinated proteins accumulate in β-cells in T2DM, also noted in β-cells of rodents with high expression of amyloidogenic hIAPP but not soluble rodent-IAPP.Citation9 In both β-cells of humans with T2DM and hIAPP transgenic rodents, the accumulation of polyubiquitinated proteins was attributed at least in part to a deficit in the deubiquinating enzyme UCHL1, a specific component of the ubiquitin-proteasome system.Citation9,Citation16
UCHL1 is abundantly expressed in neuronsCitation17 and β-cells.Citation14 UCHL1 hydrolyzes ubiquitin chains to allow the protein targeted for degradation to gain access to the proteasome, and leads to the generation and stabilization of free ubiquitin. Mutations and deficiency of UCHL1 in humans are associated with neurodegenerative diseases,Citation18 such as Parkinson and Alzheimer diseases.Citation19,Citation20 In mouse models with an intragenic deletion mutation in the Uchl1 gene, such as gracile axonal dystrophy (gad) miceCitation21 and nm3419 mice,Citation22 loss of UCHL1 expression and activity leads to neurological phenotypes and neurodegeneration.
Therefore, while it is established that there is β-cell deficiency of UCHL1 in T2DM,Citation9,Citation16 and that formation of misfolded intracellular IAPP oligomers in β-cells may induce deficiency of UCHL1,Citation9 to date it is unknown if a UCHL1 deficiency can initiate this adverse cycle. This is important as there is an increasing appreciation that widely used pesticides have the capacity to impair the function of the ubiquitin/proteasome system,Citation23 and recent studies suggest that T2DM may be more common in individuals exposed to environmental chemicals.Citation24 In the present studies, by use of the nm3419 mutant mouse model cross bred onto mice transgenic for human-IAPP, we have tested the overall hypothesis that impaired flux through the ubiquitin/proteasome system due to UCHL1 deficiency may initiate diabetes onset by accelerating β-cell loss through apoptosis.
A secondary question also addressed in these studies is whether a compromised ubiquitin/proteasome pathway for clearance of misfolded amyloidogenic IAPP is compensated for by a sufficient increased flux through the autophagy/lysosomal pathway, preventing intracellular accumulation of misfolded proteins, or rather exacerbates the defect in the autophagy/lysosomal pathway, disabling the secondary pathway for removal of misfolded aggregates.
Results
Heterozygous Uchl1nm3419 mutation results in a decrease in UCHL1 expression and function in mouse pancreatic islets
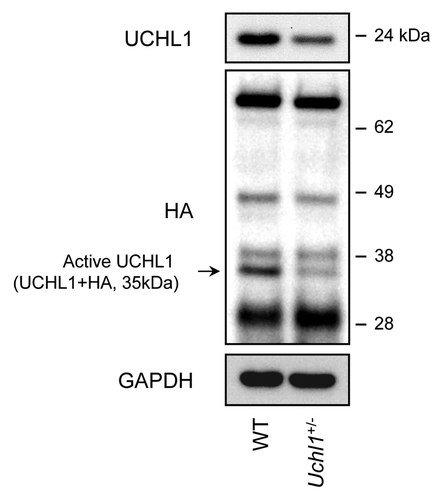
To confirm that the nm3419 mutation leads to a decrease in UCHL1 function in pancreatic islets, we analyzed islets isolated from 10-wk-old heterozygous Uchl1nm3419 mice (hereafter named Uchl1+/− mice). We first performed a western blot analysis to measure UCHL1 protein levels. Islets isolated from Uchl1+/− mice exhibited an ~50% decrease in UCHL1 protein levels in comparison to wild-type (WT) mice (). To measure UCHL1 activity, we performed labeling studies using the ubiquitin vinyl sulfone (Ub-VS) active site probe for deubiquitinating enzymes. This probe (tagged with HA) covalently binds to active deubiquitinating enzymes in the extract. Examination of the Ub-VS-treated islet extracts from WT or Uchl1+/− mice revealed several HA-reactive bands, including a 35-kDa band corresponding to UCHL1 situated with the probe.Citation25 This band was decreased by ~60% in Uchl1+/− mouse islets (), confirming a decrease in active UCHL1. Moreover, the analysis did not show any change in other labeled deubiquitinating enzymes in Uchl1+/− mouse islets as compared with WT islets, suggesting that the Uchl1nm3419 mutation predominantly alters the deubiquitinating function and expression of UCHL1 in mouse islets.
Figure 1. UCHL1 activity and expression are decreased in islets isolated from Uchl1nm3419 heterozygous mice (Uchl1+/−). Activity of UCHL1 was assessed by active-site labeling of deubiquitinating enzymes. Islet obtained from 8–10-wk-old WT and Uchl1+/− mice were incubated with HA-Ub-VS and lysates were analyzed by western blotting using anti-HA antibody. Levels of UCHL1 were analyzed by western blotting. Levels of GAPDH were shown as loading control.
A genetically induced deficit in UCHL1 accelerates the onset of diabetes in hIAPP transgenic mouse model
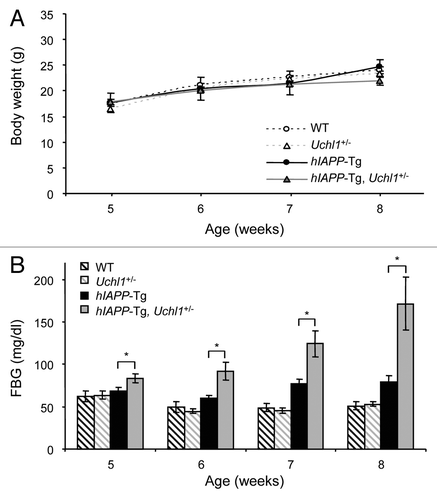
To test the hypothesis that UCHL1 deficiency would exacerbate the actions of amyloidogenic hIAPP to induce diabetes in hIAPP expressing mice, fasting blood glucose was measured weekly starting from 5 wk of age in the following 4 groups of mice: WT, Uchl1+/−, hIAPP transgenic (hIAPP-Tg) and hIAPP-Tg, Uchl1+/− mice. Body weight remained comparable in all 4 groups (). Fasting blood glucose concentrations were comparable in the WT and Uchl1+/− groups throughout the study period (). Fasting blood glucose modestly increased in hIAPP-Tg mice in comparison to control mice. In contrast, the fasting blood glucose progressively increased in hIAPP-Tg, Uchl1+/− mice from 5–8 wk of age (P < 0.05 vs. hIAPP-Tg mice; ); by 7 wk, hIAPP-Tg, Uchl1+/− mice developed impaired fasting blood glucose (124.3 ± 15.2 mg/dl vs. 77 ± 6 mg/dl; P < 0.05); and by 8 wk, hIAPP-Tg, Uchl1+/− mice developed overt diabetes (171.9 ± 30.8 mg/dl vs. 79.2 ± 7.8 mg/dl; P < 0.05; ).
