
Abstract
The autophagy receptor NBR1 (neighbor of BRCA1 gene 1) binds UB/ubiquitin and the autophagosome-conjugated MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) proteins, thereby ensuring ubiquitinated protein degradation. Numerous neurodegenerative and neuromuscular diseases are associated with inappropriate aggregation of ubiquitinated proteins and GSK3 (glycogen synthase kinase 3) activity is involved in several of these proteinopathies. Here we show that NBR1 is a substrate of GSK3. NBR1 phosphorylation by GSK3 at Thr586 prevents the aggregation of ubiquitinated proteins and their selective autophagic degradation. Indeed, NBR1 phosphorylation decreases protein aggregation induced by puromycin or by the DES/desmin N342D mutant found in desminopathy patients and stabilizes ubiquitinated proteins. Importantly, decrease of protein aggregates is due to an inhibition of their formation and not to their autophagic degradation as confirmed by data on Atg7 knockout mice. The relevance of NBR1 phosphorylation in human pathology was investigated. Analysis of muscle biopsies of sporadic inclusion body myositis (sIBM) patients revealed a strong decrease of NBR1 phosphorylation in muscles of sIBM patients that directly correlated with the severity of protein aggregation. We propose that phosphorylation of NBR1 by GSK3 modulates the formation of protein aggregates and that this regulation mechanism is defective in a human muscle proteinopathy.
Introduction
Efficient clearance of damaged or mutant proteins is essential for cellular homeostasis. In light of this, a common cytopathological feature of diseases called proteinopathies is the presence of intracellular UB/ubiquitin-positive protein aggregates.Citation1 Proteinopathies include neurodegenerative diseases like Alzheimer, Parkinson, and Huntington diseases as well as myopathies such as inclusion body myositis and myofibrillar myopathies. The pathological significance of protein aggregates is still a matter of debate and might vary among diseases. Aggregates trap potentially toxic mutant proteins but also trap proteins essential for cell homeostasis. Phenotypic amelioration of mouse models of several proteinopathies such as Huntington disease and spinocerebellar ataxia 1 tightly correlates with clearance of protein aggregates.Citation2-Citation4
Misfolded proteins are polyubiquitinated and targeted to the 2 major degradation systems in cells: the ubiquitin-proteasome and autophagy-lysosomal pathways. While the narrow barrel of the proteasome precludes entry of protein aggregates and organelles,Citation5,Citation6 such substrates can be degraded by macroautophagy, hereafter named autophagy.Citation2,Citation7 Autophagy involves the formation of double-membrane structures called autophagosomes, which fuse with lysosomes to form autolysosomes where the autophagosome content is degraded by proteases and acidic pH.Citation8 Random bulk autophagy is regulated by the phosphoinositide 3-kinase (PI3K)-AKT (v-akt murine thymoma viral oncogene homolog)-MTOR (mechanistic target of rapamycin [serine/threonine kinase]) pathway and provides amino acids for new protein synthesis and energy production. Recently, a selective autophagy pathway has been shown to ensure quality control of cellular components. Long-lived proteins, damaged organelles, some pathogens, and aggregate-prone proteins are ubiquitinated and recruited into autophagosomes by adaptor proteins among which NBR1 and SQSTM1/p62 (sequestosome 1) play a critical role. Indeed, these proteins are equipped with both a UBA (ubiquitin-associated) domain, with the capability to recognize and attach directly to polyubiquitin chains on the surface of aggregates, as well as an LC3-interacting region (LIR), able to recruit and directly bind to essential autophagosome membrane proteins.Citation9-Citation11
NBR1 and SQSTM1 are thought to have a double function during the selective autophagy of pathological ubiquitinated protein aggregates (named aggrephagy). They act as shuttles bringing ubiquitinated proteins to autophagosomes,Citation12 but they also participate in the formation of protein aggregates. Indeed, NBR1 knockdown decreases SNCA/α-synuclein aggregates in a cell model of Parkinson disease, and SQSTM1 overexpression enhances the formation of mutant SOD1 (superoxide dismutase 1, soluble) aggregates in a cell model of amyotrophic lateral sclerosis.Citation13,Citation14 NBR1 and SQSTM1 accumulate within abnormal intracellular protein aggregates in several proteinopathies including Alzheimer and Parkinson diseases, and the sporadic form of IBM.Citation9,Citation14-Citation16
Little is known about the mechanisms regulating selective autophagy and the protein aggregation process. Phosphorylation of SQSTM1 by CSNK2/CK2 (casein kinase 2) has recently been shown to increase the affinity of SQSTM1 for polyubiquitin chains resulting in increased autophagic degradation of ubiquitinated proteins.Citation12 Phosphorylation of the autophagy receptor OPTN/optineurin at Ser177 by TBK1 (TANK-binding kinase 1) enhances MAP1LC3/LC3 binding affinity and autophagic clearance of Salmonella.Citation17
Since its recent identification as an autophagy receptor for ubiquitinated proteins in selective autophagy, NBR1 has been shown to direct selective autophagic degradation of midbody derivatives and peroxisomes.Citation9,Citation11,Citation18,Citation19 The NBR1 protein is also involved in pleiotropic functions that appear unrelated to autophagy. It has been described as a scaffold protein in signaling pathways that mediates lysosomal degradation of activated FGF (fibroblast growth factor) receptors and subsequent attenuation of FGF signaling via its interaction with SPRED2 (sprouty-related, EVH1 domain containing 2).Citation20 NBR1 was also shown to inhibit receptor tyrosine kinase degradation.Citation21 NBR1 binds and is phosphorylated by the muscle giant sarcomeric protein TTN (titin).Citation22 Both proteins are part of a signaling complex that regulates SRF (serum response factor [c-fos serum response element-binding transcription factor])-dependent muscle gene expression. In humans, a mutation in TTN that disrupts its binding to NBR1 causes hereditary myopathy with early respiratory failure (HMERF).
In a yeast 2-hybrid screen, we identified an interaction between NBR1 and the kinase GSK3. GSK3 proteins, GSK3A and GSK3B, are components of the PI3K-AKT pathway. They are phosphorylated and inhibited by AKT kinases at Ser21 and Ser9, respectively.Citation23 GSK3 is involved in diverse physiological pathways ranging from metabolism, cell cycle, gene expression, and development and is linked to a large number of diseases including diabetes, cancer, and bipolar mood disorders.Citation24 Particularly, GSK3 activity is involved in several proteinopathies, making the finding of a direct interaction with NBR1 of particular interest. Indeed, GSK3 is activated in the striatum of Parkinson disease patientsCitation25 and its dysregulation has been involved in the formation of protein aggregates in Alzheimer and Huntington diseases.Citation26-Citation28 The involvement of both NBR1 and GSK3 proteins in some pathological protein aggregations led us to investigate the role of the NBR1-GSK3 interaction in this process. We chose to focus on muscle proteinopathies whose pathophysiological mechanisms of protein aggregation have not been studied extensively.
In this work, we show that NBR1 is a substrate of GSK3. NBR1 phosphorylation by GSK3 at Thr586 inhibits its adaptor function during the formation of pathological protein aggregates and the selective autophagy of ubiquitinated proteins. Interestingly, our work has established a link between NBR1 phosphorylation and protein aggregation in sIBM muscle pathology. Indeed, we have shown that NBR1 phosphorylation is strongly decreased in muscles of sIBM patients and directly correlates with the severity of protein aggregation in these patients. Our work thus connects selective autophagy defects and a human muscle pathology.
Results
NBR1 is phosphorylated by GSK3
In the context of a systematic protein-protein interaction network mapping effort for components of the PI3K-AKT-MTOR pathway, we screened a cDNA library from E10.5 mouse embryo and the hORFeome1.1 library with human GSK3A and GSK3B as baits.Citation29 We identified NBR1 as the most represented GSK3A and GSK3B interacting protein. These interactions were confirmed by co-affinity purification experiments in 293T cells (Fig. S1A). NBR1 bound to wild-type GSK3A and GSK3B and to constitutively active GSK3 mutants in which inhibitory phosphorylation sites Ser21 (GSK3A S21A) of GSK3A or Ser9 of GSK3B (GSK3B S9A) had been mutated to nonphosphorylable alanines. Similar to several GSK3A and GSK3B interaction partners,Citation29 NBR1 binding was lost with inactive GSK3 mutated in the active site (GSK3A K148A or GSK3B K85A). NBR1 possesses a N-terminal PB1 (phox and bemp1) domain, a ZZ-type zinc finger, 2 coiled-coil domains, 2 LIR, a J (juxta-UBA) domain and a C-terminal UBA domain (). Because GSK3 is a kinase, we tested if NBR1 was a substrate. In vitro radioactive 32P kinase assays showed that wild-type and constitutively active GSK3A and GSK3B efficiently phosphorylated NBR1 (). As expected, this phosphorylation was lost with inactive GSK3 mutants or upon GSK3 inhibition by 1-azakenpaullone, an inhibitor of GSK3 kinase activity (; Fig. S1B).Citation30 Altogether, our results show that GSK3A and GSK3B phosphorylate NBR1 in vitro.
