
Abstract
The autophagy protein BECN1/Beclin 1 is known to play a central role in autophagosome formation and maturation. The results presented here demonstrate that BECN1 interacts with the Parkinson disease-related protein PARK2. This interaction does not require PARK2 translocation to mitochondria and occurs mostly in cytosol. However, our results suggest that BECN1 is involved in PARK2 translocation to mitochondria because loss of BECN1 inhibits CCCP- or PINK1 overexpression-induced PARK2 translocation. Our results also demonstrate that the observed PARK2-BECN1 interaction is functionally important. Measurements of the level of MFN2 (mitofusin 2), a PARK2 substrate, demonstrate that depletion of BECN1 prevents PARK2 translocation-induced MFN2 ubiquitination and loss. BECN1 depletion also rescues the MFN2 loss-induced suppression of mitochondrial fusion. In sum, our results demonstrate that BECN1 interacts with PARK2 and regulates PARK2 translocation to mitochondria as well as PARK2-induced mitophagy prior to autophagosome formation.
Introduction
The autophagy protein BECN1 plays a central role in autophagosome formation and maturation. Together with PIK3C3 and PIK3R4, it forms the core of the phosphatidylinositol-3-kinase complex required for autophagosome formation.Citation1 BECN1 has a number of binding partners that can positively or negatively regulate the autophagy core complex. ATG14, UVRAG, and SH3GLB1 are reported to be positive regulators of autophagosome formation or maturation,Citation2-Citation5 whereas KIAA0226 may have an inhibitory role.Citation3,Citation6 BECN1 has been discovered as a BCL2-interacting protein,Citation7 and BCL2 was shown to decrease BECN1 interaction with PIK3C3 and may therefore sequester BECN1 away from the core complex.Citation8 Other important BECN1 interacting proteins including AMBRA1 and BCL2L11 have been found to affect autophagy by switching the subcellular localization of BECN1 or core complex between the cytoskeleton and endoplasmic reticulum.Citation9,Citation10
Recently, a Parkinson disease (PD)-related protein, PINK1 (PTEN-induced putative kinase 1), has also been shown to be a BECN1-interacting protein that promotes BECN1-dependent autophagy.Citation11 PINK1 is a mitochondrial serine/threonine protein kinase that is turned over rapidly, which keeps its levels low under normal conditions when the mitochondrial membrane is polarized.Citation12-Citation14 However, depolarization of the mitochondrial membrane potential inhibits the degradation of PINK1 and leads to a rapid accumulation of full-length PINK1 at the mitochondrial outer membrane.Citation15-Citation18 This leads to the recruitment of another PD-related protein, the E3 ubiquitin ligase PARK2/parkin (parkin RBR E3 ubiquitin protein ligase), to the mitochondria for ubiquitination of mitochondrial outer membrane proteins.Citation16,Citation19-Citation21 The autophagy receptor proteins SQSTM1 and/or VCP, an AAA+ ATPase, bind to polyubiquitinated proteins at the outer membrane and target these mitochondria for engulfment by autophagosomes.Citation16,Citation21
Our results demonstrate that BECN1 interacts with PARK2 and regulates PARK2 translocation to mitochondria as well as PARK2-induced mitophagy, prior to autophagosome formation. Thus, BECN1 appears to be involved in the initiation of mitophagy and could actively participate in the PINK1-PARK2 pathway thereby playing a crucial role in PD. These data suggest that BECN1 is not only required for the formation of autophagosomes but also plays a role in the initiation of mitophagy.
Results
Becn1 silencing inhibits PARK2 translocation to mitochondria
Our first task was to test whether BECN1 participates in CCCP-induced PARK2 translocation to mitochondria. PC12 cells were transfected with fluorescent YFP-PARK2, scrambled shRNA, or rat Becn1 shRNA and treated with 10 µM CCCP for 3 h. This treatment led to translocation in approximately 75% of scrambled shRNA-expressing cells but a significantly smaller proportion (~50%) showed translocation in Becn1 shRNA-expressing cells (). Atg12 shRNA, which was used as a negative control, had no inhibitory effect on CCCP-induced YFP-PARK2 translocation. We also observed that coexpression of human BECN1 with Becn1 shRNA reversed the inhibitory effect of Becn1 shRNA on CCCP-induced YFP-PARK2 translocation in PC12 cells. However, BECN1 overexpression was unable to reverse the effect of Pink1 shRNA on CCCP-induced YFP-PARK2 translocation ().
Figure 1. BECN1 is required for PARK2 translocation to mitochondria. (A and B) PC12 cells transfected with YFP-PARK2, Mito-CFP, and plasmids of interest were treated with DMSO or 10 µM CCCP for 3 h. (A) Intracellular localization of PARK2 in the presence of DMSO or CCCP in scrambled shRNA-, Becn1 shRNA-, or Pink1 shRNA-expressing cells. (B) The percentage of cells with PARK2 translocated to mitochondria was lower in the Becn1 shRNA-expressing group but not when this shRNA was coexpressed with shRNA-resistant BECN1. ***P < 0.001 compared with the DMSO-treated scrambled shRNA, $$$P < 0.001 compared with the CCCP-treated scrambled shRNA and ###P < 0.001 compared with the Becn1 shRNA group. (C and D) PC12 cells transfected with plasmids of interest were selected using G418 for 7 d and treated with DMSO or 10 µM CCCP for 3 h, after that total cell lysates, and mitochondria-enriched and cytosolic fractions were prepared. (C) CCCP increases the level of PARK2 in the mitochondrial fraction in the presence of scrambled shRNA but considerably less PARK2 was found in cells expressing Becn1 shRNA. The effect of Becn1 shRNA was reversed when coexpressed with shRNA-resistant BECN1. Reciprocal depletion of PARK2 was observed in the cytosol but PARK2 levels remained unaltered in total cell lysates. (D) Densitometric band-intensity ratio of PARK2/VDAC1 based on 3 different experiments, one of which is shown in (C). Becn1 shRNA significantly inhibits PARK2 translocation to mitochondria but not when this shRNA was coexpressed with shRNA-resistant BECN1. ***P < 0.001 compared with the DMSO-treated scrambled shRNA, $$$P < 0.001 compared with the CCCP-treated scrambled shRNA and ###P < 0.001 compared with the Becn1 shRNA group. (E) PC12 cells transfected with scrambled or Becn1 shRNA were treated with DMSO or CCCP followed by mitochondrial isolation and WB to detect the accumulation of full size 63-kDa PINK1. CCCP-induced accumulation of endogenous PINK1 was not inhibited by Becn1 shRNA. (F) PC12 cells were transfected with YFP-PARK2 and scrambled shRNA- or Becn1 shRNA-expressing plasmids with or without PINK1 overexpression. PINK1-induced YFP-PARK2 translocation to mitochondria was partly suppressed in Becn1 shRNA-expressing cells. ***P < 0.001 compared with the scrambled shRNA and ###P < 0.001 compared with the PINK1 plus scrambled shRNA.
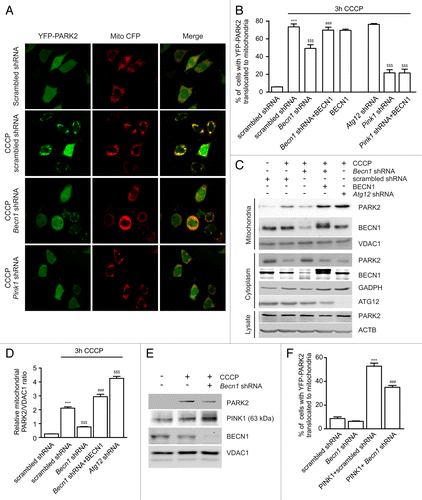
To verify that BECN1 is similarly involved in endogenous PARK2 translocation, PC12 cells were transfected with scrambled shRNA, Becn1 shRNA, and Atg12 shRNA alone or with BECN1. One scrambled shRNA group was treated with DMSO and all remaining groups were treated with 10 µM CCCP for 3 h, and western blots (WB) were performed from cytosolic and mitochondrial fractions as well as from total cell lysate. shows that CCCP treatment increased PARK2 levels in mitochondria but decreased it in cytosol. In contrast, significantly less PARK2 translocation from cytosol to mitochondria was observed in the CCCP-treated Becn1 shRNA group (). Once again, we observed that the inhibitory effect of Becn1 shRNA on PARK2 translocation was rescued by shRNA-resistant BECN1 whereas Atg12 shRNA did not inhibit PARK2 translocation.