Figure 2. UCHL1 deficiency accelerates diabetes progression in hIAPP-Tg mice. Body weight (A) and fasting blood glucose (B) were measured in the 4 groups of mice: wild-type (WT: n = 7); UCHL1 deficient (Uchl1+/−: n = 9), hIAPP transgenic (hIAPP-Tg: n = 9) and hIAPP transgenic mice deficient for UCHL1 (hIAPP-Tg, Uchl1+/−: n = 17 at 5, 6, and 7 wk; n = 7 at 8 wk). Data are expressed as mean ± SEM; *P < 0.05.
Importantly, there was an increase in both UCHL1 protein levels and mRNA in 7–8-wk-old hIAPP-Tg vs. WT mice (), whereas UCHL1 levels were decreased in 10-wk-old pre-diabetic hIAPP-Tg mice (Fig. S1). These data therefore uncover an early compensatory increase in UCHL1 expression in β-cells expressing high levels of hIAPP and reinforce the importance of UCHL1 in β-cells.Citation9 Altogether, these data support the hypothesis that a deficit in functional UCHL1 accelerates development of diabetes in mice expressing the amyloidogenic form of hIAPP.
Figure 3.Uchl1 mRNA and UCHL1 protein levels in mouse islets. (A) UCHL1 protein levels were assessed by western blotting using islet protein lysates obtained from 7–8-wk-old WT, Uchl1+/−, hIAPP-Tg, and hIAPP-Tg, Uchl1+/− mice (n = 3). GAPDH was used as a control. (B) Levels of Uchl1 mRNA were evaluated by RT-qPCR in islets isolated from 7–8-wk-old WT, Uchl1+/−, hIAPP-Tg, and hIAPP-Tg, Uchl1+/− mice (n = 2–4). Data are expressed as mean ± SEM. The mouse Ppia (peptidylprolyl isomerase A [cyclophilin A]) gene was used for the ratio.
![Figure 3.Uchl1 mRNA and UCHL1 protein levels in mouse islets. (A) UCHL1 protein levels were assessed by western blotting using islet protein lysates obtained from 7–8-wk-old WT, Uchl1+/−, hIAPP-Tg, and hIAPP-Tg, Uchl1+/− mice (n = 3). GAPDH was used as a control. (B) Levels of Uchl1 mRNA were evaluated by RT-qPCR in islets isolated from 7–8-wk-old WT, Uchl1+/−, hIAPP-Tg, and hIAPP-Tg, Uchl1+/− mice (n = 2–4). Data are expressed as mean ± SEM. The mouse Ppia (peptidylprolyl isomerase A [cyclophilin A]) gene was used for the ratio.](/cms/asset/0f50cded-ec0e-4839-8f21-cd7908543fa9/kaup_a_10928478_f0003.gif)
Deficiency in UCHL1 decreases β-cell mass but does not alter insulin sensitivity in hIAPP transgenic mice
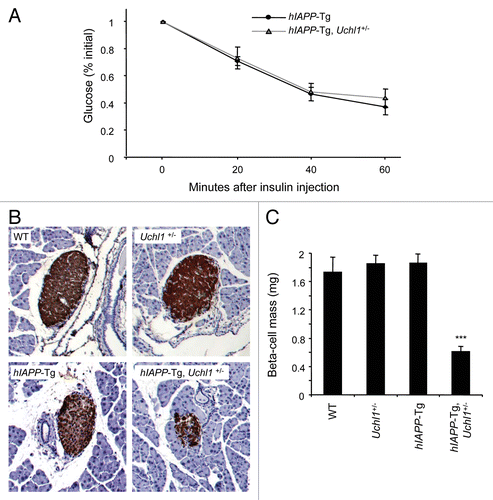
To distinguish between a β-cell deficit and/or insulin resistance to account for the acceleration of diabetes progression in hIAPP transgenic mice deficient for UCHL1, we evaluated insulin sensitivity and pancreatic β-cell mass. Insulin sensitivity was comparable in 7-wk-old hIAPP-Tg and hIAPP-Tg mice deficient for UCHL1 (). However, by 7–8 wk of age, β-cell mass in hIAPP-Tg, Uchl1+/− mice was one-third of that in hIAPP-Tg mice (P < 0.001; ) (pancreas weight was not different between all 4 groups, data not shown). In contrast, in WT mice expressing the soluble (nonamyloidogenic) form of IAPP, the deficit in UCHL1 function did not lead to either diabetes or a loss of β-cell mass (; ). In conclusion, the accelerated diabetes onset in hIAPP-Tg, Uchl1+/− mice was characterized by a marked decline in β-cell mass but with no change in insulin sensitivity. These data thus reveal the important role of UCHL1 in the maintenance of a functional β-cell mass in the context of expression of an amyloidogenic form of hIAPP in vivo.
Figure 4. Deficiency in UCHL1 decreases β-cell mass but does not alter insulin sensitivity in hIAPP-Tg mice. (A) Insulin tolerance tests were performed on 7-wk-old mice of the indicated genotypes: hIAPP-Tg (n = 10) and hIAPP-Tg, Uchl1+/− mice (n = 14). Results represent the blood glucose concentration as a percentage of the starting glucose value and are expressed as mean ± SEM (B) Representative pancreatic islets immunostained for insulin (brown) and counterstained with hematoxylin (blue) from WT; Uchl1+/−; hIAPP-Tg; and hIAPP-Tg, Uchl1+/− mice. (C) β-cell mass was evaluated in the 4 groups of 7–8-wk-old mice: WT (n = 4), Uchl1+/− (n = 4), hIAPP-Tg (n = 3) and hIAPP-Tg, Uchl1+/− mice (n = 4). Data are expressed as mean ± SEM; ***P < 0.001, significant differences vs. hIAPP-Tg mice.