Figure 1. NBR1 is phosphorylated by GSK3. (A) Schematic representation of NBR1 functional protein domains. Amino acid positions of phosphorylated threonine and serine residues (letter P in circle) on the human protein are shown. PB1, Phox, and Bemp1 domain; ZZ, ZZ-type zinc finger; CC1 and 2, coiled-coil domains; LIR1 and 2, LC3-interacting regions; J, juxta-UBA domain (membrane-interacting amphipathic α-helix), UBA, ubiquitin-associated domain. Alignment of NBR1 amino acids flanking GSK3 putative phosphorylation consensus site shows conservation of threonine and serine residues (indicated by stars) in different species. Gray color shows amino acids conserved in all depicted species, and numbers above stars indicate threonine and serine residue positions in the human NBR1 protein. H. sapiens, Homo sapiens; M. musculus, Mus musculus; G. gallus, Gallus gallus; X. tropicalis, Xenopus tropicalis; D. rerio, Danio rerio. (B) GSK3 in vitro kinase assays. GST-tagged GSK3A or GSK3B forms (wt, wild-type; inactive, GSK3A K148A or GSK3B K85A; active, GSK3A S21A or GSK3B S9A) and wild-type GST-tagged NBR1 proteins were purified from 293T cells and mixed with radioactive γ−32P ATP. The upper panel shows radioactive 32P signal and the lower panels show GST-tagged protein amounts revealed with an anti-GST antibody. Arrows indicate the bands corresponding to phosphorylation of NBR1 by GSK3, GSK3 autophosphorylation, and total proteins. (C) GSK3 in vitro kinase assays performed with purified active (S9A) or inactive (K85A) GST-GSK3B mutants mixed with wild-type or mutant GST-NBR1. Thr581, Thr586, and Ser590 were mutated to nonphosphorylable alanine (3Mut., T581A T586A S590A). Lower panel: adjusted phosphorylation signal calculated as the ratio of the relative amount of phosphorylated NBR1 (32P signal) to total GST-NBR1. (D) Western blot showing the specificity of an antibody generated against human NBR1 phosphorylated at threonine 586. Human HeLa cells and murine C2C12 cells were transfected with a control siRNA targeting luciferase or human or mouse NBR1-specific siRNA. (E) Western blot showing the level of phospho-NBR1T586 signal in gsk3a−/− MEFs cotransfected with plasmids encoding GST or GST-GSK3A forms (wt, wild-type; inactive, K148A; active, S21A) and DsRed-NBR1 forms (wt, wild-type; nonphosphorylable, T586A). Phosphorylation of NBR1 by endogenous GSK3B is detectable in the absence of wild-type and active GST-GSK3A. The phosphorylation status of this signal was confirmed through treatment of gsk3a−/− MEF protein extracts with lambda-phosphatase. (F) Confocal pictures showing the immunofluorescence staining of anti-phospho-NBR1T586 in C2C12 myoblasts transfected with wild-type DsRed-NBR1 or the nonphosphorylable DsRed-NBR1 T586A S590A double mutant. Inserts correspond to a magnification of the dotted frame in each picture. Right panels are merged pictures showing the presence or absence of colocalization. Scale bar: 15 μm. Data in this figure are representative of at least 2 independent experiments.
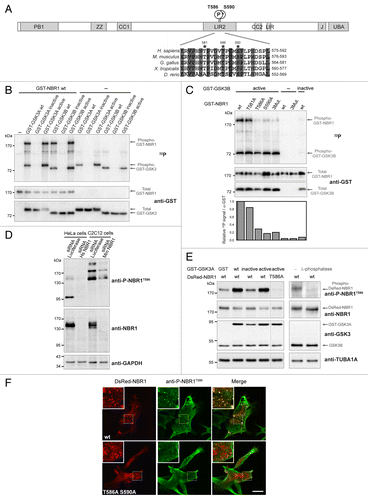
NBR1 is phosphorylated by GSK3 at Threonine 586
GSK3 preferentially phosphorylates substrates on serine and threonine residues followed by proline in a relay fashion.Citation31 The GSK3 consensus phosphorylation site is S/T-X-X-X-S/T-(P): a priming phosphorylation on a Ser/Thr residue is catalyzed by GSK3 or another kinase. This increases the rate of phosphorylation by GSK3 of another Ser/Thr residue located 4 amino acids upstream of the primed phosphorylation site. However, the phosphorylation of “unprimed” substrates by GSK3, such as AXIN and CTNNB1 (catenin [cadherin-associated protein]), β 1, 88 kDa), has also been described.Citation32 A bioinformatics search for GSK3 consensus phosphorylation sites on the NBR1 sequence with the NetPhosK 1.0 Server (http://www.cbs.dtu.dk/services/NetPhosK/) indicated human NBR1 Thr581, Thr586, and Ser590 as putative phosphorylated residues (). These 3 amino acids are followed by proline residues and are located in the LIR2 domain. Thr581 is not conserved in zebrafish, but Thr586 and Ser590 are widely conserved among animal species. Thr586 and Ser590 were confirmed to be essential amino acids for GSK3-dependent phosphorylation of NBR1, as mutations of these residues to nonphosphorylable alanines led to the loss of NBR1 phosphorylation by GSK3 in kinase assays (). Thr581 is probably not a phosphorylation site but co-affinity purification experiments showed that it was essential for GSK3 binding (Fig. S1C). To determine if NBR1 needs to be primed for GSK3 phosphorylation, we performed kinase assays with the GSK3B R96A mutant, a mutant that is unable to phosphorylate primed substrates but remains capable of phosphorylation of unprimed substrates.Citation32 NBR1 phosphorylation was reduced by 70% when NBR1 was incubated with GSK3B R96A (Fig. S1D). This strongly suggests that NBR1 is a GSK3 substrate that requires phosphorylation priming by GSK3 or another kinase at Ser590. To confirm and investigate the phosphorylation of NBR1 on Thr586 by GSK3 in vivo, we generated an antibody against human NBR1 phosphorylated on Thr586 (phospho-NBR1T586). Specificity of the antibody was verified in human HeLa cells and mouse immortalized C2C12 myoblasts using a specific siRNA (small interfering RNA) against NBR1 (). Anti-phospho-NBR1T586 detected several bands by western blot suggesting that phosphorylated NBR1 is subjected to other posttranscriptional modifications. Commercial antibody targeted against total NBR1 mostly recognized bands around 150 kDa despite a theoretical size of 108 kDa for the longest isoform of NBR1; this antibody might have less affinity for phosphorylated NBR1 forms migrating at others sizes, or these phosphorylated forms might be present in lower quantity. The level of phosphorylation of NBR1T586 detected by anti-phospho-NBR1T586 was consistent with GSK3 activity as shown with gsk3a−/− mouse embryonic fibroblasts (MEFs) expressing ectopic NBR1 and GSK3 activity mutants (). Moreover, anti-phospho-NBR1T586 did not recognize NBR1 when Thr586 was mutated to a nonphosphorylable alanine (T586A mutant, ). This confirms the specificity of our antibody for phosphorylated Thr586 and demonstrates that GSK3 phosphorylates NBR1T586 in cells. Finally, treatment of gsk3a−/− MEF protein extract with lambda-phosphatase confirmed that anti-phospho-NBR1T586 detected a residue phosphorylated by endogenous GSK3B (). Consistent with western blot results, immunofluorescence experiments performed in basal conditions in C2C12 transfected cells showed that anti-phospho-NBR1T586 recognized ectopic wild-type DsRed-NBR1 present as dots, but not the nonphosphorylable DsRed-NBR1 T586A S590A double mutant (). We also tested the level of NBR1 phosphorylation in response to stimuli that modulate GSK3 activity. Cell starvation inhibits the PI3K-AKT pathway, which leads to a decreased inhibitory phosphorylation of GSK3A at Ser21 by AKT. Accordingly, phosphorylation of NBR1T586 was increased in starved cells (Fig. S1E). Altogether, these findings show that GSK3 phosphorylates primed NBR1 at Thr586 in cells.
Phosphorylation of NBR1 by GSK3 modulates protein aggregation
Both GSK3 and NBR1 have independently been shown to modulate pathological protein aggregation.Citation14,Citation26,Citation28,Citation33 We tested if phosphorylation of NBR1 by GSK3 is involved in this process. For this purpose, we first used a drug-induced model of protein aggregation. Puromycin inhibits protein synthesis by disrupting peptide elongation on ribosomes causing premature chain termination during translation.Citation34 Resulting unfolded proteins are polyubiquitinated and form protein aggregates through binding with NBR1 and/or SQSTM1.Citation9 Indeed, puromycin treatment induced the formation of NBR1, SQSTM1 and UB-positive aggregates in C2C12 myoblasts ( and ). Following transfection with wild-type, inactive, or active GSK3, all aggregates were positive for NBR1, SQSTM1, and UB. Despite the presence of endogenous GSK3A/B, transfection of inactive GSK3 increased the proportion of cells harboring aggregates, whereas wild-type and constitutively active GSK3 reduced this proportion (). This was further confirmed by detection of endogenous NBR1, SQSTM1 and ubiquitinated proteins by western blot after the separation of puromycin-induced protein aggregates in a detergent-insoluble fraction (). Accordingly, following puromycin treatment, NBR1, SQSTM1, and ubiquitinated proteins accumulated in the pellet fractions obtained from cells transfected with inactive GSK3A. Conversely, protein amounts were reduced in pellet fractions of cells transfected with wild-type or constitutively active GSK3. GSK3 activity can thus modulate protein aggregation induced by puromycin. To confirm the impact of GSK3 on puromycin-induced aggregation and to deal only with endogenous proteins, we performed the same aggregation assay with Gsk3b+/+ and gsk3b−/− MEFs. MEFs are more resistant than C2C12 myoblasts to puromycin treatment. We left cells untreated or treated them with 7.5 μg/ml puromycin for 4 h (). Under puromycin treatment, 15% of Gsk3b+/+ MEFs showed protein aggregates that were positive for endogenous SQSTM1, UB, and NBR1. We observed a strong increase of the proportion of cells with protein aggregates in gsk3b−/− cells (44%). This clearly demonstrates the importance of GSK3 activity for puromycin-induced protein aggregation. The impact of NBR1 phosphorylation was next tested in a pathological model of protein aggregation. Desminopathies and myotilinopathies are part of myofibrillar myopathies, a group of rare genetic diseases with variable clinical features including a frequent progressive muscle weakness sometimes associated with cardiomyopathy and peripheral neuropathy.Citation35 A common morphological phenotype is the intracellular aggregation of the disease-related mutant protein i.e., myofibrillar proteins DES or MYOT (myotilin). The aggregates contain several other proteins including cytoskeletal proteins, phosphorylated MAPT/tau, and β-amyloid. In particular, DES aggregates have been described to colocalize with SQSTM1 and UB.Citation36 In cultured myoblasts, expression of the DES N342D mutant, harboring a mutation found in desminopathy patients,Citation37 led to the formation of one large protein aggregate containing round structures in half of the transfected cells (). These aggregates contained diffuse SQSTM1 and dotted UB and NBR1. Similar to puromycin-induced aggregates, the proportion of cells containing DES aggregates was modified by GSK3 activity mutants: inactive GSK3 increased the proportion of cells with DES aggregates, whereas this proportion was decreased with constitutively active GSK3 (). To test if the impact of GSK3 on aggregation was directly linked to GSK3-mediated NBR1 phosphorylation, we co-expressed GSK3 activity mutants with wild-type NBR1 or nonphosphorylable T586A S590A or phosphomimetic T586E S590E (Thr586 and Ser590 replaced with glutamic acids mimicking serine and threonine phosphorylation) NBR1 mutants. The quantification of aggregate-containing cells revealed that expression of the phosphomimetic NBR1 mutant counteracted the effect of inactive GSK3 on DES aggregation (). Consistently, the effect of active GSK3 was reversed by the expression of the nonphosphorylable NBR1 mutant. Importantly, the expression of NBR1 mutants alone recapitulated the impact of GSK3 activity mutants on protein aggregation. This places NBR1 downstream of GSK3 and shows that GSK3 prevented protein aggregation through NBR1 phosphorylation. In conclusion, phosphorylation of NBR1 by GSK3 on Thr586 modulates protein aggregation induced either by a drug or by a pathological mutant protein.