Thus, both microscopic and WB evidence suggest that Becn1 silencing inhibited ectopically expressed as well as endogenous PARK2 translocation to mitochondria following CCCP treatment whereas silencing another autophagy gene, Atg12, did not inhibit PARK2 translocation in either case.
We next aimed to differentiate whether the silencing of Becn1 inhibited PARK2 translocation per se or whether it decreased PINK1 accumulation on mitochondria, thus leading to decreased PARK2 translocation. As demonstrated in , 3 h of CCCP treatment led to the accumulation of full-length PINK1 to mitochondria. Similar or even stronger accumulation of PINK1 was observed in the Becn1 shRNA group, whereas in the same experiment, PARK2 translocation to mitochondria was inhibited. Moreover, Becn1 shRNA also inhibited PARK2 translocation induced by PINK1 overexpression (). PINK1 overexpression induced PARK2 translocation in 51% of scrambled shRNA-expressing cells but in only 33% of Becn1 shRNA-expressing cells. These results suggest that BECN1 is involved in CCCP or PINK1 overexpression-induced PARK2 translocation to mitochondria and that this effect is not related to inhibition of PINK1 accumulation at mitochondria.
BECN1 interacts with PARK2
We next asked whether BECN1 interacts with PARK2. HEK293 cells were transfected with FLAG-BECN1 and MYC-PARK2, treated with DMSO or CCCP for 3 h, and immunoprecipitated using an anti-MYC antibody. demonstrates that FLAG-BECN1 coimmunoprecipitates with MYC-PARK2 and that this interaction is stronger in CCCP-treated cells. We also performed control experiments using endogenous AMBRA1, which is known to interact with PARK2.Citation22 As expected, AMBRA1 coimmunoprecipitated with PARK2, and similar to BECN1, this interaction was increased in CCCP-treated cells. However, AMBRA1 was not required for the PARK2-BECN1 interaction because Ambra1 shRNA did not abolish it (Fig. S1A). Similarly, the PARK2-AMBRA1 interaction was also insensitive to Becn1 knockdown (Fig. S1B). Consistent with an earlier report,Citation22 we also found that Ambra1 shRNA did not inhibit YFP-PARK2 translocation in PC12 cells (Fig. S1C). Similar to AMBRA1, another BECN1-binding protein, PIK3C3, also coimmunoprecipitated with PARK2 in a CCCP-dependent manner (Fig. S1D).
Figure 2. BECN1 and PARK2 interaction does not require PARK2 ligase activity. (A) HEK293 cells transfected with FLAG-BECN1 or/and MYC-PARK2 were treated with DMSO or 10 µM CCCP for 3 h. Total cell lysates were coimmunoprecipitated with mouse anti-MYC and analyzed on WB using antibodies raised in rabbit. Overexpressed FLAG-BECN1 coimmunoprecipitated with MYC-PARK2 both in DMSO- and CCCP-treated cells. Note that endogenous AMBRA1 coimmunoprecipitates with PARK2. (B) Total cell lysates from DMSO- or CCCP-treated PC12 cells were immunoprecipitated with mouse anti-PARK2, and then BECN1 and PARK2 were detected using antibodies raised in rabbit. The panel shows that endogenous BECN1 interacts with endogenous PARK2 in a CCCP-dependent manner. (C) WT or Pink1-deficient MEFs were treated with either DMSO or 10 µM CCCP for 3 h. Endogenous PARK2 was immunoprecipitated from total cell lysates using rabbit anti-PARK2. Input and IP elutes were analyzed using antibodies raised in mouse with the exception that PINK1 in the input was detected using rabbit anti-PINK1. The panel demonstrates that the PARK2-BECN1 interaction is decreased although not lost in Pink1-deficient MEFs under basal conditions whereas CCCP treatment augmented the interaction in both WT and Pink1-deficient MEFs. (D) WT or Pink1-deficient MEFs transfected with YFP-PARK2 were quantified for YFP-PARK2 translocation at 0 h and after 4 h of 10 μM FCCP treatment. ***P < 0.001 compared with the control group. (E) HEK293 cells transfected with FLAG-BECN1 and WT, K161N- or T240R-mutated MYC-PARK2 were treated with DMSO or 10 µM CCCP for 3 h. Immunoprecipitation and WB were performed as described for (A). The panel demonstrates that BECN1 interacts with all PARK2 versions and that CCCP increased this interaction in all groups. Note also the increased levels of immunoprecipitated AMBRA1 in the CCCP-treated groups. (F) PC12 cells transfected with WT, K161N, or T240R YFP-PARK2 were treated with DMSO or 10 µM FCCP for 3 h. FCCP induced translocation in WT YFP-PARK2-transfected cells and little or no translocation was observed in the K161N or T240R YFP-PARK2-transfected cells. ***P < 0.001 compared with the respective control group. (G) HeLa cells were transfected with BECN1 and IKBKG-FLAG with empty vector, WT PARK2 or PARK2T240R. At 24 h, transfected cells were treated with 10 µM MG132 for 8 h. BECN1 and IKBKG were immunoprecipitated using antibodies raised in rabbit and then probed using antibodies raised in mouse. The panel demonstrates that only WT PARK2 led to the prominent appearance of higher molecular weight forms of IKBKG whereas not in case of BECN1. Furthermore, the UBB signal was detected only in IKBKG blots.
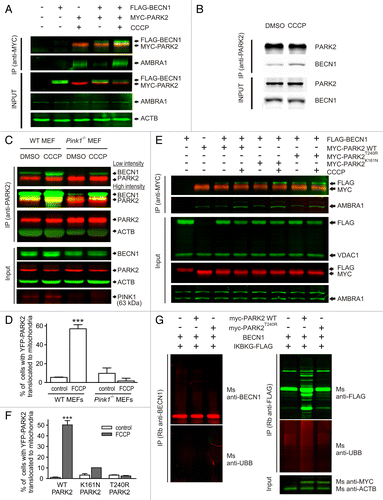
We next performed an endogenous BECN1-PARK2 coimmunoprecipitation experiment in the presence or absence of CCCP. As demonstrated in , endogenous BECN1 in PC12 cells also coimmunoprecipitated with endogenous PARK2, and this interaction increased after CCCP treatment. Similar to CCCP treatment, starvation also increased BECN1-PARK2 interaction (Fig. S2A).
An earlier report demonstrated that BECN1 interacted with full-length PINK1.Citation11 To determine whether the BECN1-PARK2 interaction requires PINK1, we immunoprecipitated endogenous PARK2 from mouse embryonic fibroblasts (MEFs) isolated from wild-type or Pink1-deficient mice. BECN1 coimmunoprecipitated with PARK2 both in wild-type and in Pink1-deficient MEFs, although visibly less in the latter (). Moreover, CCCP treatment increased this interaction in both groups. We also verified that Pink1-deficient MEFs showed no PARK2 translocation in response to FCCP (). Further, PINK1 overexpression had no effect on the amount of immunoprecipitated BECN1-PARK2 (Fig. S2B).
We next tested whether BECN1 interacts with pathogenic, ligase-dead mutants of PARK2. HEK293 cells were transfected with FLAG-BECN1 and wild-type (WT), K161N- or T240R-mutated MYC-tagged PARK2 and treated with DMSO or CCCP followed by immunoprecipitation using a mouse anti-MYC antibody (). The amount of FLAG-BECN1 coimmunoprecipitating with PARK2 increased when cells were treated with CCCP, suggesting that the ligase activity of PARK2 was not required for the BECN1-PARK2 interaction (). Note also that with all PARK2 mutants, the levels of immunoprecipitated endogenous AMBRA1 increased after CCCP treatment. These observations are consistent with an earlier report where the PARK2-AMBRA1 interaction was not found to be affected by several PARK2 mutants.Citation22 Parallel experiments with YFP-tagged PARK2 mutants demonstrated that both PARK2 mutants failed to translocate to mitochondria after FCCP treatment ().
