
Abstract
Although autophagic pathways are essential to developmental processes, many questions still remain regarding the initiation signals that regulate autophagy in the context of differentiation. To address these questions we studied the ocular lens, as the programmed elimination of nuclei and organelles occurs in a precisely regulated spatiotemporal manner to form the organelle-free zone (OFZ), a characteristic essential for vision acuity. Here, we report our discovery that inactivation of MAPK/JNK induces autophagy for formation of the OFZ through its regulation of MTORC1, where MAPK/JNK signaling is required for both MTOR activation and RPTOR/RAPTOR phosphorylation. Autophagy pathway proteins including ULK1, BECN1/Beclin 1, and MAP1LC3B2/LC3B-II were upregulated in the presence of inhibitors to either MAPK/JNK or MTOR, inducing autophagic loss of organelles to form the OFZ. These results reveal that MAPK/JNK is a positive regulator of MTORC1 signaling and its developmentally regulated inactivation provides an inducing signal for the coordinated autophagic removal of nuclei and organelles required for lens function.
Introduction
Autophagy, initially known as a form of cell deathCitation1,Citation2 is now understood to be involved in regulating cell survival, cellular homeostasis, and physiological function.Citation3,Citation4 The autophagy process also plays an essential role in tissue development,Citation5,Citation6 and cell differentiation including that of reticulocytes,Citation7,Citation8 B cells,Citation9 macrophages,Citation10 adipocytes,Citation11 and osteoclasts.Citation12 Most of our knowledge of how autophagy functions in regulating cell differentiation is based on tissue specific knockouts of autophagy-related genes.Citation7,Citation13,Citation14 Little is known about the signaling mechanisms that initiate autophagy-dependent differentiation pathways.
Autophagy is a catabolic process that begins as the phagophore, which forms at the phagophore assembly site, extends around cytoplasmic cargo such as proteins and organelles and then fuses to form the autophagosome, a double-membraned vesicle that completely surrounds its cargo. Molecules such as Atg1/ULK1, class III phosphatidylinositol 3-kinase (PtdIns3K), Vps34/PIK3C3 (phosphatidylinositol 3-kinase, catalytic subunit type 3) and Vps30/BECN1 can induce autophagosome formation and a subset of these molecules, along with others like MAP1LC3B (microtubule-associated protein 1 light chain 3 β)/LC3B, are important to both the elongation and maturation of the autophagosome.Citation5,Citation15,Citation16 Autophagosomes fuse with lysosomes that degrade their cargo and return amino acids and recycled cellular components to the cell.Citation4-Citation6 Autophagy, a highly regulated process, occurs at basal levels in all cells to mediate protein and organelle turnover to meet the cells’ energy demands. It is enhanced under conditions of stress like starvation, hypoxia, and various pathological conditions. During development, autophagy also is associated with the structural remodeling of cells.Citation4,Citation16
Studies are now beginning to reveal the existence of many types and subsets of autophagic pathways.Citation17,Citation18 In addition, the great number of molecules associated with phagophore formation, and the ability to knock out individual autophagy-associated molecules without affecting all autophagic pathways,Citation19 suggests that there is redundancy, overlap, and/or context specificity to their function. It is becoming increasingly evident that autophagy is a complex mechanism whose individual molecular players can define a particular autophagy process. Importantly, reports now show that autophagy can progress in the absence of some molecules previously believed to be required for the autophagic mechanism to proceed.Citation14,Citation20-Citation23,Citation24 This evidence clearly indicates that autophagy can be context- and tissue-specific and emphasizes the importance of defining the subset of molecules necessary to execute a particular autophagic process.