Figure 2. GSK3 reduces protein aggregation through NBR1 phosphorylation. (A) Confocal pictures of C2C12 myoblasts left untreated or treated with 7.5 μg/ml puromycin for 2 h and stained for endogenous ubiquitin (UB, green), NBR1 (red), and SQSTM1 (violet). Scale bar: 20 μm. (B) Proportion of C2C12 myoblasts harboring protein aggregates under puromycin treatment. Cells were nontransfected or transfected with EGFP empty vector or EGFP-GSK3A forms (wt, wild-type; inactive, K148A; active, S21A). Cells were left untreated (control) or treated with puromycin. Immunostainings performed with anti-SQSTM1 and anti-UB antibodies revealed protein aggregates. Since all SQSTM1-positive aggregates were found UB-positive, transfected cells with and without SQSTM1-positive aggregates were counted. At least 200 transfected cells were counted for each condition. Error bar represent the 95% confidence interval. ***P < 0.001; Pearson Chi-square test with Yates continuity correction. Five independent experiments were performed. (C) Upper panel: confocal pictures of Gsk3b+/+ and gsk3b−/− MEFs left untreated or treated with 7.5 μg/ml puromycin for 4 h and stained with DAPI (blue) and anti-SQSTM1 (gray) to reveal protein aggregates. Scale bar: 50 μm. Lower panel: Proportion of cells harboring protein aggregates. At least 200 cells were counted for each condition. Error bar represent the 95% confidence interval. **P < 0.01; ***P < 0.001; Pearson Chi-square test with Yates continuity correction. The experiment was reproduced 2 times independently. (D) Confocal pictures of C2C12 myoblasts transfected with wild-type or N342D mutant MYC-tagged DES. DES was revealed by either an anti-MYC antibody (upper panel, green) or an anti-DES antibody (lower panel, green). Endogenous UB (upper panel, red), SQSTM1 (upper panel, violet), and NBR1 (lower panel, red) stainings were also performed. Right panels are enlargements of the corresponding dotted frame. Scale bar: 20 μm. (E) Proportion of C2C12 myoblasts harboring DES N342D-MYC aggregates. Cells were cotransfected with DES N342D-MYC and EGFP or EGFP-GSK3A mutants (inactive, K148A; active, S21A) together with DsRed, wild-type or mutant DsRed-NBR1 (T586A S590A, nonphosphorylable; T586E S590E, phosphomimetic). Cells were immunostained with anti-MYC antibody to reveal DES N342D-MYC. Proportions of MYC-, GFP-, and DsRed-positive cells with and without DES-MYC aggregates were quantified. At least 200 transfected cells were counted for each condition. Error bar represent the 95% confidence interval. *P < 0.05; ***P < 0.001; Pearson Chi-square test with Yates continuity correction. Three independent experiments were performed.
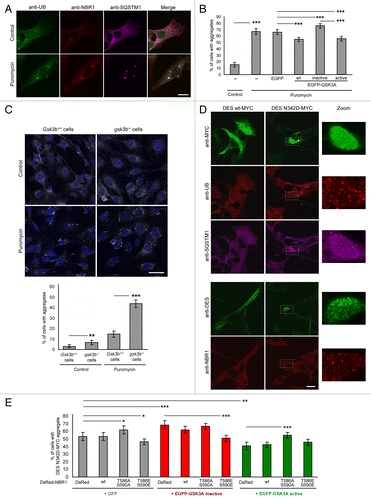
Figure 3. GSK3-dependent NBR1 phosphorylation reduces ubiquitinated protein aggregation independently of autophagic degradation. (A) Western blot of detergent-soluble (Supernatant) and detergent-insoluble (Pellet) fractions of C2C12 myoblasts transfected with GST or GST-GSK3A forms (wt, wild-type; inactive, K148A; active, S21A) and treated with DMSO (control) or with 7.5 μg/ml puromycin + DMSO or + 200 nM bafilomycin A1 for 2 h. Endogenous NBR1, SQSTM1, UB, GAPDH (loading control), and ectopic GST-GSK3A were analyzed by western blot. Ratio of protein levels quantified in pellet fractions to protein levels in supernatant fractions (Ratio P/S) were calculated and normalized to GST conditions. Supernatant and pellet fractions were run on the same gels but different film exposures are shown in upper panels for correct visualization of all fractions. In the lower panel, SQSTM1 levels are presented at the same exposure as a readout of puromycin and bafilomycin A1 treatment efficiencies: the addition of puromycin blocks protein translation and increases protein aggregation resulting in a reduced amount of proteins in supernatant fractions compared with control conditions. The addition of bafilomycin A1 blocks lysosomal degradation resulting in an enhanced quantity of proteins in supernatant and pellet fractions. (B) Microscopy pictures of muscle transverse sections of Atg7 muscle-specific knockout mice electroporated with control, inactive (K148A) or active (S21A) EGFP-GSK3A mutant expression vectors together with Histone2B-RFP expression vector. Protein aggregates were revealed with an anti-SQSTM1 antibody (red) and nuclei were stained with DAPI (blue in Merge). White stars show EGFP-GSK3-transfected cells in the anti-SQSTM1 panel. Pictures are representative data of several mice (Control, 3 mice; Inactive GSK3A, 2 mice; Active GSK3A, 5 mice). Right panel: proportion of electroporated muscle fibers without (white part of histogram bars) or with (dark gray part of histogram bars) protein aggregates. At least 180 fibers were counted for each condition. Error bar represent the 95% confidence interval. ***P < 0.001; Pearson Chi-square test with Yates continuity correction. Scale bar: 50 μm. (C) Microscopy pictures of muscle transverse sections of Atg7 muscle-specific knockout mice electroporated with control or DsRed-NBR1 forms (wt, wild-type; T586A S590A, nonphosphorylable; T586E S590E, phosphomimetic). Pictures are representative data of 3 mice for each condition. Right panel: proportion of electroporated muscle fibers with no protein aggregate (white part of histogram bars), speckles (light gray) or with protein aggregates (dark gray). Representative pictures of the 3 types of fibers are depicted below the stacked histogram. Overexpression of NBR1 induces the formation of speckles (< 1-μm diameter), independently of its phosphorylation status. NBR1 phosphorylation affects the proportion of remaining aggregates (> 1-μm diameter). At least 80 fibers were counted for each condition. Error bars represent the 95% confidence interval. N.S. = no significant difference, ***P < 0.001; Pearson Chi-square test with Yates continuity correction. Scale bar: 50 μm.
Phosphorylation of NBR1 by GSK3 modulates protein aggregation independently of autophagic degradation
Two hypotheses could explain the decrease of protein aggregation resulting from NBR1 phosphorylation by GSK3. NBR1 phosphorylation could either prevent protein aggregate formation or stimulate their degradation through autophagy. Using an inhibitor of lysosomal degradation, bafilomycin A1, we thus tested if the decrease in aggregates observed with GSK3 activation in muscle cells was due to aggregate degradation via autophagy. After transfection of C2C12 myoblasts with wild-type GSK3A and GSK3A activity mutants, cells were treated with puromycin, in the presence or absence of bafilomycin A1. We then performed a detergent separation of puromycin-induced aggregates (). In the presence of bafilomycin A1, NBR1, SQSTM1, and ubiquitinated proteins were still less accumulated in the detergent-insoluble fraction of cells expressing wild-type and active GSK3 as compared with cells expressing inactive GSK3, and control cells. Therefore, the decrease of protein aggregation in muscle cells caused by GSK3 activation is not due to their degradation in lysosomes.
To further exclude the role of autophagy in GSK3-NBR1-mediated clearance of protein aggregates, we used Atg7 muscle-specific knockout mice. ATG7 belongs to the autophagy conjugation system and is crucial for LC3 lipidation and autophagosome formation. Atg7 deletion in skeletal muscles results in severe muscle atrophy and accumulation of protein aggregates containing ubiquitinated proteins and SQSTM1.Citation38 Immunostaining with anti-NBR1 revealed that SQSTM1-positive aggregates also contained NBR1 (Fig. S2A). Interestingly, muscle fibers of Atg7 muscle-specific knockout mice, which were highly positive for phospho-NBR1T586 immunostaining, systematically contained smaller SQSTM1 aggregates compared with neighboring fibers (Fig. S2B). This is consistent with a role of phospho-NBR1T586 in aggregation inhibition. Adult muscles of autophagy-deficient mice were electroporated with inactive and active GSK3 mutants and the percentages of transfected fibers containing no aggregate or SQSTM1-positive aggregates were quantified (). Despite the context of autophagy inhibition, active GSK3 still decreased the proportion of fibers containing aggregates. Expression of the 3 different forms of NBR1 (wild type, nonphosphorylable, or phosphomimetic) by electroporation in Atg7 knockout mice induced the formation of NBR1, UB, and SQSTM1-positive speckles in a similar proportion, showing that ectopic NBR1 destabilized aggregates (size > 1-μm diameter) independently of its phosphorylation status to favor speckles (size < 1-μm diameter) (; Fig. S2D). In addition, phosphorylation of NBR1 by GSK3 had an impact on remaining aggregates. Indeed, wild-type and phosphomimetic NBR1 mutants decreased the proportion of fibers containing protein aggregates as compared with muscles transfected with nonphosphorylable NBR1. The phosphorylation status of ectopic wild-type NBR1 was confirmed by immunostaining of muscle sections performed with anti-phospho-NBR1T586 antibody (Fig. S2C). Importantly, large SQSTM1-positive aggregates almost disappeared when phosphomimetic NBR1 was expressed in adult autophagy-deficient fibers suggesting that NBR1 is able to affect pre-existing SQSTM1-positive aggregates. In conclusion, phosphorylation of NBR1 by GSK3 decreases protein aggregation both in cultured cells and in vivo in mice in an autophagy-independent manner, strongly suggesting that GSK3-mediated NBR1 phosphorylation affects the process of protein aggregation.