Furthermore, our results suggest that PARK2 does not ubiquitinate BECN1 as overexpression of WT PARK2 did not lead to the ubiquitination of BECN1. However, WT PARK2 did induce ubiquitination of IKBKG (used as a positive control, ).
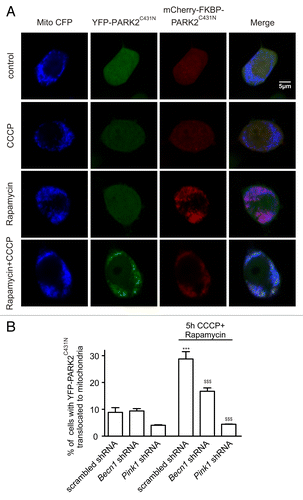
Recent evidence suggests that PINK1 induces PARK2 self-association, potentially via phosphorylationCitation23,Citation24, leading to its activation followed by attachment to mitochondrial substrates.Citation23 We therefore investigated whether BECN1 is required for PARK2 self-association and activation or for its attachment to mitochondrial substrates. We followed a previously described method using a pair of fluorescent PARK2 mutants (mCherry-FKBP-PARK2C431N and YFP-PARK2C431N) that can self-associate in response to CCCP but cannot bind to mitochondrial substrates and cannot translocate to mitochondria.Citation23,Citation25 mCherry-FKBP-PARK2C431N was first tethered to mitochondria using rapamycin-induced heterodimerization of its FKBP domain to the FRB domain, which is localized to mitochondria by the FIS1 membrane anchor (FRB-FIS1). YFP–PARK2C431N normally remained in the cytosol under these conditions (). However, YFP–PARK2C431N translocated to mitochondria after CCCP treatment, suggesting its association with mitochondrially tethered mCherry-FKBP-PARK2C431N (). Using this method, we found that mitochondrial translocation of YFP–PARK2C431N was significantly reduced in rapamycin- and CCCP-treated cells transfected with Becn1 shRNA (). In addition, silencing of Becn1 suppressed the CCCP-induced PARK2 phosphorylation, which is a modification shown to prime PARK2 for translocation (Fig. S3).Citation26 These results suggest that BECN1 is required prior to PARK2 self-association (possibly also before or during the PARK2 phosphorylation by PINK1) and activation upstream of mitochondrial translocation. Note that no YFP–PARK2C431N translocation was observed under these settings in Pink1 shRNA expressing cells ().
Figure 3. BECN1 is involved in PARK2 self-association. (A) PC12 cells transfected with mCherry-FKBP-PARK2C431N, YFP-PARK2C431N, FRB-FIS1 with scrambled-, Becn1- or Pink1 shRNA were treated with 100 nm rapamycin and 5 µM CCCP for 5 h. CCCP induced translocation of YFP-PARK2C431N to mitochondria only after rapamycin treatment, which induced heterodimerization of mCherry-FKBP-PARK2C431N to the mitochondrial FRB-FIS1. (B) The percentage of cells with YFP-PARK2C431N translocated to mitochondria was lower in the Becn1- and Pink1 shRNA-expressing groups. ***P < 0.001 compared with the DMSO-treated scrambled shRNA and $$$P < 0.001 compared with the CCCP-treated scrambled shRNA.
BECN1 and PARK2 interaction is mainly cytoplasmic
Next, we attempted to locate the site of the BECN1-PARK2 interaction in the cell. We first performed an indirect in situ proximity ligation assay (PLA). HEK293 cells overexpressing MYC-tagged WT PARK2, FLAG-tagged WT BECN1, and a GFP mitochondrial marker were first incubated with mouse anti-MYC and rabbit anti-FLAG antibodies and the interaction was visualized using specific PLA probes and red fluorophore-labeled oligonucleotides. The results demonstrate that MYC-PARK2 colocalized with FLAG-BECN1, and the majority of these interactions were in the cytoplasm (). In contrast, the negative controls, where only one of the primary antibodies was used, revealed negligible nonspecific binding of PLA probes (). We also observed a similar cytoplasmic PLA signal when we used another pair of antibodies, mouse anti-BECN1 and rabbit anti-PARK2, instead of the anti-tag antibodies ().
Figure 4. BECN1 and PARK2 interact in the cytoplasm. (A and C) HEK293 cells transfected with FLAG-BECN1, MYC-PARK2, and Mito-GFP were treated with either DMSO or 10 µM CCCP for 3 h. Fixed and permeabilized cells were first incubated with rabbit anti-FLAG and mouse anti-MYC (A) or rabbit anti-PARK2 and mouse anti-BECN1 antibodies (C). Subsequently, cells were incubated with anti-rabbit PLUS and anti-mouse MINUS PLA probes followed by ligation and amplification. The interacting proteins were visualized using red fluorophore-labeled oligonucleotides and showed cytoplasmic localization. (B and D) For controls, HEK293 cells were transfected and treated as above. Fixed and permeabilized cells were incubated with only one of the primary antibodies (either rabbit anti-FLAG or mouse anti-MYC for [B] and either mouse anti-BECN1 or rabbit anti-PARK2 for [D]). Cells were then incubated with both anti-rabbit PLUS and anti-mouse MINUS PLA probes and visualized using red fluorophore-labeled oligonucleotides, as above. The panels demonstrate that no red PLA signal was detected when one of the primary antibodies was omitted. (E) HEK293 cells were transfected with FLAG-BECN1 and MYC-PARK2 and treated as above. Fixed and permeabilized cells were incubated with mouse anti-MYC and rabbit anti-FLAG antibodies and subsequently with green anti-mouse and red anti-rabbit secondary antibodies. The panel demonstrates that both antibodies recognize the cytosolic antigens and that treatment with CCCP led to a partial translocation of MYC-PARK2 but did not affect visibly the localization of FLAG-BECN1.
![Figure 4. BECN1 and PARK2 interact in the cytoplasm. (A and C) HEK293 cells transfected with FLAG-BECN1, MYC-PARK2, and Mito-GFP were treated with either DMSO or 10 µM CCCP for 3 h. Fixed and permeabilized cells were first incubated with rabbit anti-FLAG and mouse anti-MYC (A) or rabbit anti-PARK2 and mouse anti-BECN1 antibodies (C). Subsequently, cells were incubated with anti-rabbit PLUS and anti-mouse MINUS PLA probes followed by ligation and amplification. The interacting proteins were visualized using red fluorophore-labeled oligonucleotides and showed cytoplasmic localization. (B and D) For controls, HEK293 cells were transfected and treated as above. Fixed and permeabilized cells were incubated with only one of the primary antibodies (either rabbit anti-FLAG or mouse anti-MYC for [B] and either mouse anti-BECN1 or rabbit anti-PARK2 for [D]). Cells were then incubated with both anti-rabbit PLUS and anti-mouse MINUS PLA probes and visualized using red fluorophore-labeled oligonucleotides, as above. The panels demonstrate that no red PLA signal was detected when one of the primary antibodies was omitted. (E) HEK293 cells were transfected with FLAG-BECN1 and MYC-PARK2 and treated as above. Fixed and permeabilized cells were incubated with mouse anti-MYC and rabbit anti-FLAG antibodies and subsequently with green anti-mouse and red anti-rabbit secondary antibodies. The panel demonstrates that both antibodies recognize the cytosolic antigens and that treatment with CCCP led to a partial translocation of MYC-PARK2 but did not affect visibly the localization of FLAG-BECN1.](/cms/asset/426b6ee4-243d-415f-a2f3-b16cd068b827/kaup_a_10928615_f0004.gif)
Treatment with CCCP led to the partial translocation of PARK2 but did not visibly affect the localization of BECN1 (). Nevertheless both antibody pairs demonstrated cytosolic interaction ().