Deficiency in UCHL1 leads to β-cell apoptosis in hIAPP transgenic mice
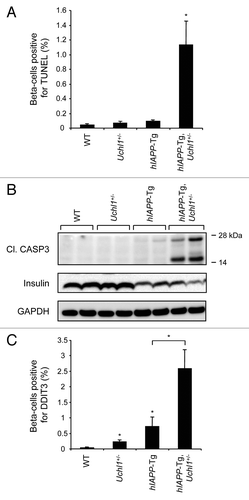
To test the hypothesis that the underlying mechanism for the decreased β-cell mass in hIAPP transgenic mice with UCHL1 deficiency was increased β-cell death by apoptosis, we quantified the frequency of TUNEL-positive β-cells in each group as well as the cleavage of CASP3/caspase 3 in isolated islets by western blotting. The frequency of TUNEL staining in β-cells was increased almost 13-fold in hIAPP-Tg, Uchl1+/− mice vs. hIAPP-Tg mice (1.14 ± 0.32% vs. 0.09 ± 0.02%, P < 0.05; ) and this was accompanied by increased cleavage of CASP3 (). We next investigated whether β-cell apoptosis in hIAPP-Tg, Uchl1+/− mice was related to ER stress. The transcription factor DDIT3/CHOP (DNA-damage-inducible transcript 3) is induced by ER stress and plays a central role in apoptosis execution.Citation26 Immunostaining analysis revealed an increased number of β-cells positive for nuclear DDIT3 in hIAPP-Tg, Uchl1+/− vs. hIAPP-Tg mice (2.59 ± 0.60% vs. 0.72 ± 0.31%, P < 0.05; ).
Figure 5. UCHL1 deficiency exacerbates β-cell ER stress and apoptosis in hIAPP-Tg mice. (A) β-cell apoptosis (percentage of β-cells positive for TUNEL) was evaluated in 7–8-wk-old WT (n = 3), Uchl1+/− (n = 3), hIAPP-Tg (n = 3) and hIAPP-Tg, Uchl1+/− mice (n = 3). (B) Protein levels of cleaved CASP3 (Cl. CASP3) were assessed by western blotting using islet protein lysates obtained from 7–8-wk-old WT (n = 2), Uchl1+/− (n = 4), hIAPP-Tg (n = 4) and hIAPP-Tg, Uchl1+/− mice (n = 5). Insulin and GAPDH were used as controls. (C) β-cell ER stress (percentage of β-cells positive for nuclear DDIT3) was evaluated in 7–8-wk-old WT (n = 3), Uchl1+/− (n = 3), hIAPP-Tg (n = 3) and hIAPP-Tg, Uchl1+/− mice (n = 3). Data are expressed as mean ± SEM; *P < 0.05, significant differences vs. hIAPP-Tg mice.
By quantifying β-cell replication using Ki-67 staining, we excluded an alteration in replication contributing to the deficit in β-cell mass in the UCHL1-deficient hIAPP-expressing mice. To the contrary, β-cell replication had a tendency to increase in hIAPP-Tg, Uchl1+/− mice vs. hIAPP-Tg mice (2.98 ± 0.27% vs. 1.82 ± 0.46%, P = 0.08; Fig. S2), presumably as a consequence of hyperglycemia,Citation27 signals released from apoptotic cellsCitation28 and/or suppression of the antiproliferative activity of UCHL1 in β-cells.Citation8
Therefore, the hypothesis that UCHL1 deficiency enhances vulnerability of hIAPP-expressing β-cells to ER stress and apoptosis is supported, confirming a key role of the ubiquitin/proteasome pathway in defense of β-cells from misfolded proteins.
UCHL1 dysfunction exacerbates the autophagy/lysosomal degradation disruption caused by high expression of hIAPP in β-cells
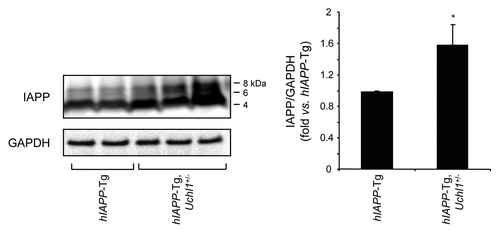
Autophagy serves as an alternative pathway for removal of misfolded proteins, particularly in long-lived cells such as pancreatic β-cells.Citation12 Autophagy is compromised in β-cells with accumulation of misfolded hIAPP.Citation15 We hypothesized that, since access of misfolded proteins to the proteasome in β-cells requires deubiquination by UCHL1, dysfunction of this enzyme would further compromise the autophagy pathway. Autophagy permits selective removal of aged or damaged organelles and degradation of ubiquitinated proteins.Citation29 In this pathway, a phagophore forms in the cytoplasm to surround such targets through the activity of specific autophagy effectors.Citation30 Among such effectors, microtubule-associated protein 1 light chain 3 (LC3) is cleaved into a soluble form known as LC3-I, which undergoes covalent conjugation to phosphatidylethanolamine to form the phagophore and autophagosome-bound LC3-II.Citation31 This lipidated form of LC3-I allows the formation of autophagosomes that then fuse with lysosomes where the sequestered material is degraded by hydrolytic enzymes. In this study, the number of autophagosomes was assessed by the conversion of LC3-I to LC3-II. In islets from hIAPP-Tg mice deficient in UCHL1, an increase in LC3-II was observed in comparison to hIAPP-Tg mice (), indicating an increased number of autophagosomes. To elucidate whether this increase is caused by an enhanced autophagosome