NBR1 phosphorylation inhibits its involvement in the formation of ubiquitinated protein aggregates
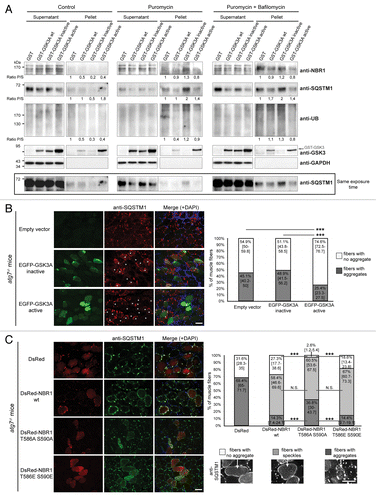
Western blot analysis of puromycin-induced protein aggregates isolated with detergents, and immunofluorescence experiments showed that the aggregation of ubiquitinated proteins in the presence of inactive GSK3 correlated with an accumulation of nonphosphorylated NBR1 in the aggregates (). This suggested that nonphosphorylated NBR1 participated in the formation of aggregates. We thus tested if protein aggregates induced by puromycin contained phospho-NBR1T586 in nontransfected HeLa cells (). While 74% of total NBR1 was found in the detergent-insoluble fraction, only 24% of phospho-NBR1T586 protein was contained in this fraction. Consistently, in muscle biopsies of sIBM and myotilinopathy patients, we observed that phospho-NBR1T586 was weakly included in protein aggregates whereas SQSTM1 and total NBR1 (most probably nonphosphorylated) strongly accumulated in aggregates (). Altogether, the exclusion of phospho-NBR1T586 from aggregates and its role as an inhibitor of protein aggregation suggests that phosphorylation of NBR1 at Thr586 inhibits its function as a cargo adaptor protein. In order to determine the molecular mechanisms of this inhibition, we tested if NBR1 phosphorylation impairs interaction with its partners involved in selective autophagy: SQSTM1, UB, LC3B, and GABARAP (GABA[A] receptor-associated protein). Wild-type NBR1, NBR1 T586A S590A, or NBR1 T586E S590E were all able to bind SQSTM1, UB, LC3B, or GABARAP in in vitro GST (glutathione S-transferase) affinity isolation experiments (Fig. S3A). We observed a slight increase of binding of SQSTM1 with NBR1 T586A S590A as compared with the phosphorylated forms of NBR1 (wild type and T586E S590E). In agreement, wild type, nonphosphorylable, and phosphomimetic NBR1 mutants were able to coimmunoprecipitate endogenous SQSTM1, LC3B, and ubiquitinated proteins (Fig. S3B). Again, nonphosphorylable NBR1 showed a slight increase of interaction with endogenous SQSTM1 compared with wild-type or phosphomimetic NBR1. However, we could not detect coimmunoprecipitated endogenous GABARAP with all NBR1 forms (not shown). We next investigated the colocalizations of the 3 forms of NBR1 with endogenous SQSTM1, UB, LC3B, or GABARAP (Fig. S4). NBR1-positive dots highly colocalized with SQSTM1 or LC3B-positive dots and weakly colocalized with UB or GABARAP-positive dots. No change of colocalization was observed using different NBR1 forms, both in basal conditions (Fig. S4) and after puromycin treatment (not shown). Phospho-NBR1T586 is thus physically able to interact with the autophagy partners tested. However, NBR1 phosphorylation might slightly reduce its interaction with SQSTM1 as shown in GST affinity isolation and coimmunoprecipitation experiments. Altogether, these results show that phosphorylated NBR1 does not participate in protein aggregation and this inhibits protein aggregate formation. NBR1 phosphorylation does not have a major influence on its interaction/colocalization with other proteins involved in autophagy.
Figure 4. GSK3-mediated phosphorylation inhibits NBR1 adaptor function and this prevents the selective autophagy of ubiquitinated proteins. (A) Western blot of detergent-soluble (Supernatant) and detergent-insoluble (Pellet) fractions of HeLa cells treated with puromycin. Endogenous phospho-NBR1T586, total NBR1, SQSTM1, and TUBA1A/α-TUB (tubulin, α) (loading control) were analyzed by western blot. Arrows show bands that have been found to be specific in NBR1-specific siRNA experiments. The proportion of total NBR1 and phospho-NBR1T586 present in the insoluble fraction was calculated as the ratio of (signal of x in pellet fraction * 100)/(signal of x in supernatant fraction + signal of x in pellet). 24% of phospho-NBR1T586 is present in the detergent-insoluble fraction whereas 74% of total NBR1 is contained in this fraction. (B) Western blot of extracts from C2C12 myoblasts transfected with GST or GST-GSK3A forms (wt, wild-type; inactive, K148A; active, S21A). Cells were treated with CHX and proteins were extracted at different time points to determine NBR1 stability. Ectopic NBR1 was detected with anti-NBR1 antibody and the same patterns were observed with anti-GFP antibody (not shown). Lower panel: quantification of the percentage of NBR1 (left) and of ubiquitinated proteins (right) degradation after 6 h of CHX treatment compared with their initial amounts. Half-life time extrapolations calculated from the quantifications of western blot are shown in rectangles. Data are mean of 3 independent experiments. Error bars represent standard deviations. *P < 0.05, Student t test. (C) Western blot of extracts from C2C12 myoblasts transfected with wild-type or mutant EGFP-NBR1 and treated with CHX. Lower panel: Quantification of the percentage of EGFP-NBR1 (left) and of ubiquitinated proteins (right) degradation after 6 h of CHX treatment compared with their initial amounts. Half-life time extrapolations calculated from quantifications of western blot are shown in rectangles. Data are mean of 3 independent experiments. Error bars represent standard deviations. *P < 0.05, Student t test.
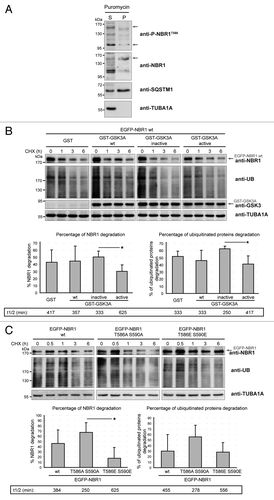
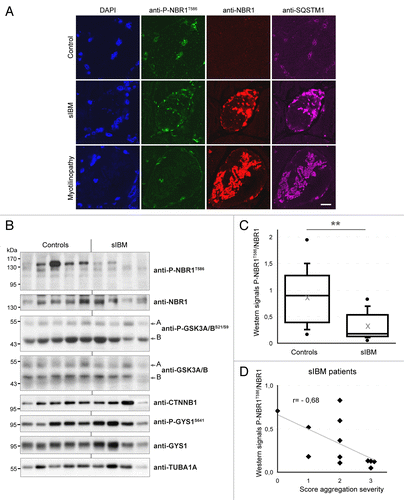
Figure 5. NBR1 phosphorylation is decreased in muscles of sIBM patients. (A) Confocal pictures of muscle sections from control, sIBM, and myotilinopathy patients stained for phospho-NBR1T586 (green), NBR1 (red), and SQSTM1 (violet). One to 5% of muscle fibers of sIBM patients show such protein aggregates. Nuclei were revealed with DAPI. All pictures have been acquired with the same laser gains. Scale bar: 20 μm. (B) Western blot analysis of endogenous phosphorylated and total NBR1, GSK3, and GYS1, and total CTNNB1 in protein extracts from control and sIBM patient muscle biopsies. (C) Tukey box plot of western blot phospho-NBR1T586 on NBR1 signal ratio. Boxes are delimited by quartiles Q1 and Q3 and crossed by median. Gray cross is the mean. Ends of bars are first and ninth deciles. Circles are minimum and maximum. Width of boxes is proportional to the effective: n = 14 controls and 13 sIBM patients. One sIBM sample with an extreme value has been excluded by Grubbs test at 5%. **P < 0.01, Student t test. (D) anti-P-NBR1T586/anti-NBR1 signals of each sIBM patient determined by western blot are represented as a function of scores of protein aggregation severity (cf Table S1). In gray is the trend line. r = Spearman rho correlation coefficient. P < 0.05: there is a significant inverse correlation between the level of remaining phospho-NBR1T586 and the severity of protein aggregation in sIBM muscle.