To confirm the cytosolic BECN1-PARK2 interaction, we performed blue native gel electrophoresis followed by western blotting analysis of cytosolic- and mitochondria-enriched fractions from HEK293 cells treated with either DMSO or CCCP. BECN1 and PARK2 showed similar migration patterns only in cytosolic fractions but not in mitochondrial fractions (Fig. S4A–S4C). Further, immunoprecipitation experiments using the same materials demonstrated that PARK2 coimmunoprecipitated with BECN1 in cytosolic but not in mitochondrial fractions (Fig. S4D). Also, on gel filtration we found endogeneous BECN1-PARK2 complex coeluted (Fig. S4E and S4F).
Functional relevance of the BECN1 in PARK2 translocation
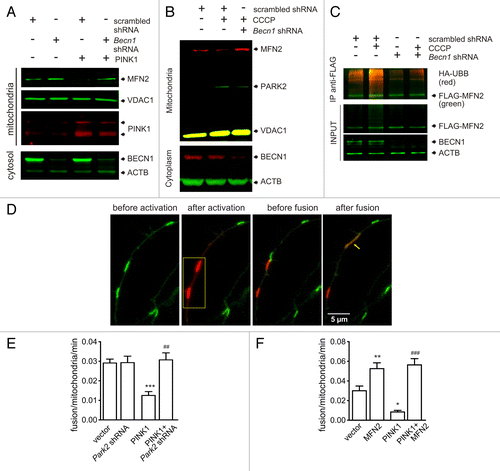
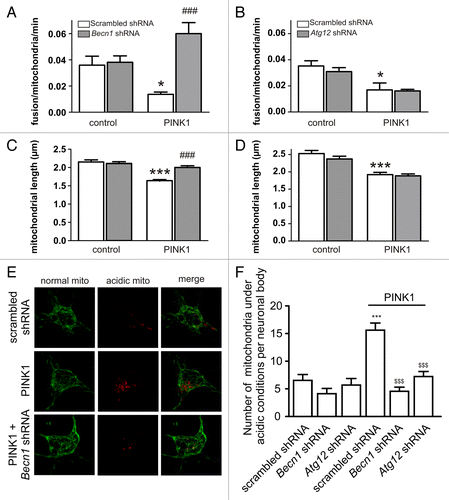
To define the role of BECN1 during PARK2 translocation, we required an endpoint that was in between PARK2 translocation and autophagosome formation. PARK2 translocation to mitochondria leads to the ubiquitination and proteasome-mediated degradation of mitochondrial outer membrane proteins including mitofusins (MFNs).Citation27-Citation29 Indeed, both PINK1 overexpression () and CCCP treatment () led to the depletion of MFN2 in PC12 cells. Interestingly, Becn1 shRNA prevented MFN2 ubiquitination and loss (). To observe the functional outcome of depleted MFN2, we quantified mitochondrial fusion events in primary cortical neurons using the mitochondria-targeted photoconvertable KikGR1 protein.Citation30 This construct labels mitochondria with green fluorescence that can be photoconverted to red by irradiation with a 405 nm laser line. Conversion of a few mitochondria to red allows visualization of their fusion with green mitochondria and enables the quantification of fusion events from a time-series of images (; Video S1). Overexpression of PINK1 led to a significant decrease in fusion rate (). Moreover, we verified that the effect of PINK1 on the fusion rate required PARK2 because no suppression was observed when we coexpressed PINK1 with Park2 shRNA (). We also confirmed that the observed effect is MFN2 dependent because coexpression of MFN2 with PINK1 abolished the negative effect of PINK1 ().
Figure 5. PINK1 overexpression suppresses MFN2-dependent mitochondrial fusions. (A) PC12 cells were transfected with scrambled shRNA or Becn1 shRNA with or without PINK1-expressing plasmid. Transfected cells were selected with G418 for 7 d, after that mitochondria-enriched and cytosolic fractions were prepared. The panel shows that PINK1 overexpression led to MFN2 depletion whereas no depletion was observed when PINK1 was coexpressed with Becn1-shRNA. (B) PC12 cells were transfected with scrambled shRNA or Becn1-shRNA. Transfected cells were selected with G418 for 7 d and then treated with DMSO or 10 µM CCCP for 6 h, after that mitochondria-enriched and cytosolic fractions were prepared. The panel shows that CCCP treatment led to MFN2 loss compared with DMSO-treated cells in scrambled shRNA transfected cells. Becn1-shRNA prevented CCCP-induced MFN2 loss. (C) PC12 cells were transfected with scrambled shRNA or Becn1 shRNA together with FLAG-MFN2 and UBB-HA. Transfected cells were selected with G418 for 7 d and then treated with 10 µM MG132 for 8 h prior to addition of DMSO or 10 µM CCCP for further 2 h incubation. Total cell protein was extracted in denaturating lysis buffer and immunoprecipitated using Rb anti-FLAG. Ubiquitination of MFN2 was detected on immunoblots using goat anti-HA antibodies. The panel shows that CCCP treatment enhanced ubiquitination of MFN2 whereas BECN1 knockdown inhibited the ubiquitination. (D) Primary cortical neurons were transfected with the photoconvertible mitochondria-targeted construct mito-KikGR1. Selected mitochondria were irradiated using a 405-nm laser line (marked with a yellow rectangle) and mito-Kikume-Green was converted into mito-Kikume-Red. Fusion events between mito-Kikume-Green and photoactivated mito-Kikume-Red mitochondria are visible when mitochondria become yellow after mixing of the contents of the red and green mitochondrial matrices (shown with arrow). (E and F) Primary cortical neurons were transfected with a plasmid expressing mito-KikGR1 and empty vector, PINK1, Park2 shRNA or MFN2-expressing plasmids as indicated. Fusion rates were estimated as described in Materials and Methods. (E) PINK1 inhibits the mitochondrial fusion rate in neurons and this effect requires PARK2. (F) The effect of PINK1 disappears in the presence of MFN2 overexpression (P = 0.027 for the interaction, 2-way ANOVA). *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the empty vector and ##P < 0.01 and ###P < 0.001 compared with the PINK1 group.
We next studied whether BECN1 is required for PINK1-induced MFN2 loss and inhibition of mitochondrial fusion. demonstrates that PINK1 overexpression did not reduce MFN2 levels in the presence of Becn1 shRNA. We also found that Becn1 shRNA reversed the inhibitory effect of PINK1 on the mitochondrial fusion rate without having any effect on the fusion rate alone (). The observed changes in fusion rate were also reflected at the level of mitochondrial length. Mitochondrial length was significantly reduced in PINK1-overexpressing neurons but not when coexpressed with Becn1 shRNA (). To confirm the specificity of these results, we performed control experiments with Atg12 shRNA. Atg12 shRNA did not reverse the reduction in fusion rate induced by PINK1 (). Atg12 shRNA also had no effect on mitochondrial length nor did it prevent PINK1-induced mitochondrial shortening () although it inhibited PINK1-induced mitophagy similar to Becn1 shRNA (). Thus, our results demonstrate that the BECN1 interaction with PARK2 has functional importance in the PINK1-PARK2 pathway.
Figure 6. PINK1-induced inhibition of mitochondrial fusions requires BECN1. (A and B) Primary cortical neurons were transfected with mito-KikGR1 and empty vector (control) or PINK1 with scrambled shRNA, Becn1 shRNA, or Atg12 shRNA as indicated in the figure. Fusion rates were estimated as described in Materials and Methods. (A) PINK1 overexpression suppresses the mitochondrial fusion rate when coexpressed with scrambled shRNA but not when coexpressed with Becn1 shRNA (P = 0.003 for interaction, 2-way ANOVA). (B) The effect of PINK1 overexpression was not reversed by Atg12 shRNA (P = 0.628 for interaction). *P < 0.05 compared with control and ###P < 0.001 compared with the PINK1-only group. (C and D) Primary cortical neurons were transfected with EGFP, mito-pDsRed2 and empty vector (control) or PINK1 with scrambled shRNA, Becn1 shRNA, or Atg12 shRNA, and mitochondrial lengths were measured. (C) PINK1 overexpression led to mitochondrial shortening that was reversed by Becn1 shRNA (P < 0.001 for interaction). (D) The effect of PINK1 was not reversed by Atg12 shRNA (P = 0.410 for interaction). ***P < 0.001 compared with control and ###P < 0.001 compared with the PINK1-only group. (E and F) Primary cortical neurons were cotransfected with mitochondrially targeted Keima and scrambled shRNA, Becn1 shRNA, or Atg12 shRNA together with empty vector or PINK1. Images were acquired with a laser scanning confocal microscope using the 458 nm (green, mitochondria at neutral pH) and 561 nm (red, mitochondria under acidic pH) laser lines. (F) Red dots were counted manually by a blinded individual. ***P < 0.001 compared with scrambled shRNA, $$$P < 0.001 compared with the PINK1+scrambled shRNA.