formation or a decrease in lysosomal degradation, we examined protein levels of SQSTM1/p62 (sequestosome 1), a marker for impaired autophagy and/or defects in lysosomal degradation.Citation32,Citation33 We observed an increase in SQSTM1 levels in islets from hIAPP-Tg mice deficient in UCHL1 vs. hIAPP-Tg mice (), in both the soluble fraction and the insoluble fraction that most likely contains aggregated SQSTM1 (Fig. S3). Immunostaining for SQSTM1 revealed the presence of SQSTM1-positive cytoplasmic inclusions in β-cells of both hIAPP-Tg mice and hIAPP-Tg mice deficient in UCHL1 (). However, the number of β-cells containing detectable labeling for SQSTM1 was markedly increased in hIAPP-Tg mice deficient in UCHL1 (29 ± 2.7% vs. 3.5 ± 2.3%, P < 0.001; ). Both the frequency (P < 0.001, ) and size (P < 0.01; ) of these SQSTM1 inclusions were increased in β-cells of UCHL1-deficient mice that express hIAPP. Confocal images revealed a massive accumulation of large SQSTM1 inclusions in close proximity to LC3-labeled areas, which may either represent autophagosomes or aggregated LC3Citation34-Citation36 in hIAPP transgenic mice deficient for UCHL1 (). Furthermore, we noticed an accumulation of polyubiquitinated (lysine 63-specific chains) proteins in hIAPP-Tg, Uchl1+/− mice in comparison to hIAPP-Tg mice (). These polyubiquitinated proteins were restricted to SQSTM1-positive inclusions. Since lysine 63-linked ubiquitination selectively facilitates clearance of inclusions via autophagy,Citation37 their accumulation in β-cells of hIAPP-Tg, Uchl1+/− mice further confirms a defect in lysosomal clearance. Under these conditions, we hypothesized that intracellular IAPP would accumulate. In islets isolated from hIAPP-Tg, Uchl1+/− mice, we indeed detected a significant increase in IAPP and proIAPP protein levels when compared with hIAPP-Tg mice (; Fig. S4), revealing a previously unrecognized role for the ubiquitin/proteasome system and autophagy in IAPP clearance in β-cells. Altogether, these data support the postulate that a restraint in the potential flux through the proteasomal pathway for degradation of misfolded proteins due to UCHL1 deficiency exacerbates the β-cell defect in lysosomal degradation induced by expression of hIAPP sufficient to compromise cell function.
Figure 6. UCHL1 deficiency exacerbates defects in lysosomal degradation in hIAPP-Tg mouse islets. Protein levels of LC3 (A) and SQSTM1 (B) were assessed by western blotting using islet protein lysates obtained from 7–8-wk-old WT (n = 2), Uchl1+/− (n = 2), hIAPP-Tg (n = 2) and hIAPP-Tg, Uchl1+/− mice (n = 3). GAPDH was used as a control. The graph represents the quantification of LC3-II protein levels. (C) SQSTM1 protein levels were assessed by immunofluorescence (SQSTM1, red; insulin, green; nuclei, blue) in pancreatic tissue from 7–8-wk-old hIAPP-Tg mice and hIAPP-Tg, Uchl1+/− mice. (D) The graph represents the quantification of β-cells positive for SQSTM1 in each group (expressed in %). (E) The graph represents the quantification of β-cell area positive for SQSTM1 aggregates in each group (expressed in %). Data are expressed as mean ± SEM; **P < 0.01; ***P < 0.001, significant differences vs. hIAPP-Tg mice.
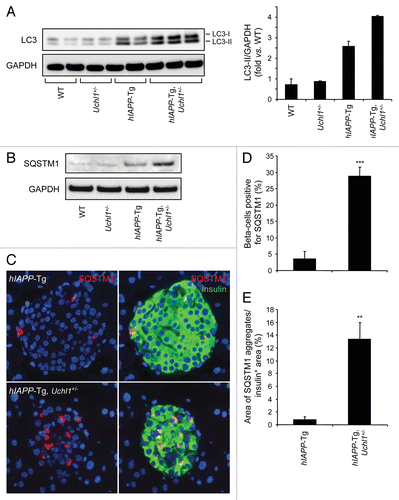
Figure 7. (A) Fluorescence confocal images of LC3 and SQSTM1 at magnification x 63 (LC3, red; SQSTM1, green; insulin, yellow; nuclei, blue) in pancreatic tissue from 7–8-wk-old hIAPP-Tg, Uchl1+/− mice. (B) The detection and localization of ubiquitin lysine 63 chains were assessed by immunofluorescence (ubiquitin K63, red; SQSTM1, green; insulin, white; nuclei, blue) in pancreatic tissue from 7–8-wk-old hIAPP-Tg mice and hIAPP-Tg, Uchl1+/− mice.
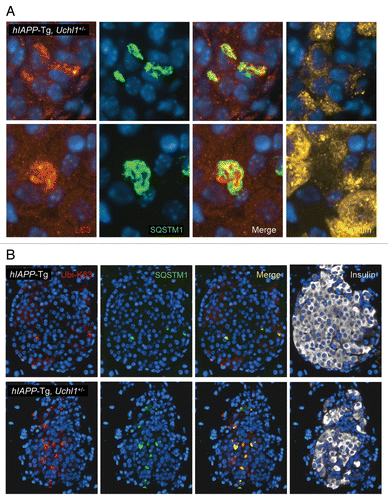
Figure 8. UCHL1 deficiency increases IAPP/proIAPP protein levels in hIAPP-Tg mouse islets. Protein levels of IAPP were assessed by western blotting using islet protein lysates obtained from 7–8-wk-old hIAPP-Tg mice (n = 3) and hIAPP-Tg, Uchl1+/− mice (n = 5). GAPDH was used as a control. The slowly migrating bands represent unprocessed and partially processed proIAPP (8 and 6 kDa, respectively). The graph represents the quantification of IAPP protein levels. Data are expressed as mean ± SEM; *P < 0.05.