NBR1 phosphorylation prevents clearance of ubiquitinated proteins
We showed that NBR1 phosphorylation inhibited aggregation of ubiquitinated proteins in aggregation-induced models. To expand this role to the selective autophagy of ubiquitinated proteins, we tested if NBR1 phosphorylation by GSK3 modulates clearance of ubiquitinated proteins in basal conditions. Indeed, selective autophagy of ubiquitinated proteins requires the function of NBR1 as an adaptor protein. NBR1 then brings ubiquitinated proteins to autophagosomes through LC3 binding, and NBR1 is degraded together with its partners within autolysosomes.Citation9 C2C12 myoblasts were transfected with wild-type GSK3A and GSK3A activity mutants, or wild-type EGFP-NBR1 or NBR1 mutants alone. Protein synthesis was blocked with cycloheximide (CHX), and the levels of ectopic NBR1 and endogenous ubiquitinated proteins were quantified by western blot after 6 h of CHX treatment ( and ). A transitory accumulation of proteins was noted after 30 min CHX treatment, suggesting that CHX temporarily blocked protein degradation (). We observed that expression of active GSK3 or phosphomimetic NBR1 reduced the clearance of ubiquitinated proteins as compared with expression of inactive GSK3 or nonphosphorylable NBR1. Ectopic NBR1 phosphorylated by active GSK3, or the phosphomimetic form of NBR1, were also more stable than nonphosphorylated forms. These results show that NBR1 phosphorylation prevents the clearance of ubiquitinated proteins as well as its own degradation in basal conditions. In conclusion, NBR1 phosphorylation inhibits protein aggregation in pathological conditions, and the elimination of ubiquitinated proteins by basal selective autophagy.
NBR1 phosphorylation is deregulated in inclusion body myositis patients
We next addressed the status of NBR1 phosphorylation in muscle proteinopathies. We found that NBR1 accumulated within aggregates of a myotilinopathy patient (). Consistent with a previous report,Citation15 NBR1 was also present in protein aggregates observed in some muscle fibers of sIBM patients (). IBM is the most frequent age-related inflammatory muscle disease characterized by progressive muscle weakness, inflammation, muscle fiber atrophy, and the presence of protein aggregates in some muscle fibers.Citation39 The primary causes of sporadic forms of IBM are unknown and no treatment is currently available. We monitored the level of phospho-NBR1T586 in protein extracts of muscle biopsies from 13 sIBM patients and 14 age-related control patients. We found that phospho-NBR1T586 was strongly decreased in sIBM patients as compared with controls (). Several groups have observed dysregulation of GSK3 activity in muscle cells of sIBM patients, although both over- and underactivation were reported.Citation40,Citation41 In our patients, variable levels of phospho-GSK3A/BS21/S9 and of the GSK3 substrates CTNNB1 and phospho-GYS1 (glycogen synthase1 [muscle]) were found, precluding any conclusion about GSK3 activity status in sIBM muscles (). sIBM patients were scored for the severity of different phenotypes: inflammation, necrosis, and fibrosis based on histological staining of muscle sections, and protein aggregation based on SQSTM1 immunostaining (Table S1; Fig. S5A). These scores were compared with the level of phospho-NBR1T586 quantified by western blot on muscle biopsies (). No correlation was found between the level of phospho-NBR1T586 and the severity of inflammation, necrosis, or fibrosis (Fig. S5B). However, a significant inverse correlation was observed between the level of phospho-NBR1T586 and the severity of protein aggregation, meaning that among sIBM patients, the level of remaining NBR1 phosphorylation is a direct readout of protein aggregation (). In conclusion, NBR1 phosphorylation is highly deregulated in sIBM, a muscle disease with protein aggregates, and inversely correlates with the severity of aggregation.
Discussion
Although several autophagy receptors have been identified, little is known about the mechanisms that control their functions in vivo. Our work describes the first regulatory mechanism of NBR1 function as an autophagy receptor. We have established that NBR1 is regulated through phosphorylation by GSK3 kinase. NBR1 phosphorylation by GSK3 at Thr586 prevents intracellular pathological protein aggregation and selective autophagy of ubiquitinated proteins. Moreover, the analysis of human muscle biopsies shows a strong reduction of NBR1 phosphorylation in muscle of sIBM patients, the most frequent inflammatory myopathy in aged people, strongly suggesting a defect of this regulatory mechanism in a human proteinopathy.
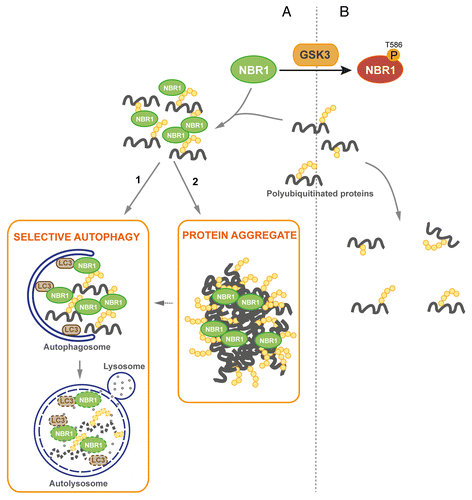
We observed that phosphomimetic NBR1 can eliminate pre-existing protein aggregates in vivo in Atg7 muscle-specific knockout mice. Two mechanisms can be considered for the mode of action of phosphorylated NBR1 on these pre-existing aggregates: it can favor their disruption or have an impact on their formation. It is not known whether aggregates of Atg7 knockout mice are stable or constantly disrupted and re-formed. The disappearance of protein aggregates following expression of phosphomimetic NBR1 could be explained by the ability of phosphorylated NBR1 to prevent the re-formation of protein aggregates. If aggregates are stable, one possibility is that overexpression of phosphomimetic NBR1 can disrupt protein aggregates. We noticed that phospho-NBR1T586 was weakly included in protein aggregates whereas nonphosphorylated NBR1 (when inactive GSK3 was expressed in cells) was strongly included in them. This observation favors the hypothesis that phosphorylation of NBR1 inhibits its adaptor function and prevents the formation of protein aggregates. We propose the following model for the regulation of protein aggregation and selective autophagy by Thr586 phosphorylation of NBR1 by GSK3 (): nonphosphorylated NBR1 binds ubiquitinated proteins in basal or pathological conditions participating in the formation of UB-bodies that are targeted to autophagosomes for degradation. In pathological conditions, saturation of degradation systems by an excess of mutant proteins leads to their accumulation and aggregation. When phosphorylated by GSK3 at Thr586, NBR1 cannot assemble ubiquitinated proteins. Consequently, UB-bodies are not formed and ubiquitinated proteins are not degraded.
Figure 6. Proposed model of regulation of protein aggregation by GSK3-mediated phosphorylation of NBR1 (A) Under its nonphosphorylated form, NBR1 acts as an adaptor for ubiquitinated proteins. Misfolded or mutant proteins are polyubiquitinated (yellow circles) and recognized by NBR1, which interacts with their polyubiquitin tail and brings them together to form UB-bodies. (1) Under normal conditions, selective autophagy occurs. NBR1 brings ubiquitinated proteins to autophagosomes through interaction with LC3 present at their membrane. Fusion with lysosomes leads to the degradation of ubiquitinated proteins and autophagy receptors by lysosomal proteases. (2) Under stress or pathological conditions, cells are overloaded with misfolded or mutant proteins and degradation pathways are saturated. Accumulation of ubiquitinated proteins leads to the formation of protein aggregates. (B) When phosphorylated by GSK3, NBR1 does not participate to the formation of UB-bodies. Consequently, ubiquitinated proteins and NBR1 undergo decreased degradation.
Phosphorylation of NBR1 does not interfere with its structural capacity to interact with its known autophagy partners, SQSTM1, UB, and LC3B, as determined with in vitro GST affinity isolation and coimmunoprecipitation experiments. However, we noted a slight reduction of phosphorylated NBR1 interaction with SQSTM1. One hypothesis is that phosphorylation of NBR1 by GSK3 could favor interactions of NBR1 with protein complexes potentially distinct from its autophagy receptor function, the existence of such complexes having been described.Citation20-Citation22 Phosphorylated NBR1 might thus have a role that is independent of selective autophagy.
The impact of GSK3 on protein aggregation that we show here is reminiscent of previous associations described between GSK3 and some pathological protein aggregation. Direct mechanistic links have been proposed for the key role of GSK3 on protein aggregation in Alzheimer and Parkinson diseases. Indeed, GSK3 interacts with the causative proteins APP (amyloid β [A4] precursor protein) and SNCA, and phosphorylation of MAPT by GSK3 has been shown to modulate MAPT aggregation in neurofibrillary tangles.Citation42 However, no direct mechanism has been described for the association described between GSK3 and Huntington disease. GSK3 inhibition by the drug SB216763 and overexpression of an inactive GSK3B mutant increases the formation of inclusions in cells expressing polyQ-mutant HTT (huntingtin) and rescues polyQ-induced cell death in cellular models of Huntington disease.Citation26,Citation28 It is not excluded that NBR1 phosphorylation might be an actor in this process.
GSK3 activity is inhibited by the PI3K-AKT pathway, a well-known regulator of nonselective autophagy. AKT activates MTOR by a cascade of phosphorylations, and MTOR inhibits autophagy through the inhibitory phosphorylation of ULK1 (unc-51 like autophagy activating kinase 1) and ATG13, proteins required for autophagy induction.Citation43 GSK3 has recently been shown to be part of this regulatory cascade. In conditions of serum deprivation, GSK3 activates autophagy through the phosphorylation and activation of the acetyltransferase KAT5/TIP60(K[lysine] acetyltransferase 5), which in turn acetylates and stimulates ULK1.Citation44 We observed that GSK3-mediated NBR1 phosphorylation was increased in serum-deprived cells and that NBR1 phosphorylation inhibited selective autophagy of ubiquitinated proteins. During starvation periods, we can speculate that GSK3 might influence a balance between activation of nonselective and inhibition of selective autophagy. This would favor random bulk autophagy that will provide the nutrients necessary for cell survival. In addition to its role in nonselective autophagy, our work opens the door to a new function for the PI3K-AKT pathway that will need to be further investigated. Through GSK3, this pathway could also modulate selective autophagy.