Discussion
BECN1 plays a key role in autophagy and is involved in the initiation of autophagosome formation and/or recruitment of membranes to form autophagosomes. Loss of BECN1 inhibits autophagy leading to various consequences in different tissues. In brain, the heterozygous deletion of BECN1 leads to neurodegeneration.Citation31 BECN1 participates in the clearance of amyloid-β,Citation32 mutant HTT/huntingtin,Citation33 and mutant ATXN3/ataxin 3.Citation34 Interestingly, a decrease in BECN1 levels has been observed in Alzheimer, Huntington, and Machado–Joseph diseases, which has been used to explain the impaired clearance of the accumulating proteins underlying these diseases.Citation32-Citation35 BECN1 overexpression activates autophagy and is protective in different neurodegenerative models. For example, administration of a lentiviral vector expressing BECN1 reduces both intracellular and extracellular amyloid pathology in transgenic mice expressing amyloid precursor protein.Citation32 Overexpression of BECN1 clears mutant ATXN3 and alleviates Machado-Joseph disease.Citation34 BECN1 overexpression has also been shown to prevent neurodegeneration in mouse models of PD.Citation36 To date, all of these effects have been linked to the nucleation step of autophagy, where BECN1 interacts with the PIK3C3 to form autophagy core complex.Citation1
This study proposes that BECN1 also plays a role in the initiation of mitophagy, prior to autophagosome formation around the mitochondrion. Our results demonstrate that BECN1 interacts with PARK2. This interaction was observed with endogenous proteins and with overexpressed proteins in different cell types. Interestingly, while this report was under revision, an article was published suggesting that functional PARK2-BECN1 interaction mediates amyloid degradation.Citation37 Besides the BECN1-PARK2 interaction, we also demonstrate that BECN1 is actively involved in the PINK1-PARK2 pathway. BECN1 depletion inhibited PARK2 translocation to mitochondria suggesting the involvement of BECN1 in PARK2 translocation. This inhibition was most evident with endogenous PARK2, suggesting insufficient endogenous BECN1 to interact with overexpressed PARK2.
The PARK2-BECN1 interaction appears to occur mostly in cytosol. We observed this interaction also with PARK2 mutants that were unable to translocate to mitochondria after mitochondrial depolarization. We also demonstrate that BECN1 is involved in PINK1-dependent PARK2 activation before PARK2 self-association.Citation23 Although weaker, the interaction was also present in Pink1-deficient MEFs, suggesting that the PARK2-BECN1 interaction is not bridged by PINK1, which has been shown to interact with BECN1.Citation11 These results suggest that BECN1 may facilitate PARK2 self-association. However, we cannot completely exclude the possibility that a small but relevant portion of the PARK2-BECN1 interaction complex in mitochondria involves PINK1. Because BECN1 appears to interact with both PARK2 and PINK1, we also cannot exclude the possibility that BECN1 itself serves as a bridge linking PARK2 and PINK1 at the mitochondrial membrane. It is important to also note that AMBRA1, previously shown to interact with BECN1 and PARK2,Citation22,Citation38 did not act as a bridge between PARK2 and BECN1 as Ambra1 silencing did not affect PARK2 translocation or the PARK2-BECN1 interaction. Interestingly, the PARK2-AMBRA1 interaction was also insensitive to Becn1 silencing.
In addition to BECN1 itself, several BECN1 binding proteins also interact with PARK2.Citation22,Citation39 We demonstrate here that another component of the autophagy core complex, PIK3C3, coimmunoprecipitates with PARK2. Consistent with earlier results, AMBRA1, which has been suggested to participate in the release of the core complex from dynein upon activation of autophagy,Citation10 coimmunoprecipitated with PARK2. These results suggest that PARK2 may form a complex with the autophagy core complex and associated proteins. If so, then PARK2 may serve as a tool to transport the autophagy core complex to sites where it is required. Close proximity of autophagy proteins would enable faster recognition of PARK2-ubiquitinated sites and facilitate mitophagy.
The PARK2-BECN1 interaction was enhanced by CCCP, the known inducer of PARK2 translocation to mitochondria, and by starvation. Consistent with a previous report,Citation22 we also observed that CCCP increased the PARK2-AMBRA1 interaction. In addition, CCCP also increased PIK3C3 interaction with PARK2. However, the effect of CCCP cannot be explained by PARK2 translocation to mitochondria because a similar increase was observed in Pink1-deficient MEFs and with PARK2 mutants unable to translocate to mitochondria.
We also demonstrate that the PARK2-BECN1 interaction is functionally important. We measured the functional activity of the PARK2 substrate MFN2. Mitochondrial PARK2 is known to ubiquitinate mitochondrial outer membrane proteins, such as MFN2, thus targeting them for degradation.Citation27-Citation29 Degradation of MFN2 is thought to inhibit mitochondrial fusion, leading to mitochondrial fragmentation and thereby facilitating mitophagy.Citation21 To our knowledge, this is the first study to demonstrate that PINK1-induced PARK2 translocation indeed suppresses the mitochondrial fusion rate. However, in the BECN1-depleted state, PINK1 overexpression did not induce MFN2 loss and failed to suppress the mitochondrial fusion rate. Because ATG12, which is known to act downstream to BECN1 in autophagosome formation, showed no such effect, the inhibition of autophagosome formation cannot be the cause. Thus, BECN1 suppression not only interferes with the clearance of dysfunctional mitochondria but also allows fusion of mitochondria irrespective of their functional status. The inability to inhibit fusion of dysfunctional mitochondria could contribute to the mitochondrial pathophysiology known to be present in PD.
In sum, our results demonstrate that BECN1 interacts with PARK2 and regulates PARK2 translocation to mitochondria in addition to PARK2-induced mitophagy prior to autophagosome formation. These data suggest that BECN1 controls mitophagy at 2 levels—at the initiation by controlling PARK2 translocation to mitochondria upon depolarization and also at the culmination where it is required for autophagosome formation for engulfment of mitochondria. This dual function of BECN1 may enable more coordinated control and balance between mitophagy and general autophagy.
Materials and Methods
Media, transfection, and reagents
Reagents were from the following manufacturers: 35-mm glass-bottomed dishes (MatTek, P35G-1.0-14-C), penicillin, streptomycin, and amphotericin B (Invitrogen, 15240-096), RPMI-1640 (Life Technologies, A10491-01), DMEM (Life Technologies, 31966-021), HBSS 1640 (Life Technologies, 14025050), Opti-MEM I (Life Technologies, 31985-047), Lipofectamine 2000 (Invitrogen, 11668-019), Metafectine (Biontex Laboratories GmbH, T040), DMSO (Sigma, 472301), CCCP (Tocris, 0452), FCCP (Tocris, 0453), G418 (Sigma, A1720), MG132 (Tocris, 1748), mitochondrial isolation kit (Mitoscience, MS852/853), Protease inhibitor cocktail, cOmplete (Roche, 04693116001) protein G-sepharose 4B beads (Invitrogen, 101243), DC Protein Assay reagent (Bio-Rad, 500-0111). The NativePAGE™ Novex® Bis-Tris Gel System was from Invitrogen.
