
Abstract
Autophagy is a dynamic cell survival mechanism by which a double-membrane vesicle, or autophagosome, sequesters portions of the cytosol for delivery to the lysosome for recycling. This process can be inhibited using the antimalarial agent chloroquine (CQ), which impairs lysosomal function and prevents autophagosome turnover. Despite its activity, CQ is a relatively inadequate inhibitor that requires high concentrations to disrupt autophagy, highlighting the need for improved small molecules. To address this, we screened a panel of antimalarial agents for autophagy inhibition and chemically synthesized a novel series of acridine and tetrahydroacridine derivatives. Structure-activity relationship studies of the acridine ring led to the discovery of VATG-027 as a potent autophagy inhibitor with a high cytotoxicity profile. In contrast, the tetrahydroacridine VATG-032 showed remarkably little cytotoxicity while still maintaining autophagy inhibition activity, suggesting that both compounds act as autophagy inhibitors with differential effects on cell viability. Further, knockdown of autophagy-related genes showed no effect on cell viability, demonstrating that the ability to inhibit autophagy is separate from the compound cytotoxicity profiles. Next, we determined that both inhibitors function through lysosomal deacidification mechanisms and ultimately disrupt autophagosome turnover. To evaluate the genetic context in which these lysosomotropic inhibitors may be effective, they were tested in patient-derived melanoma cell lines driven by oncogenic BRAF (v-raf murine sarcoma viral oncogene homolog B). We discovered that both inhibitors sensitized melanoma cells to the BRAF V600E inhibitor vemurafenib. Overall, these autophagy inhibitors provide a means to effectively block autophagy and have the potential to sensitize mutant BRAF melanomas to first-line therapies.
Introduction
Macroautophagy (autophagy) is a catabolic-salvaging pathway that cells use to segregate portions of the cytosol, including proteins and organelles, for delivery to the lysosome for degradation.Citation1 Autophagy is a very ordered process that has 4 distinct steps; initiation, nucleation, elongation, and completion.Citation2 Following initiation, nucleation generates a phagophore, or a cup-shaped double-bilipid membrane, which forms at the initiation site. The membrane subsequently elongates and eventually closes, sequestering the constituents into a mature double-membrane autophagosome. Lastly, the autophagosome fuses with the lysosome, or endocytic vesicle destined for the lysosome, for degradation of its contents and completion of the pathway.Citation3,Citation4
While autophagy is utilized to maintain homeostasis under nutrient-rich conditions, it is also activated during times of stress as a survival mechanism.Citation5,Citation6 Given this pivotal role in cell fate, autophagy is implicated in many diseases including cancer and autoimmune, inflammatory, and neurodegenerative diseases.Citation7,Citation8 Notably, emerging evidence supports a critical role for autophagy in the survival of cancer cells.Citation5,Citation6,Citation9 The same mechanism used by healthy cells to produce internal nutrients and energy is exploited by cancer cells to survive in times of metabolic, hypoxic, and therapeutic stress.Citation9-Citation11 Autophagy is particularly important in certain tumor types and in response to specific oncogenic stresses.Citation12 For example, cancer cells with high metabolic phenotypes can become ‘addicted’ to autophagy, as it provides necessary building blocks to maintain growth rates and support cell survival.Citation5,Citation6,Citation10 Emerging evidence suggests that in cancers with oncogenic activation of RAS, autophagy is often upregulated and critical for survival.Citation6,Citation13,Citation14 Further evidence shows that autophagy is not only beneficial, but is required for pancreatic tumor growth, which is frequently driven by oncogenic KRAS (Kirsten rat sarcoma viral oncogene homolog).Citation15 Similarly, oncogenic BRAF activation has been shown to upregulate autophagy. In this study, BRAF overexpression increased the autophagy protein MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) levels, and BRAF and LC3 expression positively correlated in tumors.Citation16 Taken together, autophagy inhibition represents a promising therapeutic target in tumor types where this process is upregulated and required for cell survival.
In addition to increased utilization of basal autophagy in certain tumor types, many anticancer therapeutic regimens induce autophagy.Citation9,Citation11 For example, glioma cells resistant to standard of care chemotherapy and radiotherapy display increased autophagy. Importantly, autophagy-inhibitor treatment sensitizes these resistant glioma cells to therapy, supporting a role for autophagy in glioma cell survival.Citation17 In many cases, this upregulation of autophagy contributes to survival as an unintended and counterproductive consequence of treatment. Thus, cancers targeted with a diverse set of therapeutics may be particularly vulnerable to autophagy inhibition, which consequently provides a therapeutic opportunity. Studies have demonstrated that autophagic pathway inhibition, both genetic and chemical, promotes sensitization to chemotherapy.Citation11 Along these lines, loss of key autophagic machinery proteins including BECN1, ATG5 (autophagy related 5), ATG10 (autophagy related 10), and ATG12 (autophagy related 12), confers sensitization to cell death.Citation9,Citation18-Citation20 Similar results are seen with pharmacological inhibitors that target the autophagy pathway, such as a class III phosphatidylinositol 3-kinase inhibitor (3-methlyadenine; 3-MA) and inhibitors of lysosomal function (bafilomycin A1 and CQ).Citation21
The accumulating evidence supporting autophagy-mediated cancer cell survival and the therapeutic potential for targeting autophagy, underscores the critical need to develop more effective autophagy inhibitors. Of particular interest are inhibitors that can be applied both as single agents for highly autophagic cancers, such as RAS-driven tumors, and also as adjuvants to standard chemotherapeutic regimens. Currently, the most widely used autophagy inhibitor is CQ, a well-known antimalarial drug in clinical use for more than 70 years.Citation22,Citation23 CQ functions as a freely diffusing lysosomotropic agent that enters the lysosome, is deprotonated, and becomes trapped inside as a diacidic base.Citation22,Citation24 By sequestering the free hydrogen ions required to maintain an acidic pH, CQ increases the basicity of the lysosome. This renders pH-dependent lysosomal hydrolases and proteases nonfunctional, blocks lysosomal turnover, and inhibits the final completion stage of autophagy. Consequently, autophagy-mediated cell survival is impaired, and tumor cells treated with CQ are less able to withstand therapeutic treatments and are therefore sensitized to therapy.Citation15,Citation25,Citation26 The safety profile of CQ and its ability to inhibit autophagy make this antimalarial drug a suitable starting point.
Several studies report CQ is an effective adjuvant to cancer therapeutics. In a myeloid leukemia cell line, treatment with the DNA-damaging antitumor agent, anthracycline daunorubicin (DNR), induces cytoprotective autophagy that precedes cell death.Citation27 CQ increases DNR-induced apoptotic cell death, allowing for a lower and less toxic DNR dose to be used.Citation27 In a xenograft model of triple-negative breast cancer, treatment with CQ and a histone deacetylase (HDAC) inhibitor decreases tumor burden and increases animal survival, compared with the HDAC inhibitor alone.Citation28 Furthermore, in colon cancer, CQ enhances cell death induced by topotecan, a DNA damage inducer.Citation29 These examples, and a host of others, emphasize the importance of autophagy inhibitors to increase cancer cell death in conjunction with therapeutics. Accordingly, there are currently more than 40 clinical trials in progress investigating the use of CQ or hydroxychloroquine in cancer treatment.Citation30,Citation31 One particularly promising glioblastoma multiforme clinical trial has found that when combined with conventional chemotherapy and radiotherapy, CQ prolongs the median survival of patients.Citation32 The sample size was small, thereby preventing statistical significance; however, it supports the potential use of CQ as an adjuvant therapy.
Despite the strong evidence for combining autophagy inhibitors with existing anticancer therapies, CQ is a relatively inadequate inhibitor, requiring a large effective concentration (mid-micromolar range) to disrupt autophagy. Therefore, small molecules that more potently inhibit autophagy are needed to improve efficacy, and ultimately enhance therapeutic sensitization. In this study, we established a high content cell-based assay to screen known antimalarial compounds, including a diverse set of lysosomotropic agents, for their ability to inhibit autophagy. We found the antimalarial compound, quinacrine (QN), contained greater autophagy inhibition properties than CQ, and subsequently centered our efforts on the acridine scaffold for rational chemical synthesis. We developed a series of novel acridine and 1,2,3,4-tetrahydroacridine derivatives and identified several compounds containing up to 50-fold more potent autophagy inhibition than CQ. We demonstrated that these novel compounds function by deacidifying lysosomes and impairing turnover of incoming vesicles, including autophagosomes. Intriguingly, despite similar effects on autophagy, these molecules displayed diverse cytotoxic profiles ranging from largely cytostatic (VATG-032) to more cytotoxic (VATG-027). To explore the therapeutic potential of these 2 lead molecules, we evaluated their activity in a panel of patient-derived metastatic melanoma cell lines. We discovered that both compounds reduced cell viability and anchorage independent colony growth as single agents. Furthermore, combining these molecules with vemurafenib (PLX-4032), a BRAF inhibitor selective for V600E, or the catalytic MTOR inhibitor, AZD8055, significantly reduced colony formation in a manner that exceeds additivity. Taken together, our data supports the critical role of autophagy in BRAF mutant melanoma. Moreover, the novel compounds developed here may provide utility as both single agents and adjuvant therapeutics in anticancer strategies.