Our findings reveal a novel mechanism of selective autophagy regulation by GSK3 and highlight the role of the posttranslational modification of NBR1 in controlling intracellular pathological protein aggregation and selective autophagy of ubiquitinated proteins. Consistently, we have observed a strong reduction of NBR1 phosphorylation in muscles of sIBM patients, and residual levels of phospho-NBR1T586 in sIBM patients directly correlated with the severity of protein aggregation. This is the second observation that reports NBR1 defects in a human muscle proteinopathy. Indeed, a human mutation in TTN that disrupts the binding to NBR1 causes HMERF.Citation22 HMERF patients have features similar to myofibrillar myopathies including myofibrillar lesions and intracellular protein aggregates positive for actin, MYOT and SQSTM1.Citation22,Citation45,Citation46 Importantly, experiments performed with Atg7 muscle-specific knockout mice indicated that phosphomimetic NBR1 is able to eliminate pre-existing protein aggregates in vivo. Our finding that NBR1 phosphorylation is regulated by GSK3, a druggable kinase with known inhibitors already in pre-clinical trials for diabetes and Alzheimer disease,Citation47 offers a great way to test the functional impact of protein aggregates in various proteinopathies and might potentially represent promising therapeutic strategies that would aim at modifying pathological protein aggregates. We propose that NBR1 dysregulation has a key role in the pathogenesis of proteinopathies and that modulation of NBR1 phosphorylation may provide novel opportunities for therapeutic approaches.
Materials and Methods
Study approval
Care and manipulation of mice were performed in accordance with European legislations on animal experimentation and approved by the Italian Ministry of Health. Investigations on human samples have been conducted according to the Declaration of Helsinki principles. Muscle biopsies are part of a tissue bank authorized by the French Ministry of Social Affairs and Health as DC2008-139 with cession authorization AC 2008-113. All study participants provided informed written consent.
Cell lines, culture conditions, transfections, and drugs
Murine C2C12 myoblasts (ATCC CRL-1772), human embryonic kidney 293T cells (ATCC CRL-3216), human cervix adenocarcinoma HeLa cells (ATCC CCL-2) and Gsk3b+/+, gsk3b−/−, gsk3a−/− MEFs were grown at 37 °C under 5% CO2. Gsk3 MEFs were kindly provided by Dr James Woodgett (Lunenfeld Research Institute, Toronto, Canada).Citation48 C2C12 cells were maintained as myoblasts in growth medium: Dulbecco’s modified Eagle’s medium (DMEM, PAA Laboratories-GE Healthcare Life Sciences, E15-810) supplemented with 20% fetal bovine serum (FBS, PAA Laboratories-GE Healthcare Life Sciences, A15-102). 293T and HeLa cells were cultured in DMEM supplemented with 10% FBS, and MEFs were maintained in DMEM supplemented with 10% FBS and 1 mM Na-pyruvate.
Transfections of C2C12 cells were performed with Lipofectamine2000 (Life Technologies-Invitrogen, 11668-019) according to the manufacturer’s instructions. For immunofluorescence studies, C2C12 cells were electroporated with a Microporator (Neon transfection system, Life Technologies) at 1400V during 20 ms, 1 time. Electroporated cells were then grown on glass coverslips in tissue culture plates. 293T cells were transfected using calcium chloride. siRNA transfections were performed with Stealth RNAi siRNA (Life Technologies-Invitrogen) using RNAiMAX (Life Technologies-Invitrogen, 13778-075). Effects of siRNA NBR1, whose sequences are depicted in Table S2, were compared with a control siRNA targeting Luciferase (Life Technologies-Invitrogen, 12935146). Cells were seeded in 35-mm diameter tissue culture dishes and transfected with 40 pmol siRNA. A second identical transfection was performed 24 h later. Cells were used for experiments 72 h after the first transfection. Drugs used in this study are puromycin dihydrochloride used at a final concentration of 7.5 μg/ml (Sigma-Aldrich, P8833) for 2 h with C2C12 myoblasts and for 4 h with MEFs, bafilomycin A1 at 200 nM (Sigma-Aldrich, B1793) for 2 h, 1-azakenpaullone at 1 μM (Sigma-Aldrich, A3734) and cycloheximide (Sigma-Aldrich, C7698) used at 50 μg/ml. For phosphatase treatment, 50 μg of proteins extracted from gsk3a-/- MEFs without phosphatase inhibitors were treated with 250 units of λ-phosphatase (New England Biolabs, P0753S) for 30 min at 30 °C.
Plasmids, constructs, and accession numbers
Several constructs used in this study were subcloned into eukaryotic vectors using the Gateway recombination technology (Life Technologies). The full-length open reading frames for human NBR1 (GenBank NM_005899.3), UBC (ubiquitin C; NM_01177413.1), MAP1LC3B/LC3B (NM_022818.4) and SQSTM1/p62 isoform 1 (NM_003900.4) were amplified with primers depicted in Table S2 and subcloned by restriction enzyme digestion (KpnI-XhoI or KpnI-NotI) into the Gateway pENTR1A plasmid to create entry vectors. Human full-length UBC was a kind gift of Dr Pierre Jalinot and Dr Christelle Morris (LBMC, Lyon, France). The panel of amino acid mutations was engineered by PCR-based mutagenesis from the cDNA encoding wild-type proteins using Pfu DNA polymerase (Promega, M774B) and primer sets depicted in Table S2. Double NBR1 mutants (NBR1 T586A S590A and NBR1 T586E S590E) were generated in 2 steps and the NBR1 T581A T586A S590A triple mutant was constructed in 3 steps. Most of the GSK3A (NM_019884.2) and GSK3B isoform 2 (NM_001146156.1) wild-type and mutant entry constructs were previously described.Citation29 Human GABARAP (NM_007278.1) entry vector was a kind gift of Professor Mathias Faure (CIRI, Lyon, France). All entry vectors were transferred to the pDEST27 (for mammalian expression of N-terminal GST fusions; Life Technologies, 11812-013) Gateway destination vector using the LR recombination system following the manufacturer’s instructions. NBR1 and GSK3A entry vectors were transferred to pDEST-EGFP-C1 (for mammalian expression of N-terminal EGFP fusions) and NBR1 entry vectors were transferred to pDEST-DsRed-Cter (for mammalian expression of N-terminal DsRed fusions). Human full-length wild-type and N342D DES (NM_001927.3) in the pcDNA3-MYC vector were kindly provided by Drs Jocelyn Laporte and Karim Hnia (IGBMC, Illkirch, France). All constructs were verified by sequencing.
Antibodies
Anti-phospho-NBR1T586 rabbit polyclonal antibody was raised against a phosphorylated peptide encompassing human NBR1 Thr586 (HNTPVDV-T[PO3H2]-PCMSP) and produced by Eurogentec. Sera were purified on a first column coupled with the phospho-peptide and negatively purified on a second column coupled with the nonphosphorylated peptide. The immune response evolution and final antibody solution were monitored by ELISA (enzyme-linked immunosorbent assay) screening. The following commercial antibodies were used in this study: mouse monoclonal anti-NBR1 antibody (Abnova, clone 6B11, H00004077-M01), mouse monoclonal anti-TUBA1A/α-TUB (Sigma-Aldrich, clone B-5-1-2, T6074), mouse monoclonal anti-GSK3A/B (Santa Cruz Biotechnology, clone 0011-A, sc-7291), mouse monoclonal anti-CTNNB1 (Sigma-Aldrich, clone 15B8, C7207), mouse monoclonal anti-cMYC (Covance, clone 9E10, MMS-150R), mouse monoclonal anti-GFP (Santa Cruz Biotechnology, clone B-2, sc-9996), mouse monoclonal anti-SQSTM1/p62 (used for immunohistochemistry experiments on patients; BD Biosciences, 610833), mouse monoclonal anti-GABARAP (MBL, clone 1F4, M135-3B), mouse monoclonal anti-UB/ubiquitin (Enzo Life Sciences, FK2, BML-PW8810-0100), mouse monoclonal anti-LC3B (used for immunofluorescence experiments; Enzo Life Sciences, clone 5F10, ALX-803-080-C100), rabbit polyclonal anti-LC3B (used for western blotting; Cell Signaling Technology, 2775), rabbit polyclonal anti-UB (Enzo Life Sciences, BML-UG9510), rabbit polyclonal anti-GST (Sigma-Aldrich, G7781), rabbit polyclonal anti-GFP (used for coimmunoprecipitation experiments; Life Technologies, A-11122), rabbit monoclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Cell Signaling Technology, clone 14C10, 2128), rabbit polyclonal anti-phospho-GSK3A/BS21/S9 (Cell Signaling Technology, 9331), rabbit polyclonal anti-phospho-GYS1/glycogen synthaseS641 (Cell Signaling Technology, 3891), rabbit monoclonal anti-GYS1 (Cell Signaling Technology, clone 15B1, 3886), rabbit monoclonal anti-DES (desmin; Cell Signaling Technology, clone D93F5, 5332), rabbit polyclonal anti-SQSTM1/p62 (used in mice experiments; Sigma-Aldrich, P0067), and guinea pig polyclonal anti-SQSTM1/p62 (Progen Biotechnick, GP62-C). For horseradish peroxidase-coupled secondary antibodies, sheep anti-mouse (NA931V) and sheep anti-rabbit (NA934V) antibodies were from GE Healthcare, and goat anti-guinea pig antibody was from Jackson ImmunoResearch (106-035-003). For fluorophore-coupled secondary antibodies, goat Fab’2 anti-mouse DyLight488 (115-486-072) or DyLight549 (115-506-072), donkey anti-rabbit TRITC (711-025-152), and goat anti-guinea pig TexasRed (106-076-003) or Alexa Fluor 647 (106-496-003) were from Jackson ImmunoResearch. Donkey anti-rabbit Alexa Fluor 488 was from Molecular Probes (A-21206).
Genetically modified mice and in vivo transfection
All mouse experiments were performed on 5-mo-old females of muscle-specific atg7−/− mice.Citation38 Mice were housed in an environmentally controlled room (23 °C, 12 h light/12 h dark cycle) and provided food and water ad libitum. In vivo transfection experiments were performed by intramuscular injection of DNA plasmids in the tibialis anterior muscle followed by electroporations as previously described.Citation49 We used 20 µg of EGFP-GSK3 or DsRed-NBR1 expression vectors, 5 µg of empty vectors and 5 µg of H2B-RFPI-N1 expression vector. Mice were sacrificed in the morning, 12 d after transfection, and tibialis anterior muscles were dissected and frozen in liquid nitrogen for subsequent analyses.