Plasmids
BECN1 (Addgene, 21150), PINK1 N-MYC (Addgene, 13313), IKBKG (Addgene, 11970), HA-Ubiquitin (UBB) (Addgene, 18712). Mito-Keima was from Amalgaam (AM-V0251). Plasmids expressing mitochondrial pDsRed2 (632421), EGFP (6085-1) and Mito-GFP (632432) were from Clontech. Plasmids expressing scrambled shRNA or shRNA against Park2 (KR50238N), Pink1 (KR55105N), Atg12 (KR43547N), Ambra1 (KR48097N), and Becn1 (KR06523N) were from SABiosciences. Human BECN1 siRNA was from Life Technologies (ID:s16537). MYC-PARK2, MYC-PARK2T240R and MYC-PARK2K161N were kind gifts from Dr Nobutaka Hattori,Citation40 MFN2 was from Dr S Hirose and Dr N NakamuraCitation41 and YFP-PARK2, YFP-PARK2C431N, mCherry-FKBP-PARK2C431N, and FRP-FIS1 were from Dr R Youle.Citation19,Citation23,Citation25 YFP-PARK2 point mutations T240R and K161N were generated by Mutagenex Inc. (Hillsborough, NJ, US). Mitochondrial-KikGR1 (mito-KikGR1) was constructed in our lab as described earlier.Citation30 The FLAG-tagged BECN1 construct was originally created by Dr Beth Levine.Citation7
Antibodies
Antibodies were from the following manufacturers: mouse (Ms) anti-PARK2 (Abcam, ab77924), rabbit (Rb) anti-PARK2 (Abcam, ab15954), Ms anti-BECN1 (Abcam, ab79937), Rb anti-BECN1 (Bethyl Laboratories Inc., A302-566A), sheep anti-ATG12 (Abcam, ab64208), Rb anti-AMBRA1 (Abcam, ab72098), Rb anti-VDAC1 (Abcam, ab15895), Ms anti-VDAC1 (Abcam, ab14734), Ms anti-ACTB (Sigma, A2228), Ms anti-MFN2 (Abcam, ab56889), Rb anti-MFN2 (Abcam, ab101055), Rb anti-MYC (Abcam, ab9106), Ms anti-MYC (Life Technologies, R950-25), Rb anti-FLAG (Sigma, F7425), Ms anti-FLAG (Sigma, F3165), goat anti-HA (Abcam, ab9134), Ms anti-UBB (Abcam, ab34490), Rb anti-GFP (Abcam, ab290), Rb anti-PINK1 (Sigma, HPA001931), goat anti-PINK1 (Everest Biotech, EB07940) and Rb anti-PINK1 (Novus Biological, BC100-494). Secondary antibodies used were horseradish-peroxidase (HRP)-conjugated secondary antibody (Pierce, 31460 or 31430) or goat anti-rabbit IRDye 800CW (926-32211) or 680LT (926-68021), goat anti-mouse IRDye 800CW (926-32210) or 680LT (926-68020) or donkey anti-goat IRDye 680LT (926-68024), all from Li-Cor Bioscience. Immuno-reactive bands were detected using enhanced chemiluminescence (ECL, Amersham Biosciences, 32209) or using the Odyssey®CLx Infrared Imaging System (LICOR Bioscience). The PLA probes were rabbit PLUS 100 (92002-0100) and PLA probe anti-mouse MINUS 100 (92004-0100), and Detection Reagent Red 100 (92008-0100) was used. Mounting media contained DAPI (82040-0005), and all were obtained from Olink Bioscience, Sweden.
Cell cultures
Primary cultures of rat cortical cells were prepared from neonatal Wistar rats as described earlier.Citation30 PC12 cells were grown in RPMI-1640 medium supplemented with 10% horse serum and 5% fetal bovine serum (FBS) on collagen IV (Sigma, C5533)-coated 100-mm plastic dishes or onto 35-mm glass-bottomed dishes. For starvation, PC12 cells were incubated for 24 h in an amino acid and serum-free starvation HBSS medium. HEK293 cells were cultured in DMEM-F12 nutrient mix containing glutamine and 15 mM HEPES supplemented with 10% FBS. HeLa cells were cultured in DMEM nutrient mix containing 4.5 g/l glucose, L-glutamine and supplemented with 10% FBS.
Mouse embryonic fibroblasts were isolated from wild-type and Pink1-deficient mice (C57Bl/6J and B6;129-Pink1tm1Aub/J; Jackson Laboratories) based on previously described methods.Citation42 MEF cells were cultured in DMEM containing 2 mM Glutamax (Gibco, Life Technologies, 35050-038), supplemented with 10% FBS and a 1% solution of penicillin, streptomycin, and amphotericin B. All culture media and supplements were obtained from Invitrogen unless otherwise stated.
Transfection
For transfection of cells growing on glass-bottomed dishes, the conditioned medium was replaced with 100 μl Opti-MEM I medium containing 2% Lipofectamine 2000 and 1 to 2 μg of total DNA with an equal amount of each different plasmid. The dishes were incubated for 3 to 4 h, after which fresh medium was added. For biochemical analyses, the cells were transfected in 100 mm plastic dishes as described above except that the total volume of the transfection mixture increased with proportionally adjusted Lipofectamine and DNA. The transfected DNAs were allowed to express 2 to 4 d before the experiments were performed. In some experiments with shRNA-expressing plasmids that also contained a neomycin resistance gene, the medium of PC12 cells was supplemented with 200 µg/ml G418 for 7 d. For MEFs, plasmids and Metafectine were prepared separately in PBS and then mixed at a ratio of 1:4 (w/v) DNA:Metafectine. DNA (1 μg) was added to each dish and diluted in MEF medium without antibiotics or antimycotic and incubated for 6 h. The medium was then replaced with fresh MEF medium without antibiotics or antimycotic.
Coimmunoprecipitation
For immunoprecipitation, the cells were washed twice, harvested in PBS, pelleted at 300 g for 5 min at 4 °C and then lysed in ice-cold IP lysis buffer containing 20 mM HEPES, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10% glycerol pH 7.4, and protease inhibitor cocktail. Cell lysates were passed through a 23 G needle 20 times before incubating on ice for 30 min with shaking at 350 rpm. The insoluble fraction was removed by centrifuging at 14000 g for 10 min at 4 °C. The protein concentrations were estimated using DC Protein Assay reagent according to the manufacturer’s protocol. Lysates were incubated overnight on rotary shaker at 4 °C in Pierce spin columns with 5 µg of Ms anti-PARK2 (Abcam ab77924), Rb anti-PARK2 (Abcam, ab15954), Ms anti-BECN1 (Abcam, ab79937), Rb anti-FLAG (Sigma, F7425), Rb anti-BECN1 or Ms anti-MYC (Invitrogen, 46-0603) antibodies. The following morning 150 µl protein G-sepharose 4B beads (50% suspension in IP lysis buffer) were added and further incubated for 5 h on a rotary shaker at 4 °C. Beads were then washed 5 times with IP lysis buffer and immunoprecipitates were eluted in Laemmli buffer at 99 °C for 10 min. For immunoprecipitation experiments of mitochondrial and cytosolic fractions, the cells were fractionated in advance as described below.
