
Abstract
Autophagy is essential for cellular homeostasis and its dysfunction in human diseases has been implicated in the accumulation of misfolded protein and in cellular toxicity. We have recently shown impairment in autophagic flux in the lipid storage disorder, Niemann-Pick type C1 (NPC1) disease associated with abnormal cholesterol sequestration, where maturation of autophagosomes is impaired due to defective amphisome formation caused by failure in SNARE machinery. Abrogation of autophagy also causes cholesterol accumulation, suggesting that defective autophagic flux in NPC1 disease may act as a primary causative factor not only by imparting its deleterious effects, but also by increasing cholesterol load. However, cholesterol depletion treatment with HP-β-cyclodextrin impedes autophagy, whereas pharmacologically stimulating autophagy restores its function independent of amphisome formation. Of potential therapeutic relevance is that a low dose of HP-β-cyclodextrin that does not perturb autophagy, coupled with an autophagy inducer, may rescue both the cholesterol and autophagy defects in NPC1 disease.
The homeostatic role of the proteolytic systems is of fundamental importance for cellular functioning. Autophagy, an intracellular degradation pathway for aggregation-prone proteins and damaged organelles, is implicated in diverse human physiological and disease conditions. This process is dynamic (defined as autophagic flux), encompassing the generation of autophagosomes and their fusion with late endosomes to form amphisomes, which subsequently fuse with lysosomes forming autolysosomes where the autophagic cargo is degraded. Compromised autophagy contributes to the pathology of several neurodegenerative and certain liver diseases, whereas in some conditions upregulation of autophagy is considered beneficial. One of the mediators enabling proper functioning of this process is lipids, which are essential membrane components governing vesicular trafficking. We recently reported dysfunctional autophagy during altered lipid homeostasis in Niemann-Pick type C1 disease, which is an autosomal recessive lipid/lysosomal storage disorder associated with neurodegeneration and liver dysfunction. NPC1 disease is caused primarily due to mutations in the NPC1 gene, which encodes a transmembrane protein that resides on the late endosomal/lysosomal (LE/L) compartments mediating cholesterol efflux. Disease-causing mutations impair this function of the NPC1 protein, leading to abnormal sequestration of LE/L-resident cholesterol that has been implicated in the disease pathology.
Impairment in autophagic flux in NPC1 disease is characterized by accumulation of autophagosomes and autophagy substrates (), which we observed in patient fibroblasts, and in cells and disease-affected organs of Npc1 mutant mice. Although previous studies have largely attributed the increase in autophagosomes to an induction of autophagy, autophagosome synthesis as analyzed by the bafilomycin A1 assay or by autophagosome maturation analysis with the tandem fluorescent-tagged LC3 or SQSTM1/p62 reporters indicate a block late in the pathway. We found that maturation of autophagosomes is impaired in NPC1 mutant cells, attributable to defective amphisome formation caused by a failure in the SNARE machinery that mediates fusion between autophagosomes and late endosomes. This possibly arises due to inability of late endosomes to recruit SNARE proteins, such as VAMP8 and VAMP3, leading to reduced interaction with autophagosomal STX17. Impaired SNARE functioning and autophagic flux also occur in other lysosomal storage disorders, such as multiple sulphatase deficiency and mucopolysaccharidosis type IIIA, where abnormal cholesterol sequestration in LE/L compartments is seen. Nonetheless, lysosomal proteolytic function and endocytic cargo degradation have no overt perturbations in NPC1 mutant cells, and, in contrast to a recent report, cathepsin activity is not reduced.
Figure 1. Disease mechanisms and treatment strategy for NPC1 disease. (A) Mutations in LE/L-resident NPC1 protein inhibits cholesterol efflux, leading to cholesterol accumulation. Mutant NPC1 protein also impedes autophagosome maturation, thereby causing impairment in autophagic flux associated with accumulation of autophagosomes and autophagic cargo. This is attributable to defective amphisome formation resulting from failure in the SNARE machinery due to a reduced propensity of the late endosomes for recruiting SNARE proteins. Therapeutic avenues are shown (with dashed lines) for depleting cholesterol and enhancing autophagy to rescue both of the defects. Stimulating autophagy restores its function by facilitating autophagosome maturation independent of amphisome formation, thereby mediating the clearance of autophagic cargo. (B) Mutant NPC1 protein imparts cellular toxicity through abnormal cholesterol accumulation and impairment in autophagic flux. Defective autophagy also increases intracellular cholesterol load, thereby creating a deleterious feedback in augmenting the disease pathogenesis. Combination treatment strategy can be achieved with a cholesterol-depleting agent and an autophagy enhancer to rescue both the disease phenotypes.