Results
Screening antimalarial compounds for autophagy inhibition
Autophagy inhibition can be measured by fluorescent microscopy using cells expressing a tandem fluorescent (red fluorescent protein and green fluorescent protein; RFP-GFP) labeled MAP1LC3B (microtubule-associated protein 1 light chain 3 β; LC3B) sensor (tfLC3).Citation33 Upon phagophore nucleation, LC3 localizes to the autophagic membrane and the overlapping GFP and RFP fluorescence appears as yellow puncta. After the autophagosome matures, it fuses with the lysosome, forming an autolysosome. The GFP of this sensor is pH-labile and becomes quenched by the acidic environment of the autolysosome. However, the RFP remains stable; therefore, autolysosomes are indicated by red puncta. Accordingly, when autophagy is inhibited at the final stage (completion), either by an accumulation of autophagosomes or deacidification of the autolysosome, the abundance of yellow puncta is expected to increase proportionally to the level of autophagy inhibition.
To determine if other antimalarial compounds exist which inhibit autophagy more potently than CQ, we treated U2OS cells stably expressing tfLC3 with CQ or 6 other antimalarial agents (amodiaquine, artemisinin, mefloquine, piperaquine, primaquine, and QN) for 3 h. Cells were then fixed and imaged by fluorescent microscopy.Citation34 The LC3 puncta (autophagic vesicles) from these fluorescent images were quantified and used to determine the effective concentration (EC) of autophagy inhibition (). The EC was determined as the concentration at which cells contained a statistically significant increase in puncta number compared with the vehicle control. To identify antimalarial compounds that inhibited autophagy more potently than CQ, the EC of each antimalarial was divided by the EC of CQ (ECCQ) to yield a relative potency score (EC/ECCQ) (). Accordingly, a potency score greater than one indicated a more potent autophagy inhibitor than CQ. We found that 2 antimalarial compounds, mefloquine (MQ) and QN were more potent autophagy inhibitors than CQ with relative potency scores of approximately 30 and 60, respectively.
Figure 1. Relative autophagy inhibition for each antimalarial compound. Seven known antimalarial agents were screened for the effective concentration (EC) at which they inhibit autophagy. Potency was determined by comparing the antimalarial EC to that of the EC of chloroquine.
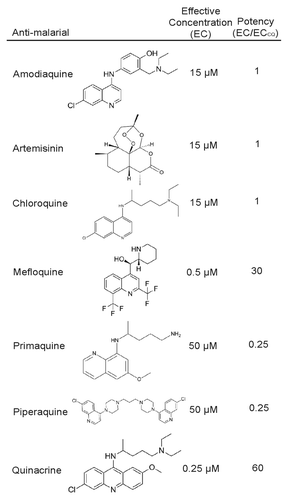
CQ, QN, MQ, and amodiaquine have been previously shown to inhibit autophagy, while the remaining antimalarial agents tested (piperaquine, primaquine, and artemisinin) have not to our knowledge.Citation35 Although these agents showed reduced potency, we confirmed that they function as autophagy inhibitors using an endogenous assay. Using immunoblotting for endogenous LC3, we demonstrated that each agent induced the accumulation of LC3-II, the form of LC3 localized to the autophagic membrane, both basally and in response to rapamycin-induced autophagy induction (Fig. S1). These data suggest that autophagy inhibition may be a common activity of antimalarial agents.
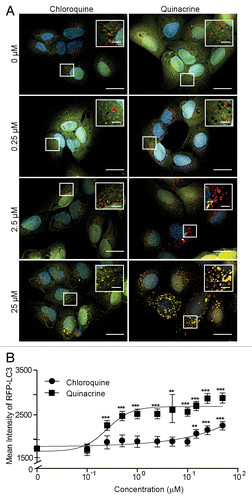
Next, we carefully characterized autophagy inhibition with QN, the most potent autophagy inhibitor in comparison to CQ. To this end, we treated U2OS-tfLC3 cells with 10-point concentration gradients of QN or QC and again, imaged LC3 puncta. While QN treatment increased the amount of LC3 puncta at nearly all concentrations, the same CQ concentrations failed to achieve an appreciable increase in puncta number (). In order to quantitatively confirm the results, we used image analysis software to determine the mean intensity of puncta at each compound concentration. We found that mean intensity more accurately measured autophagy in these experiments compared with puncta number. This was due to the large abundance of puncta that become individually indistinguishable at higher inhibitor concentrations. This autophagosome accumulation prevents accurate object separation, rendering puncta number less reliable. Mean intensity proved to correlate well with puncta number, and importantly, was not negatively affected at high concentrations (Fig. S2). Treatment with QN significantly increased the mean intensity of autophagic puncta at 0.25 μM (). However, CQ treatment was only able to produce a significant increase in mean intensity at 15 µM, a 60-fold higher concentration ().
Figure 2. Quinacrine inhibits autophagy more potently than chloroquine. (A) U2OS cells expressing tandem fluorescent LC3 (tfLC3) were treated for 3 h with chloroquine or quinacrine at the concentrations indicated, fixed, and imaged at 60× magnification. Green: GFP-LC3; Red: RFP-LC3, Blue: Hoechst (nuclei). Scale bars: 20 μm. Insets are 2× magnifications of boxed regions (scale bars: 5 μm). (B) Mean intensity of RFP-LC3-positive puncta was quantified using image analysis software following treatment with chloroquine (filled circles) or quinacrine (open circles) at the indicated concentrations (n ≥ 50 cells per condition). Error bars indicate standard deviation. Student 2-tailed t test: *P < 0.05; **P < 0.01; ***P < 0.001.
Novel autophagy inhibitor development
As previously mentioned, 2 classes of autophagy inhibitors may be useful therapeutically—those that potently inhibit autophagy and cause cytotoxicity as single agents and those agents that are potent autophagy inhibitors yet relatively cytostatic, permitting use in combination therapies (as adjuvants). To develop such compounds, we used QN as a template for rational chemical synthesis and created a series of over 60 novel small molecules. Changes were made to the acridine scaffold (6-chloro-2-methoxy-acridin) and R-group (N,1NCitation1-diethyl-NCitation4-methylpentane-1,4-diamine) of QN. These molecules were then screened for autophagy inhibition as well as effects on cell viability ().
Figure 3. Relative autophagy inhibition (EC), cytotoxicity (IC50), and chemical structure of novel autophagy compounds. The EC, IC50, and structures of the top compounds, VATG-027 and VATG-032, are shown in comparison to chloroquine and quinacrine
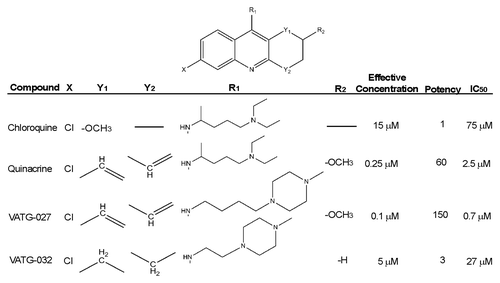
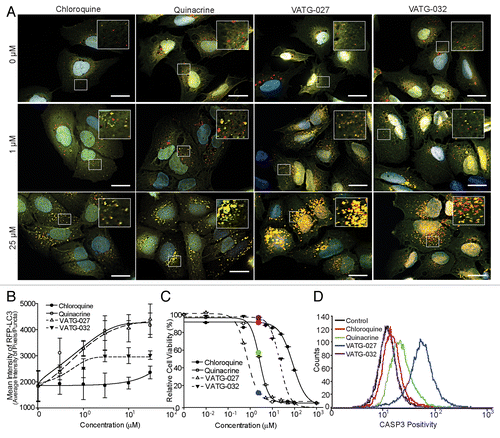
While moderate changes in autophagy inhibition and viability were seen with most chemical alterations, a few key changes had considerable impacts on cell viability (half maximal inhibitory concentration; IC50) and/or EC. From the most potent autophagy inhibitors, we chose 2 molecules for further evaluation, each with divergent effects on cell viability (IC50). While compound VATG-032 (EC = 5 μM), was less cytotoxic than QN with an IC50 equal to 27 µM, VATG-027 (EC = 0.1 μM) was considerably more cytotoxic with an IC50 of 0.7 µM. The autophagy inhibition and cell viability effects of VATG-027 and VATG-032 were carefully quantified across a concentration gradient, as described above, and compared with that of both CQ and QN (; ). We found VATG-032 to be a 3-fold-more potent autophagy inhibitor than CQ, yet 10 times less cytotoxic than QN (). The potent autophagy inhibition coupled with low cytotoxicity makes VATG-032 a candidate compound for adjuvant therapy. We also found VATG-027 to be 150-fold-more potent autophagy inhibitor than CQ (and 2× more potent than QN); however, it was also 3.5-fold-more cytotoxic than QN. To confirm autophagy inhibition independent of a fluorescent reporter, we performed a dose response and measured endogenous LC3 processing by immunoblotting (Fig. S3). Compounds VATG-027 and VATG-032 both caused an accumulation of LC3-II, consistent with the tfLC3 observationsCitation34 (; Fig. S3). Next, we evaluated the cytotoxicity of each compound by measuring activation of CASP3 (caspase 3, apoptosis-related cysteine peptidase), an executioner caspase that mediates apoptosis. We found that the reduction in cell viability caused by QN and VATG-027 is at least partially the result of apoptotic cell death ().