Immunofluorescence experiments and microscope imaging
For immunofluorescence experiments using cultured cells, cells on glass coverslips were fixed for 20 min in 4% paraformaldehyde in phosphate-buffered saline (PBS; Sigma-Aldrich, D8537) prior to permeabilization for 10 min with PBS-0.2% Triton X-100. Nonspecific sites were blocked in PBS-0.1% Triton X-100 supplemented with 10% FBS. Cells were incubated with primary antibodies diluted in PBS-0.1% Triton X-100 with 3% FBS for 2 h at room temperature. After washing with PBS-0.1% Triton X-100, cells were incubated for 45 min with a fluorophore-coupled secondary antibody and then for 10 min in a 200 nM DAPI solution that stains DNA. Immunofluorescence experiments with anti-LC3B 5F10 antibody were performed with the following protocol kindly provided by Drs Christopher Lamb and Sharon Tooze (London Research Institute, London). Cells were fixed/permeabilized with cold methanol and blocked with 1% BSA + 0.2% gelatin (Sigma-Aldrich, G8150) for 20 min. Coverslips were incubated with anti-LC3B 5F10 antibody for 30 min in block solution, washed, and incubated with anti-mouse secondary antibody for 30 min. After washing, cells were mounted onto slides. Immunofluorescence staining on human patient muscles was performed on 10-μm width isopentane frozen sections of muscle biopsies. Muscle sections were permeabilized in PBS-0.5% Triton X-100-5% normal goat serum (NGS; PAA Laboratories-GE Healthcare Life Sciences, B11-035) for 30 min at room temperature. Primary antibodies were diluted in PBS-0.1% Triton X-100-5% NGS and incubated overnight at 4 °C. After washing with PBS-0.1% Triton X-100, muscle sections were incubated for 1 h at room temperature with secondary antibodies diluted in PBS-0.1% Triton X-100-5% NGS and then with DAPI for 10 min. Slides were mounted in a Mowiol solution (Calbiochem, 475904). Fluorescence was examined with a confocal laser scanning microscope (TCS SP5, Leica). Leica confocal software (LAS AF) was used for confocal acquisition. Immunofluorescence staining on mouse anterior tibialis muscles was performed on frozen sections and then examined with a fluorescence microscope (DM 5000B, Leica). For all imaging experiments, exposure settings were identical between compared samples. Images were processed using Photoshop CS2 (Adobe) and ImageJ softwares.Citation50 Quantification of transfected fibers or cells with aggregates was performed by picturing ~200 transfected cells and by manually counting cells with and without aggregates using the “Cell counter” plugin of ImageJ software. Percentage of colocalization in immunofluorescence studies was also quantified with the “Cell counter” plugin of ImageJ. Dots were counted manually in 2 fluorescence channels and the percentage of dots positive in both channels was calculated.
Protein expression: Total extract and separation of detergent-soluble and -insoluble fractions
The entire procedures were performed at 4 °C. Total proteins were extracted from cultured cells or muscle biopsies with a lysis buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 1 mM EDTA, 10% glycerol, 2% SDS, 1% Triton X-100, protease inhibitor cocktail (SIGMAFAST; Sigma-Aldrich, S8830) and phosphatase inhibitor cocktail (PhosSTOP; Roche, 04906837001). Cell lysates were incubated for 20 min on ice, sonicated, and then centrifuged at 20,000 g for 5 min to remove cell debris. Muscle biopsy lysates were incubated for 15 min on ice, crushed with beads in a homogenizer system (FastPrep-24, MP Biomedicals) and then centrifuged at 20,000 g for 5 min. Supernatant fractions were recovered and centrifuged at 20,000 g for 10 min. Detergent -soluble and -insoluble fractions were obtained according to Hara et al. based on the insolubility of protein aggregates in a buffer containing 0.5% Triton X-100.Citation51 Protein concentrations were measured and equalized before separation of supernatant and pellet fractions and an equal volume of supernatant and pellet fractions were loaded on the SDS-polyacrylamide gel.
Protein concentration was determined with a colorimetric Lowry-like assay (DC protein assay kit, Bio-Rad, 500-0116). Equal amounts of proteins (30 μg) were then separated by SDS-PAGE and transferred onto PVDF Immobilon-P membranes (Millipore, IPVH00010). Membranes were blocked with TBST (Tris-buffered saline [50 mM Tris, 150 mM NaCl, pH 7.4] + 0.1% Tween 20) containing 3% bovine serum albumin (Euromedex, 04-100-811-E) and incubated for 90 min at room temperature with primary antibodies. Membranes were washed 3 times in TBST for 10 min and incubated for 45 min with horseradish peroxidase-conjugated secondary antibodies. After 3 washes in TBST, membranes were incubated with enhanced chemiluminescence reagents (Amersham ECL’, GE Healthcare) and revealed by autoradiography. Quantification of band intensities was made with ImageJ software. Western blot gels separated by a line in figures were run on the same gel but were noncontiguous.
In vitro kinase assays
Protein purifications and kinase assays were performed as previously described.Citation29 Briefly, 50 ng of GST-GSK3 proteins purified from 293T cells were mixed with the same amount of GST-NBR1 proteins purified from 293T cells. The solution was adjusted to final concentrations of 25 mM HEPES, pH 7.4, 10 mM MgCl2 in a final volume of 50 μl, and 20 μCi of [γ32P]ATP were added. After incubation for 30 min at 30 °C, the reaction was stopped by addition of 5x loading buffer and incubated at 95 °C for 5 min. Proteins were then loaded on SDS-polyacrylamide gels and transferred onto PVDF membranes. Membranes were exposed with a phosphorimaging system (FujiFilm FLA-5100) for radioactivity measures and were then subjected to immunoblotting with anti-GST antibody to quantify total amounts of GST-GSK3 and GST-NBR1 proteins. Band intensities were quantified in the gel with ImageJ software. Adjusted radioactive signals were calculated as the ratio of radioactive signal/total protein signal.
Co-AP, GST affinity isolation, and coIP experiments
The entire procedures were performed at 4 °C. Co-affinity purification (co-AP) experiments were performed as described previously.Citation29 Briefly, GST-GSK3, NBR1-GFP or DsRed-NBR1 vectors were co-transfected in 293T cells using calcium chloride. After 36 h, GST-GSK3 proteins were affinity isolated with glutathione agarose beads (Pierce, 16100PR). Twenty micrograms of total cell extracts and equal amounts of beads were loaded on SDS-polyacrylamide gels and transferred onto PVDF membranes. GST-GSK3 and bound NBR1 were detected after immunoblotting of the membrane with anti-GST and anti-NBR1 antibodies.
Interactions between NBR1 and SQSTM1, UB, LC3B, or GABARAP were performed by GST affinity isolation experiments. GST, GST-SQSTM1, GST-UB, GST-LC3B, or GST-GABARAP pDEST27 expression vectors were transfected in 293T cells. Twenty-four hours after transfection, cells were harvested and lysed by sonication in lysis buffer 1 (50 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Nonidet-P40 [Euromedex, UN3500], 1 mM EDTA, protease inhibitor cocktail, 200 μM NaF, 1 mM NaVa, 100 μM β-glycerophosphate) after 30 min incubation on ice. Cell lysates were precleared by centrifugation for 10 min at 10,000 g. Pre-cleared lysates were incubated with glutathione beads overnight. Beads were then washed extensively 3 times with lysis buffer and resuspended in PBS + 10% glycerol. Aliquots of beads were run on a SDS-polyacrylamide gel in parallel with a BSA scale. The gel was stained with a Coomassie solution and the quantity of GST fusion proteins was estimated. In parallel, whole cell extracts from 293T cells transfected with pEGFP-C1-EGFP or pEGFP-C1-EGFP-NBR1 (wild type and mutants) expression vectors were obtained by lysis in lysis buffer 2 containing 50 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 1% Triton X-100, protease inhibitor cocktail and phosphatase inhibitor cocktail. Cell extracts were passed through a 25G needle 5 times to disperse aggregates and lysates were pre-cleared by centrifugation at 700 g for 15 min. Total protein concentration was measured. One microgram of the purified GST fusion proteins coupled to glutathione beads were then incubated overnight with 1 mg protein extracts of cells (input) transfected with EGFP or EGFP-NBR1 expression vectors in a final volume of 1 ml lysis buffer 2. After washing beads 3 times with a buffer containing 20 mM HEPES, pH 7.3, 200 mM NaCl, 1 mM DTT), 25 μl beads (300 ng of GST-fused proteins bound to the beads) and 20 μg inputs were analyzed by SDS-PAGE. One gel was stained with Coomassie to reveal GST-tagged proteins. A second gel was transferred to PVDF membranes and bound EGFP-NBR1 was detected by immunoblotting with anti-GFP antibody.
Coimmunoprecipitations (co-IP) were performed as previously described.Citation9
Patient study
Muscle fragments of control, myotilinopathy, and sIBM patients were obtained from biopsies made for diagnostic purposes and used for research with informed written consent of patients. We used muscle biopsies from 13 sIBM (46–82 y old, mean 64 ± 9.2) and 14 age-matched (50–71 y old, mean 58.5 ± 6.7) control patients. Diagnosis was based on clinical and anatomopathological evaluations. For sIBM patients, their gender, age at the time of biopsy, affected muscles, onset of the disease, biopsied muscle, and level of creatine phosphokinase detected in blood are described in Table S1. Scores were assigned to each patient on a scale from 0 to 3 (0 for the absence of the symptom, 3 for the most severe) to evaluate the severity of 4 typical sIBM symptoms: protein aggregation, inflammation, necrosis, fibrosis (Table S1). The evaluation of the last 3 symptoms was based on the examination of 10-μm frozen muscle sections stained with conventional haemalum phloxin saffron (HPS; RAL Diagnostics; Mayer haemalum, 320550; phloxin B, 361470; saffron, 369000) staining. The presence of rimmed vacuoles and protein aggregates was assessed on immunohistochemical staining of muscle sections using anti-SQSTM1 antibody (Fig. S5A). Control patients had normal muscle biopsy with no inflammatory, necrosis, fibrosis nor vacuolar lesion and showed no aggregate with SQSTM1 immunostaining.