Discussion
We report that a genetically induced partial deficit in UCHL1 accelerates the onset of diabetes in mice expressing amyloidogenic hIAPP in β-cells, thus affirming the hypothesis that impaired flux through the ubiquitin/proteasome system due to UCHL1 deficiency may initiate diabetes onset by accelerating β-cell loss through apoptosis. A partial deficit in UCHL1 did not alter islet development or β-cell survival in the absence of expression of amyloidogenic hIAPP. Only under high expression of hIAPP, as occurs under conditions of insulin resistance, did the partial defect in UCHL1 lead to a compromise in β-cell viability. This finding that UCHL1 deficiency contributes to loss of β-cell viability under stress conditions is consistent with another report showing that UCHL1 has a protective role against lipotoxicity-induced β-cell apoptosis.Citation8 The most common risk factor for T2DM is obesity that potentially confronts β-cells with both a high burden of IAPP expression and a lipotoxic milieu.Citation38,Citation39 Therefore, UCHL1 presumably plays an important role in β-cell protection in obesity.
We previously reported that UCHL1 protein levels were decreased in β-cells of human subjects with T2DM.Citation9 Since it is not possible to evaluate islets from humans who would subsequently develop diabetes, we cannot with certainty establish if the UCHLl deficiency in T2DM precedes or follows disease onset. The current study reveals that the former is plausible. Since β-cells are sensitive to oxidative stress and ER stress,Citation40,Citation41 either condition may lead to a UCHL1 deficit in either a pre-diabetic or diabetic state in humans. Indeed, silencing of Uchl1 was mediated by transcriptional repression through promoter hypermethylationCitation42 that could result from ER stress.Citation43 In neurons, UCHL1 may be modified by oxidative stress, leading to an alteration of its function.Citation19,Citation44
Besides these acquired alterations/modifications, UCHL1 has been described as a Parkinson disease susceptibility gene.Citation20 In light of the important role of UCHL1 in pancreatic β-cell function and survival, we can thus speculate that subjects with UCHL1 mutations are more susceptible to both neurodegeneration and T2DM, providing an eventual explanation to the positive association between T2DM and neurodegenerative diseases in some epidemiological studies.Citation45-Citation48
A secondary question addressed in these studies is whether a compromised ubiquitin/proteasome pathway for clearance of misfolded amyloidogenic proteins, such as IAPP, is compensated for by a sufficient increased flux through the autophagy/lysosomal pathway, to prevent the intracellular accumulation of misfolded proteins. To the contrary, we demonstrated that UCHL1 deficiency aggravates the lysosomal degradation defect induced by a high burden of hIAPP. This is illustrated by the marked accumulation of autophagosomes and SQSTM1-positive inclusions containing polyubiquitinated proteins. These inclusions can either promote cell survivalCitation49,Citation50 or death,Citation32,Citation51 depending on their stage of maturation, environmental conditions, and cell type.Citation52 Here, we hypothesized that under conditions of increased hIAPP expression, SQSTM1 may temporarily buffer toxicity by forming intracellular inclusions that sequester amorphous aggregated proteins (like those shown by Janson et al.Citation53) and delay diabetes. Given that SQSTM1 can play a role in β-cell survival,Citation15 the longstanding sequestration of SQSTM1 into inclusions may render cells vulnerable because of loss of SQSTM1 availability for other protective functions within β-cells. Therefore, although SQSTM1 inclusions may offer temporary cytoprotection against an overload of misfolded proteins in β-cells, if this burden is sustained, the ongoing sequestration of SQSTM1 into very large cytoplasmic inclusions as seen in hIAPP-Tg, Uchl1+/− mice may then lead to increased cell vulnerability to apoptosis. Furthermore, the overload (and likely dysfunction) of the autophagy/lysosomal system under conditions of UCHL1 deficiency—by redirection of the misfolded proteins from the proteasome—may also compromise recycling of damaged organelles, such as mitochondria (mitophagy), further having an impact on β-cell function and survival.
The present study supports the previously proposed crosstalk between the UCHL1/ubiquitin system and autophagy,Citation54 extended here to β-cells. UCHL1 physically interacts with LAMP2A (lysosomal-associated membrane protein 2A), the lysosomal receptor for chaperone-mediated autophagy (CMA), HSPA8/HSC70 (heat shock 70 kDa protein 8) and HSP90, which can function as components of the CMA pathway.Citation55 This interaction of UCHL1 with CMA machinery, enhanced with mutant and carbonyl-modified UCHL1, results in inhibition of this branch of autophagy and accumulation of SNCA/α-synuclein, a risk factor for Parkinson disease.Citation55,Citation56 A similar mechanism may contribute to the accumulation of IAPP in hIAPP-Tg mice harboring the Uchl1nm3419 mutation.
In conclusion, this study further confirms some parallels between T2DM and neurodegenerative diseases, both characterized by cell death associated with accumulation of misfolded/amyloidogenic proteins. Since T2DM and neurodegenerative diseases have common risk factors,Citation45 we can speculate that these diseases have similar alterations in response to their common risk factors such as UCHL1 deficiency. In the present study, we uncover a potential early role for UCHL1 in the evolution of β-cell dysfunction and vulnerability to apoptosis in T2DM.
Materials and Methods
Mouse models
We cross-bred homozygous hIAPP transgenic mice (FVB-Tg(IAPP)6Jdm/Tg(IAPP)6Jdm)Citation53 with a mutant Uchl1nm3419 mouse model on the C57BL/6J background.Citation22 The Uchl1nm3419 mouse contains a mutation in the Uchl1 locus resulting in the deletion of the final 78 amino acids of the UCHL1 protein.Citation22 Homozygous Uchl1nm3419 mice begin to exhibit signs of motor ataxia by 4 wk of age with a progressive decline notable by 16 wk of age.Citation22 It is important to note that the mice used in the present study were heterozygous for Uchl1nm3419 and none of these signs were seen.
In the second generation, cross-breeding led to the generation of mice homozygous for hIAPP (referred to as hIAPP-Tg), and mice homozygous for hIAPP/heterozygous for Uchl1nm3419 (referred to as hIAPP-Tg, Uchl1+/−). We also studied the nontransgenic counterparts of the Uchl1+/− mice. These were generated in an identical manner except for the substitution of wild-type FVB mice (Charles Rivers Laboratory, Wilmington, MA) for the FVB-Tg(IAPP)6Jdm/Tg(IAPP)6Jdm animals, this leading to the resulting wild-type mice (referred to as WT), and mice heterozygous for Uchl1nm3419 (referred to as Uchl1+/−).