One of the principal classic regulators of autophagy is MTOR (mechanistic target of rapamycin [serine/threonine kinase]), named because the pharmacological agent rapamycin induces autophagy by blocking MTOR signaling. MTOR is a master regulator of cell growth, and is implicated in development and aging as well as in various disease states.Citation25 In the presence of growth factors and when there are ample nutrients available to the cell, MTOR signaling prevents activation of the autophagy pathway.Citation4,Citation26 This function involves MTOR’s role in the formation and activation of the highly conserved, multiprotein, MTORC1 complex, where MTOR is associated with MLST8/GβL (MTOR-associated protein, LST8 homolog [S. cerevisiae]), AKT1S1 (AKT1 substrate 1 [proline-rich]), DEPTOR (DEP domain containing MTOR-interacting protein), and RPTOR (regulatory associated protein of MTOR, complex 1). RPTOR, a scaffolding protein, has a pivotal role in the formation of the MTORC1 complex.Citation27,Citation28 A well-known target of MTOR under conditions of osmotic stress RPTORCitation29 is also reported to be targeted by MAPK8/JNK1 (c-Jun N-terminal Kinase)Citation30 and both MTOR and JNK1/2/3 (MAPK8/9/10) are implicated as factors in the regulation of autophagy.Citation25,Citation31-Citation34 The binding of RPTOR to MTOR is necessary for effective MTOR-catalyzed phosphorylation of its downstream targets in vivo,Citation35 including RPTOR itself. RPTOR’s phosphorylation is a key factor in the regulation of MTORC1 signaling.Citation29 Both MTOR and MAPK8/JNK1 phosphorylate RPTOR at Ser863, a master biochemical switch that modulates the hierarchical phosphorylation of RPTOR at Ser859 and Ser855. Ser863 phosphorylation is necessary for the MTORC1 complex to block autophagy.Citation30,Citation36,Citation37
Interestingly, while the role of MTOR in the MTORC1 complex is exclusively to inhibit autophagy, MAPK/JNK functions as both a positive and a negative regulator of autophagy, its specific action being context- and tissue-specific.Citation31,Citation38 MAPK8/JNK1 can promote autophagy through its phosphorylation of BCL2, which releases BECN1 from its association with BCL2 to function in autophagosome formation.Citation39 The majority of studies where MAPK8/9/JNK1/2 is found as a positive regulator of autophagy involve its activation as a stress signal, such as with compounds that activate reactive oxygen speciesCitation40 or following nutrient starvation.Citation41 In those situations, blocking stress-induced activation of MAPK8/9/JNK1/2 prevents induction of autophagy. On the other hand, in studies of neurons from triple MAPK/JNK knockout mice, in which endogenous MAPK8/JNK1, MAPK9/JNK2, and MAPK10/JNK3 are eliminated, MAPK8/9/10 has been reported to function as a negative regulator of autophagy.Citation31 We have examined the role of an endogenous MAPK/JNK signal in regulating autophagy for its role in development. The inhibitors used in this study block the activation of all 3 isoforms of JNK (MAPK8/JNK1, MAPK9/JNK2, and MAKP10/JNK3). Therefore, to avoid the suggestion that our results pertain to a specific isoform, and to prevent confusion with other MAPKs, we used the name MAPK/JNK throughout this study. Though both MAPK/JNK and MTOR are molecular players in autophagy regulation, prior to the studies presented here no molecular link had been established between MAPK/JNK inactivation, MTORC1 signaling, and autophagy. We have now discovered that in the context of development the suppression of MAPK/JNK activity blocks MTOR-RPTOR signaling to induce autophagy.
Our studies uncover the role of MAPK/JNK-MTOR-RPTOR regulation of autophagy using the developing ocular lens as our model, where an almost complete spectrum of cell differentiation is present at a single time point in development (modeled in Fig. S1).Citation42 The anterior aspect of the embryonic lens is lined with a monolayer of undifferentiated epithelial cells. In the anterior-most region of the equatorial epithelium is located the germinative zone, the region of lens cell proliferation. As these epithelial cells leave the germinative zone and withdraw from the cell cycle they initiate the lens differentiation program which leads to the formation of lens fiber cells. Important to this study, terminal differentiation/maturation of lens fiber cells involves the loss of cytoplasmic organelles (nucleus, Golgi apparatus, endoplasmic reticulum, mitochondria) from the central zone of the lens to form a region crucial to lens transparency called the organelle-free zone. Aberrations in the formation of the OFZ including the inability to degrade DNA during lens fiber cell maturation can lead to congenital cataracts and visual impairment.Citation43,Citation44 Although the phenomenon of OFZ formation is well documented,Citation45,Citation46 the inducing signal and the mechanism behind the loss of organelles remain unknown. Here, using both a lens organ culture system and primary lens cell cultures that mimic lens differentiation as it occurs in vivo,Citation47 we show that autophagy plays a central role in the loss of organelles to form the OFZ during lens development and reveal a novel pathway in which MAPK/JNK inactivation initiates the process by suppressing MTOR and RPTOR activation. These new findings provide insight into the signaling mechanisms behind loss of nuclei and organelles during the development of the lens, with implications for other developmental mechanisms in which autophagic processes are necessary for tissue maturation.