Western blotting
Cytosolic- and mitochondrial-enriched fractions were prepared using a mitochondrial isolation kit following the manufacturer’s instructions. RIPA-solubilized mitochondrial fractions, concentrated cytosolic fractions, RIPA-solubilized total cell lysates, inputs, and elutes from immunoprecipitation experiments were analyzed using western blotting (WB). Equivalent amounts of proteins were resolved on 10% or 12% polyacrylamide gels by SDS-PAGE. Resolved proteins were transferred to Hybond-P PVDF membranes or onto Immobilon-FL in 0.1 M Tris-base, pH 8.3, 0.192 M glycine and 10% (v/v) methanol using an electrophoretic transfer system with cold-block. The membranes were blocked using 5% (w/v) nonfat dried milk in TBS containing 0.025% (v/v) Tween-20 (Sigma, P1379) or Odyssey blocking buffer (LICOR Bioscience, 927-40000) at room temperature for 1 h. After blocking, the membranes were incubated sequentially overnight with different primary antibodies based on the requirement of the experiment. Antibody dilutions used were Ms anti-PARK2 1:1000, Rb anti-PARK2 1:1000, Ms anti-BECN1 1:1000, Rb anti-BECN1 1:1000, sheep anti-ATG12 1:1000, Rb anti-AMBRA1 1:300, Rb anti-VDAC1 1:2000, Ms anti-VDAC1 1:2000, Ms anti-ACTB 1:4000, Ms anti-MFN2 2 1:500, Rb anti-MYC 1:2000, Rb anti-FLAG 1:5000, Ms anti-FLAG 1:5000 and Rb anti-GFP 1:15000. For detection of PINK1, several antibodies from different sources were used: Rb anti-PINK1 1:300, goat anti-PINK1 1:300 and Rb anti-PINK1 1:300. Incubations were followed by washing and incubation with the appropriate horseradish-peroxidase (HRP)-conjugated secondary antibody (1:4000,) or with goat anti-rabbit IRDye 800CW or 680LT, goat anti-mouse IRDye 800CW or 680LT or donkey anti-goat IRDye 680LT (all from Li-Cor Bioscience) for 1 h at room temperature. Immuno-reactive bands were detected by enhanced chemiluminescence using medical X-ray film or using the Odyssey Infrared Imaging System (Li-Cor Bioscience, Lincoln, NE, USA).
Ubiquitination assay
The assays were performed as described earlierCitation22 with slight modifications. Briefly, for BECN1 and IKBKG ubiquitination studies, HeLa cells were transfected with BECN1 and IKBKG-FLAG together with empty vector, MYC tagged WT PARK2 or PARK2T240R. After 24 h, transfected cells were treated with 10 µM MG132 for 8 h and lysates were prepared as described below. For MFN2 ubiquitination studies, PC12 cells were transfected with scrambled shRNA or Becn1 shRNA together with Flag-MFN2 and UBB-HA. Transfected cells were selected with G418 for 7 d. Post-selection, transfected cells were first treated with 10 µM MG132 for 8 h and subsequently treated with DMSO or 10 µM CCCP for 2 h. From both types of cells, total cell proteins were extracted in denaturating lysis buffer (50 mM TRIS-HCl, pH 7.4, 5 mM EDTA, 1% SDS, 15 U/ml DNase I, and protease inhibitor cocktail) and boiled for 5 min. Protein extracts were then diluted 1:10 with non-denaturating lysis buffer (50 mM TRIS-HCl, pH 7.4, 300 mM NaCl, 5 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail), followed by preclearing and immunoprecipitation using Rb anti-BECN1 and Rb anti-FLAG (for IKBKG and MFN2). Ubiquitination was detected on immunoblots using either Ms anti-UBB or goat anti-HA antibodies.
Phos-tag gel
In order to check PARK2 phosphorylation in the presence and absence of Becn1 shRNA, Phos-tag SDS-PAGE was performed using Phos-tagTM Acrylamide from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), essentially as described by the manufacturer. Briefly, PC12 cells were transfected with scrambled shRNA or Becn1 shRNA and selected with G418 for 7 d. Post-selection, cells were treated with DMSO- or CCCP-treated and total cell lysates were prepared in RIPA buffer containing protease inhibitor cocktail supplemented with 1 mM Na3VO4. Total cell lysates were resolved by 10% SDS-PAGE using a gel containing 50 µM Phos-tag acrylamide and MnCl2 at 50 V. Before transfer, gels were incubated twice in SDS-PAGE transfer buffer containing 5 mM EDTA for 20 min. Later, gels were incubated in SDS-PAGE transfer buffer without EDTA. Transfers and immunoblotting were performed as described above.
Blue native gels and gel filtration
Samples for blue native gels were prepared as recommended for the NativePAGE™ Novex® Bis-Tris Gel System (Invitrogen). Samples were fractionated to obtain mitochondrial-enriched fractions and cytosolic fractions for either blue native PAGE as per manufacturer’s instructions or immunoprecipitation using mouse anti-BECN1. To check specificity of the antibodies, HEK293 cells were transfected with either scrambled or human BECN1 siRNA. Analysis of total cell lysates preparations by blue native gel electrophoresis was performed using the NativePAGE™ Novex® Bis-Tris Gel System (Invitrogen) as per the manufacturer’s instructions. The effect of BECN1 knockdown on native complexes was detected on immunoblots using Rb anti-PARK2 antibody.
For gel filtration, HEK293 total cell lysate was prepared in IP lysis buffer. Total cell lysates were resolved on a Superdex 200 10/300GL size exclusion column (GE Healthcare, UK) attached to an ÄKTA Purifier 100 (GE Healthcare, UK) at flow-rate of 0.35 ml/min in a mobile-phase containing 20 mM HEPES, 100 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, and 5% glycerol pH 7.4. Fractions of 0.2 ml were eluted and analyzed on dot blots for the presence of PARK2. Later, PARK2-containing fractions were resolved on SDS-page and immunoblotted with Ms anti-BECN1 and Rb anti-PARK2. Thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), and carbonic anhydrase (29 kDa) were used as markers.
In situ proximity ligation assay
The assay was performed as described earlier.Citation43 Briefly, HEK293 cells were fixed using 4% paraformaldehyde in medium containing 5% sucrose for 5 min at 37 °C and then permeabilized using 1% Triton X-100 in PBS for 5 min at room temperature. They were then blocked with 10% goat serum in PBS for 1 h at room temperature and incubated overnight with primary antibodies targeted against MYC tag or PARK2 and FLAG tag or BECN1 (mouse anti-MYC 1:1000; rabbit anti-PARK2 1:500, rabbit anti-FLAG 1:1000, mouse anti-BECN1 1:500) at 4 °C. On the following day, the cells were incubated with anti-rabbit PLUS and anti-mouse MINUS PLA probes in a humidity chamber for 1 h at 37 °C. The ligation step of the oligonucleotides was performed for 30 min at 37 °C followed by an amplification step using red fluorophore-labeled oligonucleotides (excitation maxima 598, emission maxima 634) for 100 min at 37 °C to enable the visualization of the interaction of the 2 proteins as red dots using a confocal microscope. For negative controls, the above-mentioned method was followed with the exception that only 1 primary antibody was used at a time.
PARK2 translocation studies
PC12 cells or MEFs transfected with YFP-PARK2 and plasmids of interest were treated with DMSO or 10 µM CCCP/FCCP for 3 h. In other experiments YFP-PARK2C431N was transfected together with mCherry-FKBP-PARK2C431N and FRP-FIS1 into PC12 cells, and they were treated with 100 nM rapamycin and 5 µM DMSO/CCCP for 5 h. Cells were visualized before and after treatment using an Olympus IX70 inverted microscope (Tokyo, Japan) equipped with a WLSM PlanApo 40×/0.90 (20× for MEFs) water immersion objective and Olympus DP70 CCD camera (Tokyo, Japan). For each experimental condition, at least 30 fields per dish from at least 3 dishes were randomly captured for further analysis. Control experiments were performed to ensure localization of YFP-PARK2 aggregates with mitochondrial marker CFP-Mito.
Mitochondrial fusion rate analysis
For mitochondrial fusion rate analysis, cortical neuronal cultures were transfected with mito-KikGR1 plasmid and plasmids of interest as described earlier.Citation30 A laser scanning confocal microscope (LSM 510 Duo, Carl Zeiss Microscopy GmbH, Göttingen, Germany) equipped with a LCI Plan-Neofluar 63×/1.3 water immersion DIC M27 objective (Carl Zeiss Microscopy GmbH, Göttingen, Germany) was used. The temperature was maintained at 37 °C using a climate chamber. For fusion acquisition, mito-KikGR1 was illuminated with a 488 nm argon laser line to visualize the intense green mitochondrial staining. Selected mitochondria were then photoconverted to red using a 405 nm diode laser and illuminated using a 561 nm DPSS laser. The images were taken at 10 s intervals for 10 min, the fate of all activated mitochondria was followed throughout the time-lapse, and the fusion and fission events were recorded. In experiments that compared different conditions, 4 fields per dish were imaged and at least 4 dishes per condition were used. Mitochondrial length measurements were performed as described earlier.Citation30
Mitophagy assay
Primary cortical neurons were cotransfected with mitochondrially targeted Keima plus scrambled shRNA, Becn1 shRNA and Atg12 shRNA alone or together with N-MYC PINK1. The excitation spectrum of Keima shifts from 440 to 586 nm when mitochondria are delivered to acidic lysosomes, which enables quantification of mitophagy.Citation44,Citation45 Images were acquired using a laser scanning confocal microscope using the laser lines 458nm (green, mitochondria at neutral pH) and 561nm (red, mitochondria under acidic pH), and red dots were counted manually and blindly.