Figure 4. VATG-027 and VATG-032 show greater autophagy inhibition than chloroquine. (A) U2OS-tfLC3 cells were treated for 3 h with chloroquine, VATG-027, or VATG-032 at the indicated concentrations, fixed, and imaged at 60× magnification. Green: GFP-LC3; Red: RFP-LC3; Blue: Hoechst (nuclei). Scale bars: 20 μm. Insets are 2.5× magnifications of boxed regions (scale bars: 8 μm). (B) Mean pixel intensity of RFP-LC3 (red) puncta over a dose response with chloroquine (filled circles, solid line), quinacrine (open circles, solid line), VATG-027 (closed triangles, dashed line), and VATG-032 (open triangles, dashed line). Error bars indicate standard deviation. (C) Relative cell viability (as a percent of DMSO control) determined 48 h after treatment with chloroquine, quinacrine, VATG-027 or VATG-032. Error bars indicate standard deviation. Circles indicate 3 μM concentrations and colors correlate with treatment colors in (D). (D) FACS analysis of cleaved CASP3 after treatment with 3 μM chloroquine, quinacrine, VATG-027, and VATG-032.
Cytotoxicity of compounds separate from autophagy inhibition
To determine whether the cytotoxicity observed was dependent on the ability of these compounds to inhibit autophagy, cells were treated with concentration gradients of CQ, QN, VATG-027, and VATG-032 in the presence or absence of small interfering RNA (siRNA) targeted to ATG5 and ATG12 and ULK1 (unc-51 like autophagy activating kinase 1), core proteins required for autophagy. Effective knockdown of ATG5 and ATG12 and ULK1 was measured by quantitative real-time polymerase chain reaction (qRT-PCR) and autophagy impairment was confirmed as a lack of LC3 lipidation by immunoblot (). Depletion of ATG5 and ATG12 or ULK1 did not significantly change the sensitivity of cells to any compound, but CQ and the less cytotoxic VATG-032 did appear to have minor protective curve shifts. However, the lack of significant change in cytotoxicity to targeted gene knockdown suggests that autophagy does not play a role in the acute cytotoxicity of these compounds (Fig. S4C). Further, the cytotoxicity of each compound (at 3 μM and 30 μM) was again tested for CASP3 activation following transfection with control (non-targeting) siRNA or siRNA targeted to ATG5 and ATG12 and ULK1. CASP3 activation was not significantly altered by either ATG5 and ATG12 or ULK1 knockdown, further supporting the acute reduction in cell viability is likely the result of apoptotic cell death and primarily independent of autophagy inhibition (Fig. S4D).
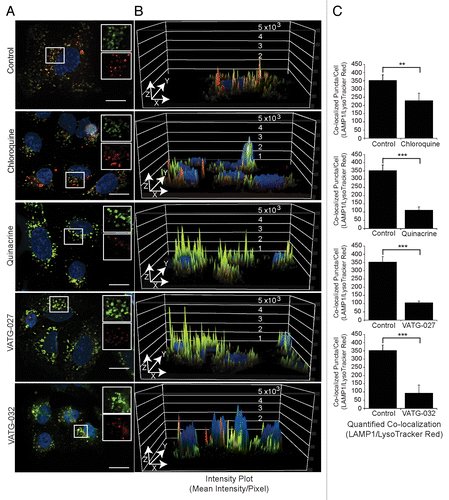
Figure 5. Autophagy inhibitors decrease lysosomal pH and impair lysosomal turnover. (A) U2OS cells were treated for 3 h with vehicle control or 3 μM autophagy inhibitor (chloroquine, quinacrine, VATG-027, or VATG-032). Cells were stained with 100 nM LysoTracker Red for 1 h prior to fixation. Cells were stained with endogenous LAMP1 antibodies and fluorescently conjugated secondary antibodies (green), Hoechst (nuclei; blue), and imaged at 60× magnification. Scale bars: 20 μm. Upper right insets are red and green channels separated and magnified 1.5× from boxed regions. (B) Red and green channel intensity plots were generated using image analysis software and displayed on the z axis (peaks) of a 3D representation of the images from (A). (C) Quantification of colocalized LAMP1 and LysoTracker Red as described in Materials and Methods. Student 2-tailed t test: **P < 0.01; ***P < 0.001 and Mander colocalization coefficient (MCC).
Mechanism of action: Lysosomal inhibition
Since both CQ and QN are both known to inhibit autophagy by deacidifying lysosomes and preventing the turnover of their constituents, we hypothesized that these VATG compounds function by the same mechanism. To test this, we first visualized lysosomes using transmission electron microscopy (TEM). We treated U2OS cells with a CQ concentration above the effective concentration (100 μM) for 3 h as a positive control, fixed and imaged cells. While few vesicles were observed in vehicle-treated cells, we found an accumulation of large, electron-dense and electron-lucent vesicles, consistent with lysosomes and endosomes, upon addition of CQ (). Once this phenotype was established, we treated cells with a lower CQ concentration (3 μM) to compare with cells treated with each compound at the same concentration. This concentration of QN, VATG-027, or VATG-032, but not CQ, was previously shown to inhibit autophagy (). As expected, we found that 3 μM CQ was insufficient to cause a noticeable increase in either vesicle size or number compared with the vehicle control. However, QN, VATG-027, and VATG-032 treatments caused a significant increase in the number of electron-lucent and electron-dense vesicles detected (). In addition, VATG-027 and VATG-32 treatment resulted in the appearance of several electron-opaque structures, consistent with lipid droplets, which are known to accumulate following autophagy impairment ().Citation36,Citation37
Figure 6. Autophagy inhibitors inactivate lysosomes and cause accumulation of cytosolic vesicles. (A) U2OS cells were treated for 3 h with a vehicle control or 100 μM chloroquine, fixed, and analyzed by transmission electron microscopy (TEM). Accumulation in both size and number of electron dense and lucent vesicles, consistent with lysosomes and endosomes (black arrows), is observed following chloroquine treatment. Scale bars: 2 μm. Right panels are magnifications of the boxed regions (scale bars: 1.14 μm and 500 nm, respectively). (B) U2OS cells were treated for 3 h with 3 μM chloroquine, quinacrine, VATG-027, or VATG-032, fixed, and analyzed by TEM. Electron-dense and electron-lucent vesicles are indicated with black arrows. Scale bars: 2 μm (left images). Right panels are magnified images of the boxed regions indicated by number (panel 1 scale bars: 1.2 μm; panel 2 scale bars: 500 nm). (C) Mean vesicle number per cell for treatments shown in B (20 images per treatment). Error bars indicate standard deviation. Student 2-tailed t test to control or chloroquine treatment as indicated: **P < 0.01; ***P < 0.001. (D) Immunoblot of U2OS cells treated with 3 μM and 30 μM of chloroquine, quinacrine, VATG-027, and VATG-032 for 6 h. Cell lysates were probed for pro and active forms of CTSB (pro and active forms indicated). Tubulin was included as a loading control.
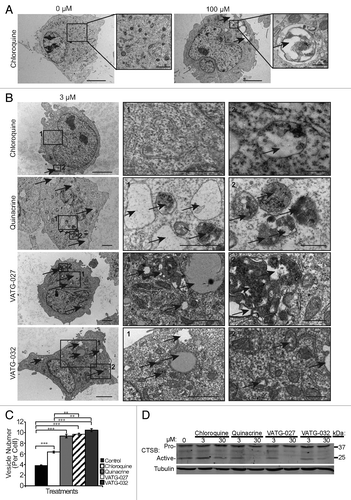
Deacidification of lysosomes would be expected to not only prevent the maturation and turnover of the lysosomes, but also affect the functionality of lysosomal enzymes and consequently, the turnover of lysosomal constituents. To determine if lysosomal activity was inhibited, we measured pro and active forms of the lysosomal protease, CTSB (cathepsin B), by immunoblotting. Lysates were harvested from cells following treatment with CQ, QN, VATG-027, and VATG-032 at either 3 μM or 30 μM for 6 h. QN and VATG-027 showed a nearly complete loss of active CTSB at 30 μM, while VATG-032 showed a significant decrease at the same concentration (). In contrast, CQ showed little effect on active CTSB at either concentration (). Taken together, this suggests that the acridine derivatives are considerably more potent than CQ at blocking lysosomal activity and turnover.