Results
Temporal loss of endoplasmic reticulum and Golgi in the developing chick embryo lens
During development of the ocular lens, the central lens fiber cells lose their nuclei and organelles, including endoplasmic reticulum (ER) and Golgi, in a tightly programmed manner to form the organelle-free zone. In the chick embryo this loss of organelles begins as a wave that progresses from the center of the lens to the periphery, and by embryonic d (E)18 the loss of organelles and nuclei is complete.Citation48,Citation49 We investigated the timing of organelle loss in our developing lens model by examining lenses at multiple stages of chick embryo development, the results shown here at E13 and E15. Cryosections prepared from the embryonic lenses were immunolabeled for the Golgi marker, FTCD (formiminotransferase cyclodeaminase),Citation50 or the ER marker, HSPA5/Bip/GRP78.Citation51 Immunoblot analysis for FTCD and HSPA5 was performed on cortical fiber (FP) and central fiber (FC) regions of the embryonic lenses obtained by microdissection (regions modeled in Fig. S1). Our studies showed that both ER () and Golgi () were still widely present throughout the embryonic lens at E13, including in the central lens fiber zone where elongated cisternae stretched across the cells. However, by E15 both ER (HSPA5; ) and Golgi (FTCD; ) were greatly diminished in the central fiber cell zone. Similar results were obtained for another ER marker, CALR3 (calreticulin 3; Fig. S1). In contrast, the lens-specific water channel protein AQP0/aquaporin-0 remained highly expressed at the membranes of differentiated lens fiber cells in the central lens fiber zone (see Fig. S2). The ER and Golgi that remained in the central lens fiber cells at E15 were fragmented, and vesicular in appearance (, i and D, i), leading us to investigate whether an autophagic process is involved in their removal from the center of the visual field.
Figure 1. Temporal loss of organelles and nuclei in the embryonic chick lens. (A–D) Immunoblot and immunostaining analysis for presence of the ER marker HSPA5/Bip and the Golgi marker FTCD in chick embryo lenses at E13 (A and B) and E15 (C and D). (A–D) Immunostaining of mid-sagittal lens cryosections for ER (HSPA5; A and C, red) and Golgi (FTCD; B and D, red), colabeled with the nuclear dye TO-PRO-3 (blue). Images were obtained by confocal microscopy. Low magnification images in the left panels represent projections of acquired z-stacks; scale bar, 500 μm. Boxed areas are shown at higher magnification to the right, each a 1-μm optical slice selected from an acquired z-stack; scale bar, 20 μm. Nuclei are from the same field as the ER or Golgi marker at their left. Organelle markers are designated as (i), nuclear marker as (ii). Immunoblots with accompanying densitometric analyses for HSPA5 (A and C) and FTCD (B and D) was performed on microdissected fractions of the cortical lens fiber cell zone (FP) and the central lens fiber cell zone (FC), quantification is presented as a ratio to GAPDH. Both immunostaining and immunoblotting results show that significant loss of ER and Golgi had occurred between E13 and E15. The remaining organelles in the FC zone at E15 had a vesicular presentation suggesting that they are being degraded. With the same temporal sequence as organelle loss nuclei become highly pyknotic and began to disappear. All of these processes occur in the central region of the lens where lens fiber cells mature and the organelle-free zone is established. These data are representative of at least 3 independent studies. Error bars represent SE. See Figure S1 for immunostaining of the ER protein CALR3 at E15.