Statistics
Data are presented as the mean ± SEM. The D'Agostino-Pearson omnibus test was used to test the normality of distribution. One-way ANOVAs followed by Newman-Keuls posthoc test, or Kruskal-Wallis tests followed by the Dunn test, were used to compare differences between experimental samples and control groups. Two-way ANOVAs were used to analyze mitochondrial fusion experiments. P values of less than 0.05 were considered statistically significant.
| Abbreviations: | ||
| ACTB | = | actin, beta |
| AMBRA1 | = | autophagy/Beclin 1 regulator 1 |
| ATG12 | = | autophagy-related 12 |
| BECN1 | = | Beclin 1, autophagy-related |
| CCCP | = | carbonyl cyanide m-chlorophenylhydrazone |
| DMSO | = | dimethyl sulfoxide |
| FCCP | = | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| FBS | = | fetal bovine serum |
| FIS1 | = | fission1 (mitochondrial outer membrane) homolog (S. cerevisiae) |
| GFP | = | green fluorescent protein |
| IKBKG | = | inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma |
| MEF | = | mouse embryonic fibroblast |
| MFN2 | = | mitofusin 2 |
| Mito-GFP | = | mitochondria-targeted green fluorescent protein |
| Mito-CFP | = | mitochondria-targeted cyan fluorescent protein |
| Ms | = | mouse |
| PARK2 | = | parkin RBR E3 ubiquitin protein ligase |
| PD | = | Parkinson disease |
| PIK3C3 | = | phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| PIK3R4 | = | phosphoinositide-3-kinase, regulatory subunit 4 |
| PINK1 | = | PTEN-induced putative kinase 1 |
| Rb | = | rabbit |
| SQSTM1 | = | sequestosome 1 |
| UBB | = | ubiquitin B |
| VDAC1 | = | voltage-dependent anion channel 1 |
| YFP | = | yellow fluorescent protein |
Additional material
Download Zip (3.8 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr S Hirose, Dr N Nakamura, Dr N Hattori, and Dr R Youle for providing the plasmids used in this study. We thank Ulla Peterson for her preparation of primary neurons. We thank Pratyush Kumar Das, University of Tartu, for his help in gel-filtration experiments. This work was supported by grants from the Estonian Research Council (IUT2-5 and PUT513), the European Community, and the European Regional Development Fund. VC was supported by MOBILITAS (grant number MJD120).
References
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571 - 80; http://dx.doi.org/10.1038/cdd.2010.191; PMID: 21311563
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360 - 72; http://dx.doi.org/10.1091/mbc.E08-01-0080; PMID: 18843052
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468 - 76; http://dx.doi.org/10.1038/ncb1854; PMID: 19270693
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10:776 - 87; http://dx.doi.org/10.1038/ncb1740; PMID: 18552835
- Takahashi Y, Meyerkord CL, Wang HG. Bif-1/endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ 2009; 16:947 - 55; http://dx.doi.org/10.1038/cdd.2009.19; PMID: 19265852
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385 - 96; http://dx.doi.org/10.1038/ncb1846; PMID: 19270696
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, Rubinsztein DC. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell 2012; 47:359 - 70; http://dx.doi.org/10.1016/j.molcel.2012.05.040; PMID: 22742832
- Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155 - 68; http://dx.doi.org/10.1083/jcb.201002100; PMID: 20921139
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 2010; 17:962 - 74; http://dx.doi.org/10.1038/cdd.2009.200; PMID: 20057503
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 2008; 106:464 - 74; http://dx.doi.org/10.1111/j.1471-4159.2008.05398.x; PMID: 18397367
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010; 191:933 - 42; http://dx.doi.org/10.1083/jcb.201008084; PMID: 21115803
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011; 20:867 - 79; http://dx.doi.org/10.1093/hmg/ddq526; PMID: 21138942
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189:211 - 21; http://dx.doi.org/10.1083/jcb.200910140; PMID: 20404107
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; http://dx.doi.org/10.1371/journal.pbio.1000298; PMID: 20126261
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378 - 83; http://dx.doi.org/10.1073/pnas.0911187107; PMID: 19966284
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795 - 803; http://dx.doi.org/10.1083/jcb.200809125; PMID: 19029340
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6:1090 - 106; http://dx.doi.org/10.4161/auto.6.8.13426; PMID: 20890124
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367 - 80; http://dx.doi.org/10.1083/jcb.201007013; PMID: 21173115
- Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 2011; 31:10249 - 61; http://dx.doi.org/10.1523/JNEUROSCI.1917-11.2011; PMID: 21753002
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol 2013; 200:163 - 72; http://dx.doi.org/10.1083/jcb.201210111; PMID: 23319602
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2012; 2:120080; http://dx.doi.org/10.1098/rsob.120080; PMID: 22724072
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320 - 33; http://dx.doi.org/10.1016/j.devcel.2011.12.014; PMID: 22280891
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2012; 2:1002; http://dx.doi.org/10.1038/srep01002; PMID: 23256036
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 2010; 5:e10054; http://dx.doi.org/10.1371/journal.pone.0010054; PMID: 20383334
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19:4861 - 70; http://dx.doi.org/10.1093/hmg/ddq419; PMID: 20871098
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A 2010; 107:5018 - 23; http://dx.doi.org/10.1073/pnas.0913485107; PMID: 20194754
- Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, Veksler V, Kaasik A. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci 2013; 126:2187 - 97; http://dx.doi.org/10.1242/jcs.118844; PMID: 23525002
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; http://dx.doi.org/10.1172/JCI20039; PMID: 14638851
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190 - 9; PMID: 18497889
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 2006; 281:14474 - 85; http://dx.doi.org/10.1074/jbc.M600364200; PMID: 16522639
- Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, Dufour N, Colomer Gould VF, Koeppen A, Déglon N, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011; 134:1400 - 15; http://dx.doi.org/10.1093/brain/awr047; PMID: 21478185
- Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis 2011; 43:68 - 78; http://dx.doi.org/10.1016/j.nbd.2010.11.003; PMID: 21081164
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 2009; 29:13578 - 88; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009; PMID: 19864570
- Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med 2013; 5:1247 - 62; http://dx.doi.org/10.1002/emmm.201302771; PMID: 23737459
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121 - 5; PMID: 17589504
- Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, Wang G. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J Biol Chem 2010; 285:38214 - 23; http://dx.doi.org/10.1074/jbc.M110.101469; PMID: 20889974
- Shiba K, Arai T, Sato S, Kubo S, Ohba Y, Mizuno Y, Hattori N. Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun 2009; 383:331 - 5; http://dx.doi.org/10.1016/j.bbrc.2009.04.006; PMID: 19358826
- Honda S, Hirose S. Stage-specific enhanced expression of mitochondrial fusion and fission factors during spermatogenesis in rat testis. Biochem Biophys Res Commun 2003; 311:424 - 32; http://dx.doi.org/10.1016/j.bbrc.2003.10.008; PMID: 14592431
- Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol 2005; Chapter 28:Unit 28 1.
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3:995 - 1000; http://dx.doi.org/10.1038/nmeth947; PMID: 17072308
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 2011; 18:1042 - 52; http://dx.doi.org/10.1016/j.chembiol.2011.05.013; PMID: 21867919
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490