Next, we sought to confirm lysosomal turnover inhibition induced by VATG compounds thru evaluating endogenous lysosomal protein, LAMP1, abundance. In addition, we assessed lysosome acidity by costaining cells with LysoTracker Red, a dye that localizes to the lysosome based on low pH. Cells were treated with 3 μM CQ, QN, VATG-027, or VATG-032 for 3 h, with LysoTracker Red supplemented for the final hour. Next, cells were fixed and stained with endogenous LAMP1 antibodies and both LAMP1 and LysoTracker Red imaged. We found that CQ failed to yield an appreciable change in LAMP1-positive membranes and LysoTracker Red staining at 3 μM (). In contrast, QN, VATG-027, and VATG-032 treatments all caused substantial increases in LAMP1 staining and essentially eliminated LysoTracker Red staining (). To quantify this phenotype, the colocalization of LAMP1 and LysoTracker Red was measured using image-analysis software. The ratio of signal intensity across pixels of individual vesicles was measured and displayed using a colorimetric scale, where red indicates the presence of LAMP1-only, purple indicates the presence of LysoTracker Red-only, and green indicates the presence of both (Fig. S5). In addition, Mander colocalization coefficient values were determined for each treatment (). Mander colocalization coefficient measures pixel by pixel the co-occurrence of each channel or the proportion of pixels with positive values for both channels. We confirmed that not only does the presence of LAMP1-positive membranes increase, but the intensity of LAMP1 staining also increases with QN, VATG-027, and VATG-032 treatment (). We also show the inverse holds true for LysoTracker Red staining; treatment with QN, VATG-027, or VATG-032 decreased LysoTracker Red staining more so than CQ, suggesting a more substantial loss of lysosomal acidity at these lower concentrations. Collectively, these results suggest that these compounds function by deacidifying lysosomes and impairing their turnover.
Autophagic flux determination in BRAF mutant melanoma lines
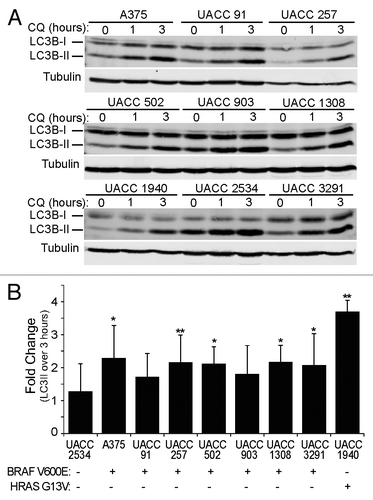
Oncogenic BRAF V600E, a genetic driver in greater than 50% of melanomas, can increase autophagy, potentially as a cell survival mechanism.Citation26,Citation38,Citation39 Accordingly, we evaluated autophagy inhibition in the A375 melanoma cell line, as well as 8 metastatic patient-derived melanoma lines (UACC-91, UACC-257, UACC-502, UACC-903, UACC-1308, UACC-1940, UACC-2534, and UACC-3291), 7 of which contain the BRAF V600E mutation as determined by Sanger sequencing. First, basal autophagic flux was measured in each cell line. To do this, we evaluated LC3, an ubiquitin-like molecule that translocates from the cytosol (LC3-I) to autophagic membranes (LC3-II) during autophagy. LC3-II is turned over in the lysosome along with autophagic cargo, therefore, autophagic flux can be determined by measuring the accumulation of LC3-II in response to lysosome inhibition over a short period of time.Citation33 Each cell line was treated with CQ (to inhibit the lysosomes) for 0, 1, or 3 h and quantitative immunoblotting used to measure the fold-change in LC3-II levels with CQ treatment. Importantly, we found that cell lines expressing BRAF V600E had a high level of autophagic flux, resulting in a ≥ 2-fold LC3-II accumulation by 3 h (). In addition, while one cell line expressing wild-type BRAF (UACC-1940) exhibited high autophagic flux, another wild-type BRAF cell line (UACC-2534) did not. Upon further investigation into the mutational status of the 2 cell lines, we found that UACC-1940 cells contain a HRAS (G13V) mutation, which activates the MAPK pathway similar to BRAF (). Taken together, all melanoma lines showed measurable levels of basal autophagic flux; however, the UACC 2534 cell line not driven by either oncogenic BRAF or RAS showed the lowest level of autophagic flux.
Figure 7. Melanoma cell lines have active basal autophagy. (A) Nine patient-derived melanoma cell lines were treated with 50 μM CQ for 0, 1, or 3 h. Cell lysates were probed by immunoblotting for endogenous LC3B (LC3B-I: cytosolic; LC3B-II: membrane-bound). (B) The levels of LC3B-II and tubulin α were measured using quantitative infrared imaging system (Odyssey) and immunoblotting. The fold change was determined by the change in LC3B-II normalized to tubulin α (LC3-II/tubulin α) from 0 to 3 h (y axis). Error bars indicate standard deviation. Student 2-tailed t test: *P < 0.05; **P < 0.01 compared with UACC2534 cells. Mutational status of BRAF and HRAS is indicated as mutant by (+) and wild type by (−).
Next, we determined the sensitivity of each melanoma cell line to CQ, QN, VATG-027, and VATG-032 (Table S1). CQ reduced cell viability with IC50 values ranging from 13 μM to 40 μM. VATG-032 affected cell viability in a similar manner, yielding IC50 values between 15 μM and 42 μM. Consistent with observations from U2OS cells, QN was considerably more cytotoxic than CQ, with IC50 values between 1.9 μM and 3.9 μM. VATG-027 treatment produced IC50 values that closely matched those of QN, between 0.4 μM to 2.7 μM. Overall, the 4 inhibitors affected viability of the 9 melanoma cell lines comparable to that observed in U2OS cells (Table S1; ).
Figure 8. The BRAF V600E inhibitor, PLX-4032, does not alter autophagic flux. (A) A375 cell viability was determined after 48 h treatment with CQ (filled circles), QN (open circles), VATG-027 (filled triangles), VATG-032 (open triangles), or PLX-4032 (filled squares). (B) Immunoblot of A375 cells treated with 0 μM, 10 nM, 100 nM, and 1000 nM of PLX-4032 in the presence (+) or absence (−) of CQ (50 μM). Cell lysates were probed for total MAPK1/3, phospho-MAPK1/3, and LC3B (LC3B-I, cytosolic; LC3B-II, membrane-bound). Tubulin was included as a loading control. (C) Soft agar colony formation assays using A375 cells treated every other day for 3 wk with 3 μM of CQ, QN, VATG-032, and 1 μM VATG-027 in the presence or absence of PLX-4032 (400 nM). Colonies were stained with crystal violet and quantified using image analysis software. Three replicates were averaged and standard deviation indicated by error bars. Student 2-tailed t test: *P < 0.05, **P < 0.01, and ***P < 0.001.
Combination treatment—vemurafenib and autophagy inhibitors
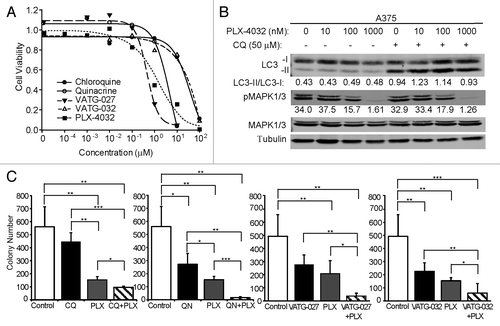
Since autophagy is active in melanoma cell lines and they are sensitive to autophagy inhibition, we questioned whether autophagy inhibitors could improve the efficacy of the latest approved drug for advanced metastatic melanoma.Citation40 This drug, vemurafenib (PLX-4032), selectively targets V600E mutant BRAF and it is unknown how this drug may affect autophagic flux. To address this, we first determined whether PLX-4032 induces autophagy, as has been observed with other targeted agents.Citation11 To do this, we measured LC3-II accumulation in response to lysosome inhibition by quantitative immunoblotting. A375 cells were treated with PLX-4032 (10 nM, 100 nM, and 1 μM) for 3 h in the presence or absence of CQ. While PLX-4032 effectively blocked oncogenic BRAF signaling, as measured by reduced phosphorylation of the downstream effector ERK1/2 (MAPK3/1; mitogen-activated protein kinase 3/1), autophagy was not significantly altered (). Next, A375 cells were treated with PLX-4032 in combination with CQ, QN, VATG-027, or VATG-032. Again, autophagic flux was not significantly altered by PLX-4032 (data not shown). These data suggest that while mutant BRAF V600E-expressing cell lines undergo a higher level of basal autophagy, chemical inhibition of oncogenic MAPK signaling does not significantly alter autophagic flux.