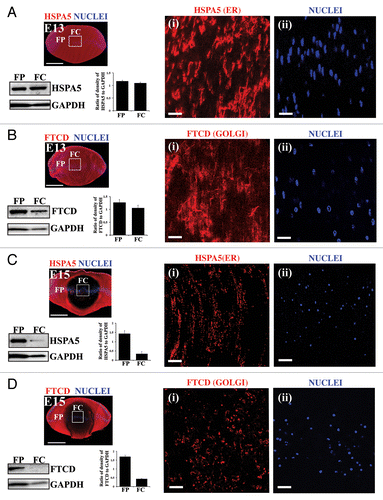
We also examined the developmental timing of nuclear loss in these embryonic lenses. At E13 the nuclei of differentiating fiber cells in the central region of the lens still had a normal oval, elongated morphology (, ii and B, ii). Coincident with the loss of ER and Golgi at E15, the nuclei in the center of the lens became pyknotic (condensed) (, ii and D, ii). This finding was consistent with previous observations that reported chick embryo lens nuclei become pyknotic before they disappear from the central lens fiber cells at E18.Citation49 Our results demonstrate that the formation of the OFZ occurs around E15 of development, when ER and Golgi have become fragmented and vesicular, and nuclei pyknotic, while at E13 much of the organelle composition remains intact. Therefore, the signals that initiate formation of the OFZ must be enhanced after E13 of development in this system.
Autophagy regulators LC3B and BECN1 are expressed in the E15 lens and associated with autophagic vesicles containing lens organelles
There are many markers of the autophagic process, each having specific functions during autophagy. Recent studies show that the classic autophagy markers LC3B and BECN1 are expressed in lens fiber cells of the postnatal mouse,Citation52 suggesting the involvement of autophagic processes in lens development. BECN1 is required for the formation of the phagophore assembly site, the initiation step in autophagy,Citation53-Citation56 while LC3B promotes expansion and fusion of phagophore membranes and directs cargo to this sequestering compartment.Citation57-Citation59 It is the cleaved lipid modified form of LC3B, LC3B-II, which associates with the phagophore and autophagosomal membrane.Citation60 Using immunostaining analysis we examined the expression and localization of these autophagic markers in the E15 lens. Their localization to vesicles that also labeled for an ER marker was used as a measure of whether an autophagic process was responsible for the temporal removal of organelles during lens development that results in formation of the OFZ. At E15, when significant loss of organelles has occurred in the central region of the lens, LC3B () and BECN1 () were expressed in both lens epithelial cells and differentiating lens fiber cells. Across the fiber cell zones their staining intensity was differentiation-state specific. Both molecules were prominently expressed in cortical lens fiber cells (, i and B, i), and diminished in the central region of the lens where they had a distinctly vesicular staining pattern (, ii and B, ii) that is typically associated with autophagy. Co-immunostaining for the ER marker CALR3 with either LC3B (, i and ii) or BECN1 (, iii) revealed that organelles (or fragments thereof) were present within autophagic vesicles, particularly in the FC zone. To determine whether there was an increase in association of ER with LC3B-positive vesicles in the FC region of the developing lens during the time of OFZ formation at E15, we compared the percent CALR3-positive vesicular-like structures labeled with LC3B in the FP zone to the FC zone. While 28% of the CALR3-containing vesicles were LC3B-positive in the FP zone, this increased to 52% in the FC zone (Fig. S3). Co-immunostaining for the mitochondrial marker TOMM20 (translocase of outer mitochondrial membrane 20 homolog [yeast]) and LC3B, showed that mitochondria also were localized to LC3B-positive vesicular structures in the central lens fiber cells of E15 lenses, further evidence of the involvement of autophagy in the removal of organelles during lens fiber cell maturation (Fig. S4).