Statistical analysis
Statistical analyses were performed using unpaired Student t test for means, Pearson Chi-square test with Yates continuity correction for proportions and Spearman rank correlation for correlations. Grubbs test was used to detect outliers in sIBM data samples. Tests were performed using R software (R Core Team, http://www.R-project.org) and statistical significance was set at *P < 0.05.
| Abbreviations: | ||
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| ATG7 | = | autophagy-related 7 |
| CHX | = | cycloheximide |
| DES | = | desmin |
| GABARAP | = | GABA(A) receptor-associated protein |
| GSK3 | = | glycogen synthase kinase 3 |
| GST | = | glutathione S-transferase |
| GYS1 | = | glycogen synthase 1 (muscle) |
| HMERF | = | hereditary myopathy with early respiratory failure |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| LIR | = | LC3-interacting region |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NBR1 | = | neighbor of BRCA1 gene 1 |
| PI3K | = | phosphoinositide 3-kinase |
| sIBM | = | sporadic inclusion body myositis |
| siRNA | = | small interfering RNA |
| SQSTM1 | = | sequestosome 1 |
| UB | = | ubiquitin |
| UBA domain | = | ubiquitin-associated domain |
Additional material
Download Zip (1,008.9 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the patients for participation in this study. We thank Dr James Woodgett for the kind gift of Gsk3a, Gsk3b+/+, and gsk3−/− MEFs, Drs Jocelyn Laporte, Karim Hnia, Pierre Jalinot, Christelle Morris, and Mathias Faure for kindly providing us with plasmids; Drs Yann-Gaël Gangloff, Vincent Mocquet, and April Reedy for critical reading of the manuscript. We also thank the microscopy facility (Plateau technique Imagerie/Microscopie) of the SFR Biosciences Gerland–Lyon Sud (UMS3444/US8) and its staff Christophe Chamot, Claire Lionnet, and Olivier Duc for technical assistance with imaging. This work was supported by a grant from the Association Française contre les Myopathies (AFM 16084). M Sandri was supported by a grant from the European Research Council (ERC 282310-MyoPHAGY). AS Nicot was a recipient of fellowships from AFM (14496) and from AXA Research Fund.
References
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science 2002; 296:1991 - 5; http://dx.doi.org/10.1126/science.1067122; PMID: 12065827
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 95; http://dx.doi.org/10.1038/ng1362; PMID: 15146184
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000; 101:57 - 66; http://dx.doi.org/10.1016/S0092-8674(00)80623-6; PMID: 10778856
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 2004; 24:8853 - 61; http://dx.doi.org/10.1523/JNEUROSCI.2978-04.2004; PMID: 15470152
- Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J 2004; 23:4307 - 18; http://dx.doi.org/10.1038/sj.emboj.7600426; PMID: 15470501
- Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 2004; 14:95 - 104; http://dx.doi.org/10.1016/S1097-2765(04)00151-0; PMID: 15068806
- Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010; 38:265 - 79; http://dx.doi.org/10.1016/j.molcel.2010.04.007; PMID: 20417604
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/10.1016/j.molcel.2009.01.020; PMID: 19250911
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Waters S, Marchbank K, Solomon E, Whitehouse C, Gautel M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett 2009; 583:1846 - 52; http://dx.doi.org/10.1016/j.febslet.2009.04.049; PMID: 19427866
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279 - 89; http://dx.doi.org/10.1016/j.molcel.2011.07.039; PMID: 22017874
- Gal J, Ström AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem 2007; 282:11068 - 77; http://dx.doi.org/10.1074/jbc.M608787200; PMID: 17296612
- Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in α-synucleinopathy. Acta Neuropathol 2012; 124:173 - 86; http://dx.doi.org/10.1007/s00401-012-0975-7; PMID: 22484440
- D’Agostino C, Nogalska A, Cacciottolo M, Engel WK, Askanas V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol 2011; 122:627 - 36; http://dx.doi.org/10.1007/s00401-011-0874-3; PMID: 21935636
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160:255 - 63; http://dx.doi.org/10.1016/S0002-9440(10)64369-6; PMID: 11786419
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228 - 33; http://dx.doi.org/10.1126/science.1205405; PMID: 21617041
- Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 2013; 126:939 - 52; http://dx.doi.org/10.1242/jcs.114819; PMID: 23239026
- Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S, Weismann CM, Xu P, Houghton JM, Gao FB, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 2011; 13:1214 - 23; http://dx.doi.org/10.1038/ncb2332; PMID: 21909099
- Mardakheh FK, Yekezare M, Machesky LM, Heath JK. Spred2 interaction with the late endosomal protein NBR1 down-regulates fibroblast growth factor receptor signaling. J Cell Biol 2009; 187:265 - 77; http://dx.doi.org/10.1083/jcb.200905118; PMID: 19822672
- Mardakheh FK, Auciello G, Dafforn TR, Rappoport JZ, Heath JK. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol Cell Biol 2010; 30:5672 - 85; http://dx.doi.org/10.1128/MCB.00878-10; PMID: 20937771
- Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005; 308:1599 - 603; http://dx.doi.org/10.1126/science.1110463; PMID: 15802564
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378:785 - 9; http://dx.doi.org/10.1038/378785a0; PMID: 8524413
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004; 29:95 - 102; http://dx.doi.org/10.1016/j.tibs.2003.12.004; PMID: 15102436
- Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol 2010; 225:210 - 8; http://dx.doi.org/10.1016/j.expneurol.2010.06.017; PMID: 20599975
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem 2002; 277:33791 - 8; http://dx.doi.org/10.1074/jbc.M204861200; PMID: 12097329
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 2001; 65:391 - 426; http://dx.doi.org/10.1016/S0301-0082(01)00011-9; PMID: 11527574
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101 - 11; http://dx.doi.org/10.1083/jcb.200504035; PMID: 16186256
- Pilot-Storck F, Chopin E, Rual JF, Baudot A, Dobrokhotov P, Robinson-Rechavi M, Brun C, Cusick ME, Hill DE, Schaeffer L, et al. Interactome mapping of the phosphatidylinositol 3-kinase-mammalian target of rapamycin pathway identifies deformed epidermal autoregulatory factor-1 as a new glycogen synthase kinase-3 interactor. Mol Cell Proteomics 2010; 9:1578 - 93; http://dx.doi.org/10.1074/mcp.M900568-MCP200; PMID: 20368287
- Kunick C, Lauenroth K, Leost M, Meijer L, Lemcke T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3 beta. Bioorg Med Chem Lett 2004; 14:413 - 6; http://dx.doi.org/10.1016/j.bmcl.2003.10.062; PMID: 14698171
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2001; 2:769 - 76; http://dx.doi.org/10.1038/35096075; PMID: 11584304
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell 2001; 7:1321 - 7; http://dx.doi.org/10.1016/S1097-2765(01)00253-2; PMID: 11430833
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002; 34:509 - 19; http://dx.doi.org/10.1016/S0896-6273(02)00706-7; PMID: 12062036
- Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, Gatti E, Pierre P. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 2004; 164:667 - 75; http://dx.doi.org/10.1083/jcb.200312073; PMID: 14981091
- Selcen D. Myofibrillar myopathies. Neuromuscul Disord 2011; 21:161 - 71; http://dx.doi.org/10.1016/j.nmd.2010.12.007; PMID: 21256014
- Olivé M, van Leeuwen FW, Janué A, Moreno D, Torrejón-Escribano B, Ferrer I. Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol Appl Neurobiol 2008; 34:76 - 87; PMID: 17931355
- Dalakas MC, Dagvadorj A, Goudeau B, Park KY, Takeda K, Simon-Casteras M, Vasconcelos O, Sambuughin N, Shatunov A, Nagle JW, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord 2003; 13:252 - 8; http://dx.doi.org/10.1016/s0960-8966(02)00271-7; PMID: 12609507
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Askanas V, Engel WK. Sporadic inclusion-body myositis: conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-β42 oligomers and phosphorylated tau. Presse Med 2011; 40:e219 - 35; http://dx.doi.org/10.1016/j.lpm.2010.11.024; PMID: 21392932
- Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA, Ricci E, Mirabella M. Increased aging in primary muscle cultures of sporadic inclusion-body myositis. Neurobiol Aging 2010; 31:1205 - 14; http://dx.doi.org/10.1016/j.neurobiolaging.2008.08.011; PMID: 18823681
- Terracciano C, Nogalska A, Engel WK, Askanas V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J Neurochem 2010; 112:389 - 96; http://dx.doi.org/10.1111/j.1471-4159.2009.06461.x; PMID: 19878439
- Lei P, Ayton S, Bush AI, Adlard PA. GSK-3 in Neurodegenerative Diseases. Int J Alzheimers Dis 2011; 2011:189246; http://dx.doi.org/10.4061/2011/189246; PMID: 21629738
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 91; http://dx.doi.org/10.1091/mbc.E08-12-1248; PMID: 19211835
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012; 336:477 - 81; http://dx.doi.org/10.1126/science.1217032; PMID: 22539723
- Ohlsson M, Hedberg C, Brådvik B, Lindberg C, Tajsharghi H, Danielsson O, Melberg A, Udd B, Martinsson T, Oldfors A. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 2012; 135:1682 - 94; http://dx.doi.org/10.1093/brain/aws103; PMID: 22577218
- Pfeffer G, Barresi R, Wilson IJ, Hardy SA, Griffin H, Hudson J, Elliott HR, Ramesh AV, Radunovic A, Winer JB, et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J Neurol Neurosurg Psychiatry 2014; 85:331 - 8; http://dx.doi.org/10.1136/jnnp-2012-304728; PMID: 23486992
- Kramer T, Schmidt B, Lo Monte F. Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer’s Disease Models. Int J Alzheimers Dis 2012; 2012:381029; http://dx.doi.org/10.1155/2012/381029; PMID: 22888461
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 2007; 6:329 - 37; http://dx.doi.org/10.1016/j.cmet.2007.08.013; PMID: 17908561
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117:399 - 412; http://dx.doi.org/10.1016/S0092-8674(04)00400-3; PMID: 15109499
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671 - 5; http://dx.doi.org/10.1038/nmeth.2089; PMID: 22930834
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204