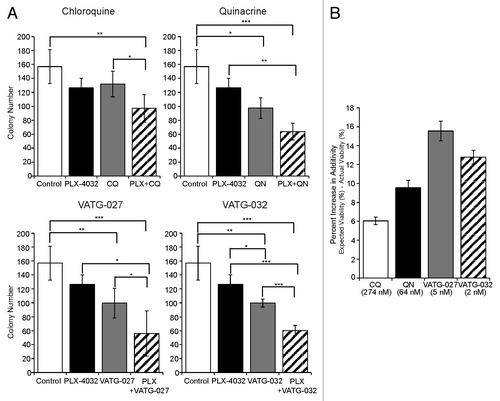
Despite the lack of autophagy induction by PLX-4032, the autophagic capacity retained in cells during treatment suggests that autophagy may potentially mediate cell survival. Therefore, we hypothesized that autophagy inhibitors may be effective in combinatorial treatment regimens with PLX-4032. To evaluate this, we performed soft agar colony formation assays to assess anchorage independent growth, one hallmark of cellular transformation and tumor growth. First, A375 cells were plated in soft agar for 3 wk and treated every other day with a range of concentrations of PLX-4032, CQ, QN, VATG-027, or VATG-032 (). In addition, we evaluated how autophagy inhibitors affect efficacy of a known autophagy stimulus, the catalytic MTOR inhibitor, AZD8055 (Fig. S6A). The IC10 for single compounds was determined to evaluate combinatorial effects. Following, we treated cells plated in soft agar with the IC10 of PLX-4032 alone or in combination with the IC10 of CQ, QN, VATG-027, or VATG-032 (). These same combinatorial soft agar experiments were also completed with AZD-8055 in place of PLX-4032 (Fig. S6B). To determine if the effects of each combination were more than, less than, or equal to additive, we made predictions for additivity using the Bliss Independence model (see Materials and Methods and Table S2).Citation41-Citation43 We found that CQ and PLX-4032 reduced colony formation by 38%, slightly greater than the effect predicted if these agents interact additively (33%) (; Table S2). Similarly, combinatorial treatment of QN and PLX-4032 reduced colony formation by 59%, just greater than the expected value of 50%. Both VATG-027 and VATG-032 were more significant than QN at increasing the efficacy of PLX-4032, reducing colony formation by 64% and 62%, compared with expected values of 49%, respectively (; Table S2). Similar results were obtained with combinatorial treatments of AZD-8055 and each autophagy inhibitor (Fig. S6B and S6C). We confirmed that PLX-4032 combination treatments with QN or VATG-032 exceeded additive effects in a second BRAF V600E mutant melanoma cell line, UACC 91 (Fig. S7A). In contrast, while single-agent autophagy inhibitor treatments reduced colony formation in the UACC 1940 (wild-type BRAF; HRAS G13V mutant) cell line, the same concentration of PLX-4032 in A375 cells (BRAF V600E) showed to be more effective as a single agent at growth inhibition. However, combined treatment with autophagy inhibitors did not exceed additivity (Fig. S7B and S7C). Taken together, these results suggest that autophagy inhibitors may have utility in melanoma treatment, both as single agents or in combination with BRAF inhibitors.
Figure 9. Autophagy inhibitors reduce A375 colony formation alone and in combination with PLX-4032. (A) A375 cells were grown in soft agar and treated every other day for 3 wk with the IC10 concentration of PLX-4032 (1.3 nM) in the presence or absence of the IC10 concentration for chloroquine (CQ; 274 nM), quinacrine (QN; 64 nM), VATG-027 (5 nM), or VATG-032 (2 nM). Colonies were stained with crystal violet and quantified using image analysis software. Colony numbers from 3 independent experiments were averaged and standard deviation indicated by error bars. Student 2-tailed t test: Significant *P < 0.05, **P < 0.01, and ***P < 0.001. (B) The percent change in response (colony reduction) exceeding the expected additive effect as determined by the Bliss Independence Model was determined for each autophagy inhibitor in combination with PLX-4032. Error bars represent standard deviation.
Discussion
In this study, we utilized melanoma cell models to evaluate the therapeutic potential of autophagy inhibitors. Melanoma is an aggressive cancer that has several well-identified oncogenes and tumor suppressors and mutations in these genes are known to upregulate autophagy and survival in melanoma.Citation26,Citation38,Citation44 Three common genes recurrently mutated in melanoma are RAS, BRAF, and PTEN, which in turn activate PtdIns3K-AKT-MTOR and RAS-RAF-MEK-MAPK pathways known to dysregulate autophagy.Citation45,Citation46 In a recent report, inhibition of both the MTOR pathway (temsirolimus) and autophagy (hydroxychloroquine) produces synergistic effects in melanoma cell death.Citation47 Additional reports reveal a hyperactivated MAPK signaling prevents MTOR-mediated nutrient sensing, specifically its inhibition due to the lack of leucine.Citation48 The role of autophagy and nutrient sensing was further assessed in vivo using human melanoma xenografts, and the combination of a leucine-free diet and an autophagy inhibitor dramatically reduced tumor volume.Citation48 Taken together, there is mounting evidence for autophagy inhibition in melanoma tumorigenesis.
Consistent with these findings, we confirmed active basal autophagy in a panel of 9 melanoma cell lines and found that these lines were sensitive to autophagy inhibition. Through the use of quantitative microscopy and rational chemical synthesis, we have further identified novel autophagy inhibitors with up to a 50-fold increase in autophagy inhibition compared with that of CQ. Our data also suggest that these compounds function to deacidify the lysosome and thus, inhibit the delivery and degradation of autophagic vesicles, similar to CQ. Importantly, we demonstrate that autophagy inhibitors decrease cell viability, both as single agents and in combination, supporting the hypothesis that autophagy promotes melanoma cell survival. This is consistent with evidence that many therapeutics, including targeted agents, can benefit from the addition of an autophagy inhibitor as an adjuvant.
Several studies have found that autophagy inhibition may sensitize cancers to therapeutic treatments that were otherwise ineffective. In colon cancer cells containing a RAS mutation, it has been shown that inhibition of autophagy using bafilomycin A1 increased cell death; in addition, the combination of the chemotherapeutic 5-fluorouracil with CQ leads to a further increase in cell death than when used alone.Citation49,Citation50 The effectiveness of autophagy inhibition in colon cancer is particularly exciting as 18% of colon cancers share the BRAF V600E mutation common in melanoma, suggesting the work presented here could be applicable to additional cancer types.Citation39 Similarly, inhibition of autophagy using both CQ and MQ, another antimalarial agent, is able to induce cell death in breast cancer lines expressing RAS and BRAF mutations.Citation51
Many chemotherapeutic treatment strategies upregulate autophagy, a counterproductive result, as upregulated autophagy can promote aberrant cell survival. This was demonstrated in a study of lung cancer with EGFR (epidermal growth factor receptor) tyrosine kinase inhibitors, gefitinib, and erlotinib. Treatment with these tyrosine kinase inhibitors confers a marked increase in autophagy activation, and cytotoxicity is significantly enhanced upon the addition of CQ.Citation52 Similarly, in a model of cervical cancer, cisplatin treatment induces autophagy, and CQ enhances the cytotoxicity of cisplatin.Citation53 Increasing therapeutic efficacy with autophagy inhibitors demonstrates the value of targeting autophagy in future treatment regimens.
The role of autophagy in cancer is complex and context-dependent; this is especially true in models with BRAF mutations. As indicated earlier, a correlation between increased autophagy and mutant BRAF in cell lines has been reported, suggesting autophagy inhibition may be effective in mutant BRAF tumor types.Citation16 Despite this, others have suggested that while supporting high basal autophagy, mutant BRAF confers resistance to autophagy activation by MTOR complex I (MTORC1) inhibition.Citation54 This discrepancy underscores that not only the tumor stage, but the subtype and collective mutations of a tumor, may contribute to the role of autophagy on cell viability.Citation46 Our data highlights the importance of measuring autophagic flux and the need for more potent autophagy inhibitors in aggressive cancers. Furthermore, the fact that we observed combination treatment effects (i.e., vemurafenib and autophagy inhibitors) that exceeded additivity using soft agar colony formation assays suggest that this approach may be better for observing antitumor effects, as opposed to acute drug toxicity in standard 2D cultures. Interestingly while vemurafenib (PLX-4032) is a selective inhibitor for BRAF V600E, we observed a more potent effect on viability in a mutant HRAS cell line (with wild-type BRAF) than the BRAF V600E cell line, A375, when treated at similar concentrations. However, the fewer number of colonies in the mutant HRAS context appeared larger with vemurafenib treatment. This is consistent with previous findings that vemurafenib is not an initiator of carcinogenesis, but accelerates growth in cells driven by mutant HRAS.Citation55
Prior studies have explored the development of CQ analogs; however, these analogs were primarily investigated for efficacy in malaria treatments and have not been explored as cancer therapeutics.Citation56,Citation57 Consequently in addition to the therapeutic potential in cancer, these novel autophagy inhibitors may prove useful in the treatment of malaria. Malaria is endemic in several regions of the world and resistance to current antimalarial drugs, including CQ, is an ever-growing problem.Citation58,Citation59 The development of new treatment strategies effective for such resistant strains is critical for the management of this disease. Accordingly, these modified chemicals could be investigated for activity against malaria strains resistant to current therapeutics.
Studies that have investigated the use of CQ analogs in cancer treatment have primarily focused on their ability to induce cell death as single agents.Citation60 Even though cytotoxic compounds are valuable, potent autophagy inhibition alone does not necessarily elicit cytotoxic effects. Here, we have demonstrated that inhibition of autophagy can be accomplished with compounds that are relatively well-tolerated by cells (i.e., VATG-032). The development of such potent autophagy inhibitors provides an opportunity for use as adjuvants in treatment strategies, effectively blocking autophagy-mediated cancer cell survival without significantly increasing toxicity as a single agent. This type of compound provides an exciting outlet for sensitization of cancer cells to the latest anticancer therapeutics.