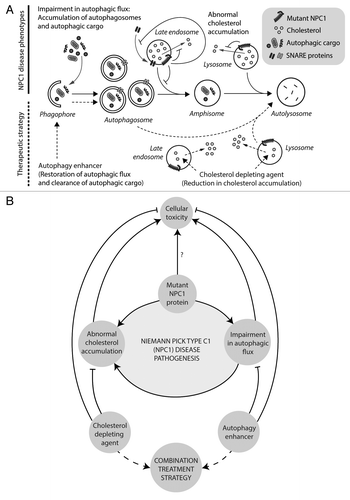
Cholesterol accumulation is a causative factor in NPC1 disease, and, therefore, cholesterol depletion strategy with HP-β-cyclodextrin is considered a potential treatment that is currently evaluated in clinical trials. However, we found that HP-β-cyclodextrin exhibits an unanticipated side effect in blocking autophagic flux in a dose-dependent manner, which is mechanistically distinct from that caused by the mutant NPC1 protein. This is likely due to alterations in membrane lipid composition caused by HP-β-cyclodextrin–mediated substantial cholesterol efflux that retards vesicle fusion events, as reported previously. Consistently, high doses of HP-β-cyclodextrin are neurotoxic, whereas lower doses that do not perturb autophagy offer cytoprotection. Thus, careful dosing of this drug is necessary for therapeutic application.
Expression of the functional NPC1 protein in Npc1 mutant cells, however, can rescue both the cholesterol and autophagy defects. It also increases autophagosome synthesis, which is possibly due to a feedback signal for its effect on positively regulating autophagic flux and autophagosome clearance/maturation. Since cholesterol depletion treatment could not rescue the autophagy defects in Npc1 mutant cells, it is likely that the NPC1 protein mediates autophagic traffic under basal conditions independently of its effect on cholesterol handling. Our data suggest that the NPC1 protein regulates the formation of amphisomes, which acts as a sink for cargo trafficked through both the autophagic and endocytic pathways, and its loss of function predominantly retards autophagic substrate clearance.
Conversely, since autophagy regulates lipid metabolism, inhibition of autophagy causes accumulation of cholesterol that resembles a mutant NPC1-like cellular phenotype. Moreover, chemical or genetic impairment of autophagy exacerbates the neurodegenerative phenotype in transgenic disease models. Notably, brain- and liver-specific abrogation of basal autophagy in normal mice is associated with degeneration in the affected organs, which may imply similar pathogenic consequences of defective autophagy in NPC1 patients. Thus, impairment of autophagic flux in NPC1 disease will not only impart its deleterious effects as a primary causative factor, but will also positively act in building up of intracellular cholesterol, thereby creating a vicious cycle contributing to the disease pathogenesis ().
Can we rescue a block in autophagic flux by enhancing autophagy? Although stimulation of autophagy is well documented to be beneficial in experimental models of several neurodegenerative diseases, such an approach is not clear for lipid/lysosomal storage disorders. Our data suggest that stimulating autophagy overcomes its block in Npc1 mutant cells and partly restores the function of the pathway by facilitating autophagosome maturation and cargo degradation possibly through direct autophagosome–lysosome fusion independent of amphisome formation (). Our findings argue against inhibition of autophagy as a therapeutic strategy in NPC1 disease as has been suggested recently. Such an approach may interfere with the critical housekeeping functions of autophagy in maintaining energy and cellular homeostasis. However, upregulating autophagy does not rescue the cholesterol phenotype in Npc1 mutant cells, possibly because cholesterol is trapped in late endosomal compartments that remain unaffected. We have shown that simultaneous application of a cholesterol depleting agent (0.2% HP-β-cyclodextrin that partially depletes cholesterol without affecting autophagic flux) with an autophagy inducer can rescue both the cholesterol and autophagy defects, thereby providing a rational combination treatment for NPC1 disease (). Our study points to treating conditions arising due to stalled basal autophagy through small molecules upregulating this process, which warrants further investigation in other lipid/lysosomal storage disorders.
Disclosure of Potential Conflicts of Interest
RJ is an advisor to Stemgent and Fate Therapeutics.
Acknowledgments
We thank NNPD Foundation (DM) and BBSRC New Investigator Award (VIK) for funding and Tom DiCesare (BARC, Whitehead Institute) for help with illustrations. This work is supported by US NIH grants R37-CA084198 and R01-CA087869 (RJ). SS and VIK are also former fellows of Hughes Hall, University of Cambridge, UK.