Mice were bred and housed at the University of California, Los Angeles (UCLA), animal housing facility. The UCLA Institutional Animal Care and Use Committee approved all experimental procedures. Animals were maintained on a 12 h day/night cycle with Harlan Teklad Rodent Diet 8604 and water ad libitum. Males were used for the experiments.
Metabolic analysis
Fasted weight and blood glucose values were obtained in the morning after an overnight fast (clean cages and bedding, no food, water ad libitum). Blood glucose values were measured on a tail-tip blood sample with a FreeStyle Freedom Lite Glucometer (Abbott, Alameda, CA). The pancreata were then rapidly dissected from euthanized animals for morphological studies (see below).
An insulin tolerance test was performed on 7-wk-old mice after a 6 h fast. Blood glucose was measured before intraperitoneal injection of insulin (0.75 mU/g body wt; Lilly, NDC 0002-8215-17) and then 20, 40, and 60 min after injection. No anesthesia was used during the experiment.
Islet isolation
After an overnight fast, animals were euthanized using isoflurane. The bile duct was clamped at the entrance to the duodenum, cannulated, and the pancreas was perfused with 2 ml of collagenase solution (HBSS [Invitrogen, 14065] supplemented with 25 mM HEPES [Invitrogen, 15630], 0.23 mg/ml liberase [Roche, 05401020001], and 0.1 mg/ml DNase [Roche, 10104159001]). The pancreas was removed, transferred into a glass vial containing 2 ml of ice-cold collagenase solution, digested for 19 min at 37 °C, then dispersed by shaking for 30 s. Islets were manually picked, washed with ice cold PBS (154 mM NaCl [Sigma, S7653], 8.1 mM Na2HPO4 × 7 H2O [Sigma, S9390], 1.9 mM NaH2HPO4 × H2O [Sigma, S9638]) and lysed in lysis buffer (50 mM HEPES, 1% Nonidet P-40 [Sigma, I8896], 2 mM Na3VO4 [Sigma, S6508], 100 mM NaF [Sigma, S7920], 10 mM PyrPO4 [Sigma, P8010], 4 mM EDTA, 1 mM PMSF [Sigma, P7626], 1 μg/ml leupeptin [Sigma, L2023], and 1 μg/ml aprotinin [Sigma, A6103]). After 20 min incubation in lysis buffer on ice, islets were sonicated for 10 s and centifuged at 10,000 rpm at 4 °C for 10 min. The supernatant fraction was stored at −20 °C until use for subsequent protein determination by BCA assay (Bio-Rad, 500-0112) and western blotting.
Activity of deubiquitinating enzymes
Mouse islets were lysed in Nonidet P-40 lysis buffer (150 mM NaCl, 50 mM Tris [pH 7.4], 5 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 2 mM ATP [Sigma, A3377] and 0.5% Nonidet P-40) and rocking at 4 °C for 1 h. Lysates were centrifuged at 12,000 × g for 20 min at 4 °C. The protein concentration of lysates was established by the BCA assay, and adjusted to 5 μg/μl. To label active deubiquitinating enzymes, 50 μg of lysate was incubated with 0.8 μg of probe HA-Ub-VS (HA-ubiquitin vinyl sulfone; BostonBiochem, U-212) in a total volume of 60 μl of homogenization buffer (50 mM Tris [pH 7.5], 5 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 2 mM ATP and 250 mM sucrose [Sigma, S9378]). Samples were incubated for 30 min at room temperature and analyzed by western blotting. The labeled deubiquitinating enzymes were detected with the 12CA5 anti-HA monoclonal antibody (Roche, 11583816001).
Western blotting
Proteins (25–50 μg/lane) were separated on a 4–12% Bis-Tris NuPAGE gel and blotted onto a PVDF membrane (FluoroTrans; VWR, 29301856). Membranes were probed overnight at 4 °C with primary antibodies against the cleaved forms of CASP3 (Cell Signaling Technology, 9661S), LC3B (Cell Signaling Technology, 2775S), SQSTM1/p62 (Progen Biotechnik, GP62-C), GAPDH (Cell Signaling Technology, 2118S), insulin (Cell Signaling Technology, 3014S), IAPP (25–37 aa; Bachem, T-4157) and UCHL1 (Millipore, AB1761). Horseradish peroxidase-conjugated secondary antibodies were from Invitrogen (anti-rabbit: 65-6120, anti-rat: 62-9520, anti-mouse: 62-6420) or Jackson Immunoresearch Laboratories (anti-guinea pig: 706-035-148). Proteins were visualized by enhanced chemiluminescence (Millipore, WBKLS0500) and protein expression levels were quantified using the Labworks software (UVP).
Immunohistochemistry and immunofluorescence
Pancreata were fixed in 4% paraformaldehyde for 24 h at 4 °C and embedded in paraffin. Four-μm sections of pancreas were then taken through the fixed tissue in the plane of embedding so that a near complete section of pancreas (head, body, and tail) was obtained with each section. Sections were stained as described.Citation5
β-cell mass
Pancreatic sections were stained for insulin (Invitrogen, 18-0067) and hematoxylin. The β-cell mass was calculated by multiplying β-cell fractional area (insulin-positive area /total area of pancreatic section) by the pancreatic weight.
β-cell apoptosis
Sections were costained by immunofluorescence for terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL method; Roche, 12156792910) and insulin. At least 20 islets per animal (with a minimum of 20 β-cells per islet in plane of section) were examined in detail for the total number of TUNEL-positive β-cells. The frequency of TUNEL was presented as the percentage of stained β-cells among the total number of β-cells.
β-cell replication
Sections were costained by immunofluorescence for Ki-67 (BD PharMingen, 550609) and insulin. At least 20 islets per animal (with a minimum of 20 β-cells per islet in plane of section) were examined in detail for the total number of Ki-67-positive β-cells. The frequency of Ki-67 was presented as the percentage of stained β-cells among the total number of β-cells.