Materials and Methods
Chemical synthesis
Antimalarial drugs shown in are all commercially available: amodiaquine (Chempacific, Corp. 35393), artemsinin (Sigma, 361593), chloroquine (Sigma, C6628), mefloquine (Amplachem, Inc., AA-90157), primaquine (OChem, Inc., 598P906), piperaquine (AK Scientific, H853), and quinacrine (TCI America, Q0056). VATG025 and VATG032 were synthesized as follows: Synthesis of VATG027: A mixture of 6,9-dichloro-2-methoxyacridine (100 mg, 0.36 mmol) and phenol (approximately 1.5 g) was heated to 100 °C under nitrogen atmosphere and stirred for 1 h. To this mixture was added 4-(4-methylpiperazin-1-yl)butan-1-amine (123 mg, 0.72 mmol). The reaction was stirred at 100 °C for 5 h, cooled to 25 °C, and diluted with dichloromethane. The mixture was washed twice with sodium hydroxide solution (1 N) and twice with ammonium chloride solution. The phases were separated, and the organic layer was dried and concentrated. The residue was purified by Biotage column chromatography using triethylamine (5%) and methanol (0 to 10%) in dichloromethane to give VATG027 (92 mg, yield: 62%); MS (Found M+1 = 413); 1H NMR (CD3OD, 300 Hz): 8.32–8.30 (d, 1H J = 8.2 Hz), 8.30–7.85 (m, 1H), 7.58–7.57 (d, 1H), 7.47–7.44 (m, 1H), 7.34–7.32 (m, 1H), 4.00 (s, 3H), 3.92–3.89 (t, 2H, J = 6Hz), 2.54–2.51 (b, 4H), 2.41–2.29 (m, 6H), 2.25 (s, 3H), 1.85–1.77 (m, 2H), 1.60–1.53 (m, 2H). Synthesis of VATG032: Following the procedure of the synthesis of VATG027 but using 6,9-dichloro-1,2,3,4-tetrahydroacridine and 1-ethylpiperidin-4-amine as the starting material. MS (Found M+1 = 344).
Autophagy inhibition screen
U2OS cells stably expressing tfLC3 (Addgene, plasmid 21074)Citation61 were seeded at 5,000 cells per well in 5A McCoy’s medium (Invitrogen, 16600-082) with 10% fetal bovine serum (FBS [CellGro, 35-101-CV]) in 96-well glass bottom tissue culture plates for 24 h at 37 °C and 5% CO2. Cells were treated with a selection of commonly used antimalarial compounds (amodiaquine, artemisinin, chloroquine, mefloquine, primaquine, piperaquine, and quinacrine) in a 6-point dose response for 3 h, fixed with 3.7% formaldehyde, and nuclei were stained with Hoechst 33342 (2 µg/mL: Invitrogen, H1399). Cells were visualized using a 60× oil-immersion objective on a Nikon Eclipse Ti fluorescent microscope (Melville, NY). Doses were qualitatively scored for effective concentration, defined as the concentration at which there was a statistically significant accumulation of tfLC3-labeled puncta over vehicle controls. Cells were later treated with novel VATG compounds in a 6-point dose response for 3 h, fixed, and visualized. An EC was established for each VATG compound.
Quantification of autophagy inhibition
U2OS cells stably expressing tfLC3 were seeded at 50,000 cells per well in 5A McCoy’s with 10% FBS on number 1.5 coverglass in 24-well tissue culture dishes. After 24 h, cells were treated with rapamycin (100 nM) (Millipore, 553210-10mg), bafilomycin A1 (100 nM) (AG Scientific, B-1183), AZD-8055 (100 nM) (Selleck Chemicals, S1555), or CQ (50 µM) as well as autophagy inhibitors (CQ, QN, VATG-027, and VATG-032) at concentrations of 0.1 µM, 0.25 µM, 0.5 µM, 1 µM, 5 µM, 15 µM, 25 µM, and 50 µM for 3 h. Cells were washed with 1× PBS, fixed with 3.7% formaldehyde, and nuclei were stained with Hoechst-33342 (2 µg/mL). Coverglass was inverted onto microscope slides using mounting gel. Cells were imaged using a 60× oil-immersion objective on a Nikon Eclipse Ti fluorescent microscope and 10 images at each concentration were taken for quantification. Image processing and quantification were completed with the NIS Elements software (Nikon). To quantify, images were deconvolved using a 2D blind deconvolution function with one iteration and settings of normal cell thickness and normal noise level. Regions of interest (ROI) were drawn around the edges of each cell. Intensity thresholds were set to include all pixels equal to or greater than the intensity of the mean background fluorescence using the separation feature and restrictions set for puncta size (Fig. S2). Objects within the threshold for each ROI were quantified using an automated object count function and exported for analysis. Although other parameters were also collected, the mean intensity of the objects was averaged between the 10 images of each concentration, or approximately 50 cells (Fig. S2B and S2C). Representative images were chosen for each concentration and the lookup table (LUT) brightness’ were set based on the mean intensity of the DMSO control ( and ). The mean intensity of each image was divided by the mean intensity of the DMSO control to control for brightness and the LUTs were adjusted by the percent difference to avoid background and for consistent visualization. All other settings (gain, exposure time, and lamp strength) were kept constant across all conditions. Puncta number was used to determine an EC, which is the statistically significant (P ≤ 0.05) increase in RFP-GFP-LC3-labeled puncta number compared with DMSO control, determined by the Student t test. Mean intensity was further chosen for quantification as it accurately represents both the increase in puncta number and area when the accumulation of autophagosomes partially fuse (Fig. S2C and S2D). Quantification of the red channel (RFP-LC3 puncta) was performed to determine the total autophagic vesicle population (both autophagosomes and autolysosomes). Autofluorescence of each compound was tested using wild-type U2OS cells to confirm that compound autofluorescence did not interfere with ptfLC3 quantification (Fig. S2E).
Cell viability (IC50) screen
U2OS cells were seeded at 500 cells per well in 5A McCoy’s with 10% FBS in 96-well clear bottom, black-walled tissue culture plates. All melanoma cell lines (A375, UACC-91, UACC-257, UACC-502, UACC-903, UACC-1308, UACC-1940, UACC-2534, and UACC-3291) were seeded at 5,000 cells per well in RPMI 1640 with 10% FBS in 96-well clear bottom, black-walled tissue culture plates. After 24 h incubation, cells were treated with VATG compounds in triplicate with a 10-point half log dose response from 0.001 μM to 1000 μM for 24 and 48 h. Medium was removed and 2× CellTiter-Glo (Promega, G7571) reagent mixed 1:1 with Opti-MEM (Invitrogen, 31985062) was added at 100 µL per well and incubated at room temperature for 15 min while rocking. Seventy-five microliters per well were moved to a white-walled 96-well plate and luminescence quantified using the 96 LUM program on an EnVision plate reader (PerkinElmer, Waltham, MA) and exported for analysis. All triplicate data points were averaged and luminescent readings for each treatment were normalized to vehicle control for change in viability. Dose response curves were analyzed in SigmaPlot and IC50 values determined (Systat Software Inc.).
Flow cytometry
U2OS cells were seeded in a 6-well plate in 5A McCoy’s with 10% FBS at 100,000 cells per well. After 24 or 48 h incubation, cells were treated at 1 µM, 3 µM, 10 µM, and 30 µM with CQ, QN, VATG-027, or VATG-032 for 48 h. Media was collected and spun down to collect floating cells. Wells were treated with 250 µL of 0.25% trypsin-EDTA and cells were again collected, spun down, and supernatant discarded. Cells were then fixed in 5 mL of 70% ethanol and stored at −20 °C for 24 h. Cells were centrifuged at 300 g for 5 min and resuspended in 1 mL of 90% chilled methanol. After 30 min, cells were washed twice in 3 mL incubation buffer (0.5 g BSA in 100 mL 1× PBS) and resuspended in 100 µL of incubation buffer for 10 min. Cells were then incubated with the primary cleaved CASP3 antibody (Cell Signal Technology, 9661S) at 1:1000 in incubation buffer for 1 h. Cells were washed (incubation buffer) and secondary anti-rabbit Alexa 546 antibody added 1:1000 for 30 min. Cells were then washed and resuspended in 100 µL 1× PBS and acquired using a FACS-Calibur (BD Biosciences, San Jose, CA).
ATG5, ATG12, and ULK1 knockdown
U2OS cells were seeded for either the cell viability screen or FACS analysis in 5A McCoy’s with 10% FBS. The next day, cells were transfected with either control (nontargeting) siRNA (Qiagen 1027281) or a pool of 2 siRNAs targeting ATG5 (ATG5: Qiagen SI00069251 and SI02655310), ATG12 (ATG12: Qiagen SI00069251 and SI02655310), or ULK1 (ULK1: Qiagen SI02223270 and SI02223277) at a final concentration of 50 nM (total siRNA) using 0.2 μl Oligofectamine (Invitrogen,12252-011) in 20 μl Opti-MEM (Invitrogen, 31985-062) and 80 μl 5A McCoy’s with 10% FBS. Cell viability assays and FACS analysis were completed as described above 24 h after transfection. Knockdown was measured using RNA extracts from siRNA-transfected cells and qRT-PCR with ATG5, ATG12, and ULK1 specific primers and an endogenous HPRT control. Delta-Delta Ct method was used to determine relative mRNA levels from control, ATG5 and ATG12, and ULK1 siRNA-transfected cells.