ER stress assessment
Sections were stained using antibodies against DDIT3/CHOP/GADD153 (Santa Cruz, sc-575) and insulin. The β-cells containing nuclear DDIT3 labeling and the total number of β-cells in each islet were counted. Results were expressed as percentage of β-cells positive for DDIT3.
Autophagy assessment
Sections were stained using the following antibodies: anti-SQSTM1/p62 (Progen Biotechnik, GP62-C), anti-polyubiquitin lysine 63-linkage-specific (Enzo Life Sciences, BML-PW0600-0025), anti-LC3B (Cell Signaling Technology, 2775S) and anti-insulin antibody. Slides were mounted with Vectashield with DAPI (Vector Laboratories, H-1200), viewed using a Leica DM6000 microscope (Leica Microsystems). Images were acquired using Openlab software (Improvision).
For detailed evaluation of islet β-cell area and number of β-cells containing SQSTM1 aggregates, 15 islets per animal (with a minimum of 20 β-cells per islet in plane of section) were selected randomly. The β-cell was considered to be positive if it had any SQSTM1 labeling with intensity above background staining of exocrine pancreas. For the computation of the area of SQSTM1 aggregates/insulin+ area, 15 representative islets were identified and analyzed. For each islet, the islet cross-sectional area positive for SQSTM1 was quantified and divided by the islet cross-sectional area positive for insulin using Image Pro Plus (Media Cybernetics).
Statistical analysis
Results are expressed as the means ± SEM for n independent experiments, as indicated in the figure legends. Statistical analyses were performed by the Student t test or ANOVA followed by Fisher post hoc test for multiple comparisons using Statistica (Statsoft). A P value of < 0.05 was taken as evidence of statistical significance. *P < 0.05, **P < 0.01, ***P < 0.001.
| Abbreviations: | ||
| CASP3 | = | caspase 3, apoptosis-related cysteine peptidase |
| CMA | = | chaperone-mediated autophagy |
| DDIT3 | = | DNA-damage-inducible transcript 3 |
| ER | = | endoplasmic reticulum |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| IAPP | = | islet amyloid polypeptide |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| SQSTM1 | = | sequestosome 1 |
| T2DM | = | type 2 diabetes mellitus |
| Tg | = | transgenic |
| TUNEL | = | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| UCHL1 | = | ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) |
| WT | = | wild type |
Additional material
Download Zip (195.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
These studies were supported by grants from the National Institutes of Health (DK059579) and the Larry L Hillblom Foundation (2007-D-003-NET and 2008-D-027-FEL to S.C.). We thank Chang Liu, Rosibel Hernandez, and Anahita Eskandari for their excellent technical help. We thank Mark Murata and Daniel Johansen for their help with image analysis. We thank Dr SM Wilson at the University of Alabama, Birmingham for kindly providing the Uchl1nm3419 mouse line.
References
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52:102 - 10; http://dx.doi.org/10.2337/diabetes.52.1.102; PMID: 12502499
- Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 2008; 29:303 - 16; http://dx.doi.org/10.1210/er.2007-0037; PMID: 18314421
- Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O’Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol 2010; 176:861 - 9; http://dx.doi.org/10.2353/ajpath.2010.090532; PMID: 20042670
- Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, Soeller WC, Butler PC. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab 2007; 293:E1656 - 62; http://dx.doi.org/10.1152/ajpendo.00318.2007; PMID: 17911343
- Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007; 56:2016 - 27; http://dx.doi.org/10.2337/db07-0197; PMID: 17475933
- Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, et al. Atomic view of a toxic amyloid small oligomer. Science 2012; 335:1228 - 31; http://dx.doi.org/10.1126/science.1213151; PMID: 22403391
- Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, Leibowitz G. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 2013; 62:1227 - 37; http://dx.doi.org/10.2337/db12-1474; PMID: 23274896
- Chu KY, Li H, Wada K, Johnson JD. Ubiquitin C-terminal hydrolase L1 is required for pancreatic beta cell survival and function in lipotoxic conditions. Diabetologia 2012; 55:128 - 40; http://dx.doi.org/10.1007/s00125-011-2323-1; PMID: 22038515
- Costes S, Huang CJ, Gurlo T, Daval M, Matveyenko AV, Rizza RA, Butler AE, Butler PC. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011; 60:227 - 38; http://dx.doi.org/10.2337/db10-0522; PMID: 20980462
- Costes S, Vandewalle B, Tourrel-Cuzin C, Broca C, Linck N, Bertrand G, Kerr-Conte J, Portha B, Pattou F, Bockaert J, et al. Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 2009; 58:1105 - 15; http://dx.doi.org/10.2337/db08-0926; PMID: 19223597
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008; 8:325 - 32; http://dx.doi.org/10.1016/j.cmet.2008.08.009; PMID: 18840363
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8:318 - 24; http://dx.doi.org/10.1016/j.cmet.2008.08.013; PMID: 18840362
- Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007; 56:930 - 9; http://dx.doi.org/10.2337/db06-1160; PMID: 17395740
- López-Avalos MD, Duvivier-Kali VF, Xu G, Bonner-Weir S, Sharma A, Weir GC. Evidence for a role of the ubiquitin-proteasome pathway in pancreatic islets. Diabetes 2006; 55:1223 - 31; http://dx.doi.org/10.2337/db05-0450; PMID: 16644676
- Rivera JF, Gurlo T, Daval M, Huang CJ, Matveyenko AV, Butler PC, Costes S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ 2011; 18:415 - 26; http://dx.doi.org/10.1038/cdd.2010.111; PMID: 20814419
- Bugliani M, Liechti R, Cheon H, Suleiman M, Marselli L, Kirkpatrick C, Filipponi F, Boggi U, Xenarios I, Syed F, et al. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic beta cell dysfunction. Mol Cell Endocrinol 2013; 367:1 - 10; http://dx.doi.org/10.1016/j.mce.2012.12.001; PMID: 23246353
- Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 1989; 246:670 - 3; http://dx.doi.org/10.1126/science.2530630; PMID: 2530630
- Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A 2013; 110:3489 - 94; http://dx.doi.org/10.1073/pnas.1222732110; PMID: 23359680
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 2004; 279:13256 - 64; http://dx.doi.org/10.1074/jbc.M314124200; PMID: 14722078
- Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Krüger R, Hattori N, Mellick GD, Quattrone A, Satoh J, et al, UCHL1 Global Genetics Consortium. UCHL1 is a Parkinson’s disease susceptibility gene. Ann Neurol 2004; 55:512 - 21; http://dx.doi.org/10.1002/ana.20017; PMID: 15048890
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet 1999; 23:47 - 51; http://dx.doi.org/10.1038/12647; PMID: 10471497
- Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, et al. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci 2008; 39:539 - 48; http://dx.doi.org/10.1016/j.mcn.2008.07.028; PMID: 18771733
- Wang XF, Li S, Chou AP, Bronstein JM. Inhibitory effects of pesticides on proteasome activity: implication in Parkinson’s disease. Neurobiol Dis 2006; 23:198 - 205; http://dx.doi.org/10.1016/j.nbd.2006.02.012; PMID: 16626962
- Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect 2012; 120:779 - 89; http://dx.doi.org/10.1289/ehp.1104597; PMID: 22296744
- Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol 2002; 9:1149 - 59; http://dx.doi.org/10.1016/S1074-5521(02)00248-X; PMID: 12401499
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13:184 - 90; http://dx.doi.org/10.1038/ncb0311-184; PMID: 21364565
- Weir GC, Bonner-Weir S. A dominant role for glucose in beta cell compensation of insulin resistance. J Clin Invest 2007; 117:81 - 3; http://dx.doi.org/10.1172/JCI30862; PMID: 17200709
- Bonner C, Bacon S, Concannon CG, Rizvi SR, Baquié M, Farrelly AM, Kilbride SM, Dussmann H, Ward MW, Boulanger CM, et al. INS-1 cells undergoing caspase-dependent apoptosis enhance the regenerative capacity of neighboring cells. Diabetes 2010; 59:2799 - 808; http://dx.doi.org/10.2337/db09-1478; PMID: 20682686
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805 - 12; http://dx.doi.org/10.1242/jcs.01131; PMID: 15169837
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 63; http://dx.doi.org/10.1016/j.cell.2007.10.035; PMID: 18083104
- Waguri S, Komatsu M. Biochemical and morphological detection of inclusion bodies in autophagy-deficient mice. Methods Enzymol 2009; 453:181 - 96; http://dx.doi.org/10.1016/S0076-6879(08)04009-3; PMID: 19216907
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262
- Shvets E, Elazar Z. Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 2008; 4:1054 - 6; PMID: 18776740
- Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K. Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 2008; 4:131 - 4; PMID: 18000393
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 2008; 17:431 - 9; http://dx.doi.org/10.1093/hmg/ddm320; PMID: 17981811
- Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell failure in type 2 diabetes: a case of asking too much of too few?. Diabetes 2013; 62:327 - 35; http://dx.doi.org/10.2337/db12-1326; PMID: 23349537
- Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995; 44:863 - 70; http://dx.doi.org/10.2337/diab.44.8.863; PMID: 7621989
- Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol Metab 2011; 22:266 - 74; PMID: 21458293
- Robertson RP. Beta-cell deterioration during diabetes: what’s in the gun?. Trends Endocrinol Metab 2009; 20:388 - 93; http://dx.doi.org/10.1016/j.tem.2009.05.004; PMID: 19748794
- Kagara I, Enokida H, Kawakami K, Matsuda R, Toki K, Nishimura H, Chiyomaru T, Tatarano S, Itesako T, Kawamoto K, et al. CpG hypermethylation of the UCHL1 gene promoter is associated with pathogenesis and poor prognosis in renal cell carcinoma. J Urol 2008; 180:343 - 51; http://dx.doi.org/10.1016/j.juro.2008.02.044; PMID: 18499164
- Bartoszewski R, Rab A, Twitty G, Stevenson L, Fortenberry J, Piotrowski A, Dumanski JP, Bebok Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem 2008; 283:12154 - 65; http://dx.doi.org/10.1074/jbc.M707610200; PMID: 18319256
- Kabuta T, Setsuie R, Mitsui T, Kinugawa A, Sakurai M, Aoki S, Uchida K, Wada K. Aberrant molecular properties shared by familial Parkinson’s disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum Mol Genet 2008; 17:1482 - 96; http://dx.doi.org/10.1093/hmg/ddn037; PMID: 18250096
- Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G. Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2008; 31:2003 - 5; http://dx.doi.org/10.2337/dc08-0688; PMID: 18599528
- Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007; 30:842 - 7; http://dx.doi.org/10.2337/dc06-2011; PMID: 17251276
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474 - 81; http://dx.doi.org/10.2337/diabetes.53.2.474; PMID: 14747300
- Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci 1993; 69:125 - 30; http://dx.doi.org/10.3109/00207459309003322; PMID: 8082998
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 2008; 106:107 - 20; http://dx.doi.org/10.1111/j.1471-4159.2008.05340.x; PMID: 18346206
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 27; http://dx.doi.org/10.1016/j.molcel.2009.01.021; PMID: 19250912
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001 - 4; http://dx.doi.org/10.1016/j.cell.2009.05.023; PMID: 19524504
- Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, Butler PC. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci U S A 1996; 93:7283 - 8; http://dx.doi.org/10.1073/pnas.93.14.7283; PMID: 8692984
- Bifsha P, Landry K, Ashmarina L, Durand S, Seyrantepe V, Trudel S, Quiniou C, Chemtob S, Xu Y, Gravel RA, et al. Altered gene expression in cells from patients with lysosomal storage disorders suggests impairment of the ubiquitin pathway. Cell Death Differ 2007; 14:511 - 23; http://dx.doi.org/10.1038/sj.cdd.4402013; PMID: 16888648
- Kabuta T, Wada K. Insights into links between familial and sporadic Parkinson’s disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 2008; 4:827 - 9; PMID: 18635949
- Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem 2008; 283:23731 - 8; http://dx.doi.org/10.1074/jbc.M801918200; PMID: 18550537