Transmission electron microscopy
U2OS cells were seeded in 10-cm plates in 5A McCoy’s with 10% FBS at 1 × 106 cells per plate. After 24 h incubation, cells were treated with DMSO (vehicle control), CQ (3 or 100 µM), quinacrine (3 µM), VATG-027 (3 µM), or VATG-032 (3 µM) for 3 h. Following that, cells were trypsinized, washed, pelleted, and resuspended in 2% glutaraldehyde fixative (Sigma, G5882). Cell pellets were embedded in 2% agarose, post-fixed in osmium tetroxide, and dehydrated with an acetone series. Cell samples were infiltrated and embedded in Poly/Bed 812 resin and polymerized at 60 °C for 24 h. Ultrathin sections of 70 nm were generated with a Power Tome XL (Boeckeler Instruments, Tucson, AZ) and placed on copper grids. Sections were examined using a JEOL 100C× Transmission Electron Microscope (Peabody, MA) at 100kV. Lysosomal structures were identified as a single membrane structure either containing or lacking cytosolic components, in addition to lipid droplets that are electron-opaque and lack a defined limiting membrane (). Transmission electron microscopy services were performed by the Michigan State University Center for Advanced Microscopy (East Lansing, MI).
Lysosome analysis by fluorescent microscopy
U2OS cells were seeded at 5 × 104 cells per well in 5A McCoy’s with 10% FBS on number 1.5 coverglass discs in 24-well tissue culture dishes. After 24 h, cells were treated with 3 μM CQ, QN, VATG-027, or VATG-032 for 3 h. An hour prior to fixation, media was supplemented with LysoTracker Red DND-99 added at 100 nM (Invitrogen, L7528). Cells were washed with 1× PBS, fixed with 3.7% formaldehyde, permeabilized with 0.2% Triton-X 100, and blocked with 3% bovine serum albumin (BSA) in PBS. LAMP1 antibody (Santa Cruz, sc-18821) was added at 1:1000 for 16 h at 4 °C followed by Alexa-Fluor-488-conjugated anti-mouse IgG (Invitrogen, A11008) 1:5000 for 1 h at room temperature. Nuclei were stained with Hoechst-33342 (2 µg/mL). Coverglass discs were inverted onto a microscope slide using mounting gel. The microscope slides were imaged using a 60× oil-immersion objective on a Nikon Eclipse Ti fluorescent microscope. Intensity of the red and green channels were visualized using the intensity plot on the Nikon NIS Elements software (). Colocalization was determined by using the ratio feature which ratios the intensity of the green channel (LAMP1) over the red channel (LysoTracker Red) per pixel and displays it on a colorimetric scale (Fig. S5). The RGB threshold of only the color (green) indicating both LAMP1 and LysoTracker Red positivity was performed and data for analysis. The Mander colocalization coefficient was produced from the Nikon NIS Elements software.
Immunoblot analyses
For immunoblotting, U2OS cells were seeded in 10 cm plates in 5A McCoy’s with 10% FBS at 1 × 106 cells per plate. All melanoma lines were seeded in 10cm plates in RPMI 1640 with 10% FBS at 1 × 106 cells per plate. After 24 h, cells were treated with CQ, QN, VATG-032, or VATG-027 in a dose response of 0.3 µM, 1 µM, 3 µM, 10 µM, and 30 µM for 3 h. A375 were treated with PLX-4032 (Selleck Chemicals, S1267) at 10 nM, 100 nM, and 1 μM for 3 h with and without CQ, QN, VATG-027, and VATG-032 at 3 μM and 30 μM. After treatment, cells were lysed (10 mM KPO4, 1mM EDTA, 10 mM MgCl2, 5 mM EGTA, 50 mM bis-glycerophosphate, 0.5% NP40 [USB Corporation, 68987-9-6], 0.1% Brij35 [Pierce, 20150], 0.1% sodium deoxycholate [Alfa Aesar, 1320759], 1 mM NaVO4, 5 mM NaF, 2 mM DTT, and complete protease inhibitors [Sigma, P8340]) and 50 µg of protein was resolved by SDS-PAGE. Proteins were transferred to PVDF membranes and probed with primary antibodies (LC3B [Sigma, L7543], anti-tubulin [Sigma, T6199], CTSB [Santa Cruz, sc-13985]) for 16 h at 4 °C followed by a secondary antibody (HRP-linked rabbit or mouse IgG [GE Healthcare, NA934 or NA931] or Odyssey IRDye 680CW Goat anti-rabbit IgG [LI-COR, 926-32221] or IRDye 800CW Goat anti-mouse IgG [LI-COR, 926-32210]) for 1 h at room temperature. Proteins were detected with enhanced chemiluminescence or using an Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE) and quantified (–; Figs. S1 and S3).
Soft agar colony formation assay
In a 6-well plate, a solidified base was created using a 1:1 solution of RPMI 1640 (with 20% FBS) and 1% agarose solution. This was overlaid with A375, UACC 1940, or UACC 91 cells at 40,000 cells per well in RPMI 1640 (with 10% FBS) mixed 1:1 with 0.7% agarose. The soft agarose containing cells was then overlaid with 0.75 mL RPMI 1640 (with 10% FBS) and incubated at 37 °C in 5% CO2. After 24 h, cells were treated in a dose response or at the determined LD10 with PLX-4032, CQ, QN, VATG-027, or VATG-032 every other day for 3 wk. After 3 wk of treatment, cells were fixed and stained in 1% paraformaldehyde in 1× PBS containing 0.005% crystal violet overnight. Cells were destained with multiple washes of 1× PBS to remove background staining. Plates were scanned and images quantified using NIS Elements software (Nikon). To quantify, each image was sharpened and a region of interest (ROI) of equal size drawn around each well. Intensity thresholds were set to include all pixels greater than the intensity of the mean background fluorescence. Object counts within the threshold of each ROI were quantified using an automated object count function to denote total colony formation. Additivity was determined using the fractional product concept or Bliss Independence model: Exy = Ex + Ey − (ExEy) where EXY is the additive effect of the 2 compounds x and y as calculated by the product of the individual effect of the 2 compounds, Ex and Ey.Citation41,Citation43 Additivity was established when the expected colony count determined by the Bliss Independence model was equal to the actual colony count. This model was chosen since the effects of both compounds are mutually nonexclusive and follow first order Michaelis-Menten kinetics.Citation42
| Abbreviations: | ||
| 3-MA | = | 3-methlyadenine |
| ATG | = | autophagy-related |
| BRAF | = | v-raf murine sarcoma viral oncogene homolog B |
| CASP3 | = | caspase 3, apoptosis-related cysteine peptidase |
| CQ | = | chloroquine |
| CTSB | = | cathepsin B |
| DNR | = | anthracycline daunorubicin |
| EC | = | effective concentration |
| EGFR | = | epidermal growth factor receptor |
| FBS | = | fetal bovine serum |
| GFP | = | green fluorescent protein |
| HDAC | = | histone deacetylase |
| KRAS | = | Kirsten rat sarcoma viral oncogene homolog |
| HRAS | = | Harvey rat sarcoma viral oncogene homolog |
| LAMP | = | lysosomal-associated membrane protein |
| LUT | = | look-up table |
| MAP1LC3 (LC3) | = | microtubule-associated protein 1 light chain 3 |
| MAPK1/3 | = | mitogen-activated protein kinase 1/3 |
| MQ | = | mefloquine |
| MTOR | = | mechanistic target of rapamycin |
| MTORC1 | = | MTOR complex 1 |
| QN | = | quinacrine |
| RFP | = | red fluorescent protein |
| ROI | = | region of interest |
| siRNA | = | small interfering ribonucleic acid |
| TEM | = | transmission electron microscopy |
| UACC | = | University of Arizona Cell Culture |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| VATG | = | Van Andel-TGen |
Additional material
Download Zip (2.6 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Alicia Pastor at the Michigan State University Center for Advanced Microscopy for her help with processing samples and imaging of TEM. We would also like to thank the Van Andel Institute Systems Biology lab for advice, analysis, and reagents. This work was supported by the Department of Defense Prostate Cancer Research Program of the Office of Congressionally Directed Medical Research Programs PC081089 to JP MacKeigan. JP MacKeigan is also supported by Award Number R01CA138651 from the National Cancer Institute. JM Trent is supported in part by a Stand up to Cancer/Melanoma Research Alliance Dream Team Award.
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 7; http://dx.doi.org/10.1038/nrm2245; PMID: 17712358
- Kohli L, Roth KA. Autophagy: cerebral home cooking. Am J Pathol 2010; 176:1065 - 71; http://dx.doi.org/10.2353/ajpath.2010.090850; PMID: 20150434
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186:773 - 82; http://dx.doi.org/10.1083/jcb.200907014; PMID: 19797076
- Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009; 11:e36; http://dx.doi.org/10.1017/S1462399409001306; PMID: 19951459
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401 - 10; http://dx.doi.org/10.1038/nrc3262; PMID: 22534666
- Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol 2011; 76:389 - 96; http://dx.doi.org/10.1101/sqb.2012.76.011015; PMID: 22442109
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, Yu J, Sung JJ. The autophagic paradox in cancer therapy. Oncogene 2012; 31:939 - 53; http://dx.doi.org/10.1038/onc.2011.295; PMID: 21765470
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 34; http://dx.doi.org/10.1038/nrc1692; PMID: 16148885
- Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N, Tamada Y, Yokoyama T, Kondo S. Stage-specific effect of inhibition of autophagy on chemotherapy-induced cytotoxicity. Autophagy 2009; 5:537 - 9; http://dx.doi.org/10.4161/auto.5.4.8164; PMID: 19270491
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447 - 61; http://dx.doi.org/10.1101/gad.219642.113; PMID: 23824538
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J Invest Dermatol 2010; 130:1657 - 67; http://dx.doi.org/10.1038/jid.2010.26; PMID: 20182446
- Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal 2010; 3:ra81; http://dx.doi.org/10.1126/scisignal.2001017; PMID: 21062993
- Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025 - 40; http://dx.doi.org/10.1128/MCB.25.3.1025-1040.2005; PMID: 15657430
- Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, Kroemer G. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 2009; 1793:1524 - 32; http://dx.doi.org/10.1016/j.bbamcr.2009.01.006; PMID: 19371598
- Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta Pharmacol Sin 2010; 31:1154 - 64; http://dx.doi.org/10.1038/aps.2010.118; PMID: 20694022
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 2008; 68:1485 - 94; http://dx.doi.org/10.1158/0008-5472.CAN-07-0562; PMID: 18316613
- Foley M, Tilley L. Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther 1998; 79:55 - 87; http://dx.doi.org/10.1016/S0163-7258(98)00012-6; PMID: 9719345
- Loeb RF. Activity of a new antimalarial agent, chloroquine (SN7618). JAMA 1946; 130:1069 - 70; http://dx.doi.org/10.1001/jama.1946.02870160015006
- Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009; 625:220 - 33; http://dx.doi.org/10.1016/j.ejphar.2009.06.063; PMID: 19836374
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478 - 89; http://dx.doi.org/10.1158/1078-0432.CCR-10-2372; PMID: 21325076
- Han W, Sun J, Feng L, Wang K, Li D, Pan Q, Chen Y, Jin W, Wang X, Pan H, et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS One 2011; 6:e28491; http://dx.doi.org/10.1371/journal.pone.0028491; PMID: 22164300
- Rao R, Balusu R, Fiskus W, Mudunuru U, Venkannagari S, Chauhan L, Smith JE, Hembruff SL, Ha K, Atadja P, et al. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol Cancer Ther 2012; 11:973 - 83; http://dx.doi.org/10.1158/1535-7163.MCT-11-0979; PMID: 22367781
- Li DD, Sun T, Wu XQ, Chen SP, Deng R, Jiang S, Feng GK, Pan JX, Zhang XS, Zeng YX, et al. The inhibition of autophagy sensitises colon cancer cells with wild-type p53 but not mutant p53 to topotecan treatment. PLoS One 2012; 7:e45058; http://dx.doi.org/10.1371/journal.pone.0045058; PMID: 23024792
- National Library of Medicine US. Clinical trials investigating the use of chloroquine in cancer: NCT01575782, NCT00969306, NCT01446016, NCT01023477, NCT01469455, NCT01438177, NCT01727531, NCT00224978. clinicaltrials.gov: Bethesda, MD, 2013
- National Library of Medicine US. Clinical trials investigating the use of hydroxychloroquine in cancer: NCT01273805, NCT00933803, NCT01006369, NCT01480154, NCT00765765, NCT00728845, NCT01266057, NCT00813423, NCT01494155, NCT01023737, NCT00786682, NCT00726596, NCT01417403, NCT00809237, NCT00909831, NCT00714181, NCT00714181, NCT01206530, NCT01026844, NCT00486603, NCT01649947, NCT00977470, NCT01506973, NCT01128296, NCT00568880, NCT01144169, NCT00962845, NCT01292408, NCT01689987, NCT01550367, NCT01396200, NCT01227135, NCT01602588, NCT01510119, NCT00031824, NCT01548768, NCT00908089, NCT01687179, NCT00405275, NCT00771056, NCT01709578. clinicaltrials.gov: Bethesda, MD, 2013
- Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2006; 144:337 - 43; http://dx.doi.org/10.7326/0003-4819-144-5-200603070-00008; PMID: 16520474
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Martin KR, Barua D, Kauffman AL, Westrate LM, Posner RG, Hlavacek WS, Mackeigan JP. Computational model for autophagic vesicle dynamics in single cells. Autophagy 2013; 9:74 - 92; http://dx.doi.org/10.4161/auto.22532; PMID: 23196898
- Qiao S, Tao S, Rojo de la Vega M, Park SL, Vonderfecht AA, Jacobs SL, Zhang DD, Wondrak GT. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy 2013; 9:2087 - 102; http://dx.doi.org/10.4161/auto.26506; PMID: 24113242
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131 - 5; http://dx.doi.org/10.1038/nature07976; PMID: 19339967
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 2012; 23:896 - 909; http://dx.doi.org/10.1091/mbc.E11-09-0785; PMID: 22219374
- Chen B, Tardell C, Higgins B, Packman K, Boylan JF, Niu H. BRAFV600E negatively regulates the AKT pathway in melanoma cell lines. PLoS One 2012; 7:e42598; http://dx.doi.org/10.1371/journal.pone.0042598; PMID: 22880048
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 54; http://dx.doi.org/10.1038/nature00766; PMID: 12068308
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Yan H, Zhang B, Li S, Zhao Q. A formal model for analyzing drug combination effects and its application in TNF-alpha-induced NFkappaB pathway. BMC Syst Biol 2010; 4:50; http://dx.doi.org/10.1186/1752-0509-4-50; PMID: 20416113
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947; PMID: 20068163
- Hiss DC, Gabriels GA, Folb PI. Combination of tunicamycin with anticancer drugs synergistically enhances their toxicity in multidrug-resistant human ovarian cystadenocarcinoma cells. Cancer Cell Int 2007; 7:5; http://dx.doi.org/10.1186/1475-2867-7-5; PMID: 17439664
- Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer 2012; 12:349 - 61; http://dx.doi.org/10.1038/nrc3218; PMID: 22475929
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528 - 39; http://dx.doi.org/10.1038/nrclinonc.2011.71; PMID: 21587219
- Corazzari MaL, Penny E. Harnessing Autophagy for Melanoma Benefit. Cell Biol 2013; 2
- Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One 2013; 8:e55096; http://dx.doi.org/10.1371/journal.pone.0055096; PMID: 23383069
- Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell 2011; 19:613 - 28; http://dx.doi.org/10.1016/j.ccr.2011.03.012; PMID: 21575862
- Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:370; http://dx.doi.org/10.1186/1471-2407-10-370; PMID: 20630104
- Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, Li HT, Sung JJ, Cho CH. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun 2009; 382:451 - 6; http://dx.doi.org/10.1016/j.bbrc.2009.03.051; PMID: 19289106
- Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA, Schönthal AH, Louie SG. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett 2012; 326:143 - 54; http://dx.doi.org/10.1016/j.canlet.2012.07.029; PMID: 22863539
- Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 2011; 6:e18691; http://dx.doi.org/10.1371/journal.pone.0018691; PMID: 21655094
- Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J, Su J, Li H, Sun L. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett 2012; 314:232 - 43; http://dx.doi.org/10.1016/j.canlet.2011.09.034; PMID: 22019047
- Armstrong JL, Corazzari M, Martin S, Pagliarini V, Falasca L, Hill DS, Ellis N, Al Sabah S, Redfern CP, Fimia GM, et al. Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin Cancer Res 2011; 17:2216 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-10-3003; PMID: 21270111
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012; 366:207 - 15; http://dx.doi.org/10.1056/NEJMoa1105358; PMID: 22256804
- De D, Krogstad FM, Byers LD, Krogstad DJ. Structure-activity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. J Med Chem 1998; 41:4918 - 26; http://dx.doi.org/10.1021/jm980146x; PMID: 9836608
- Iwaniuk DP, Whetmore ED, Rosa N, Ekoue-Kovi K, Alumasa J, de Dios AC, Roepe PD, Wolf C. Synthesis and antimalarial activity of new chloroquine analogues carrying a multifunctional linear side chain. Bioorg Med Chem 2009; 17:6560 - 6; http://dx.doi.org/10.1016/j.bmc.2009.08.003; PMID: 19703776
- Anderson T, Nkhoma S, Ecker A, Fidock D. How can we identify parasite genes that underlie antimalarial drug resistance?. Pharmacogenomics 2011; 12:59 - 85; http://dx.doi.org/10.2217/pgs.10.165; PMID: 21174623
- Muregi FW, Wamakima HN, Kimani FT. Novel drug targets in malaria parasite with potential to yield antimalarial drugs with long useful therapeutic lives. Curr Pharm Des 2012; 18:3505 - 21; PMID: 22607143
- Solomon VR, Hu C, Lee H. Design and synthesis of chloroquine analogs with anti-breast cancer property. Eur J Med Chem 2010; 45:3916 - 23; http://dx.doi.org/10.1016/j.ejmech.2010.05.046; PMID: 20561720
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139