Figure 2. Active autophagy is involved in the removal of nuclei and organelles from the lens fiber cells in the E15 chick lens. (A and B) E15 lens cryosections were immunostained for the autophagy markers LC3B (A, red) and BECN1/Beclin 1 (B, red) and colabeled with the nuclear dye TO-PRO-3 (blue). Images were acquired by confocal microscopy. Images shown in the left panel of (A and B) are low magnification images represented as projections of acquired z-stacks; scale bar, 200 μm. Boxed areas are shown at higher magnification to the right, each a 1-μm optical slice selected from an acquired z-stack; scale bar, 20 μm. The cortical lens fiber cell region (FP) is designated as (i) and the central lens fiber cell region (FC) as (ii), diagrammed in the model in (C). The results show that the intensity and staining pattern of autophagy markers LC3B and BECN1 in the E15 lens is differentiation-state specific, with a vesicular appearance in the FC zone. (C) Cryosections of E15 lenses were co-immunostained either for (i and ii) LC3B (red), CALR3 (ER marker, green), and TO-PRO-3 (nuclear dye, blue) or for (iii) BECN1 (red) and CALR3 (ER marker, green) and imaged in the (i) FP and (ii) FC zones. Boxed (red), regions on the model designate areas from where images were acquired. Images are each a 1-μm optical slice selected from an acquired z-stack; scale bar, 20 μm. Confocal images are representative of 3 independent studies. In the FC zone, both LC3B and BECN1, which localize to autophagic vesicles, showed significant overlap with the ER marker CALR3, suggesting the removal of organelles during lens development occurs through an autophagic process. See also Figures S2–S4. (D) Immunoblot studies showing expression levels of SQSTM1/p62 in differentiation-specific fractions isolated by microdissection from E15 lenses. EC, undifferentiated lens epithelial cells; EQ, equatorial epithelium, the zone of differentiation initiation; FP, cortical fiber cells, a region of lens morphogenesis; and FC, central fiber cells, the region of fiber cell maturation. Quantification of SQSTM1 expression relative to GAPDH is shown in the panel on the right. Data represent 3 independent studies. This result supported the conclusion that autophagy is occurring in lens fiber cells as the loss of SQSTM1 is correlated with active autophagy. Error bars represent S.E., **P ≤ 0.01, t test. n.s., nonsignificant. See Figure S5 for immunolocalization of SQSTM1 at E15. (E, i and ii) Electron micrographs showing double-membrane-bound autophagosomes surrounding a degrading organelle that is likely a mitochondria and cytoplasmic debris in the cortical fiber cell zone close to the region of organelle loss. Boxed insets in (i and ii) are shown at higher magnification to the right. Data show structural evidence of autophagy in the region in which organelles are lost during lens development; scale bar, 500 nm. Results are representative of 4 independent studies.
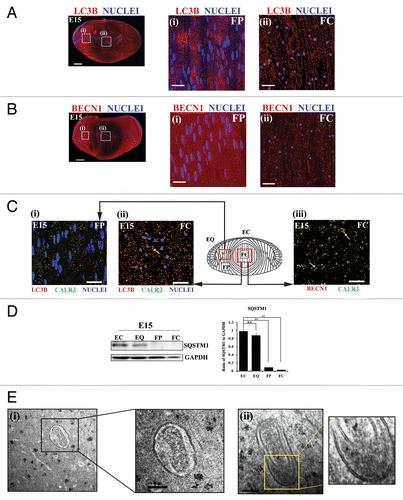
SQSTM1/p62 is a selective receptor that directly links cargo to the phagophore through LC3A/B, following which SQSTM1 is degraded. Factors that lead to a block in autophagy result in an accumulation of SQSTM1.Citation61,Citation62 Therefore, a general relationship has been established between an active autophagic process and the loss of SQSTM1.Citation63,Citation64 We examined the expression levels of SQSTM1 in E15 lenses following their microdissection into 4 differentiation state-specific regions (modeled in Fig.S1): EC, undifferentiated lens epithelial cells; EQ, equatorial epithelial cells in the zone of differentiation initiation; FP, the region of lens fiber cell morphogenesis; and FC, the zone of fiber cell maturation and organelle loss. SQSTM1 expression was high in the undifferentiated lens epithelial cells and progressively decreased in expression as the lens fiber cells differentiated, with little to no expression of SQSTM1 detected in the central lens fiber cells (). Similar results were observed when E15 lens sections were immunostained for SQSTM1 (see Fig. S5). This finding provides further support that autophagy is a significant element of the process of organelle removal from lens fiber cells during formation of the OFZ.
To validate the presence of an active autophagic process during the time period of removal of organelles from the central region of the lens, we performed electron microscopy analysis at E14 along the border of OFZ formation. This analysis revealed that double-membraned autophagosomal vesicles surrounding organelles (, i and ii), or fragments of organelles, were present in these cells. Our findings show a role for autophagy in the removal of organelles during lens development.
Inactivation of MAPK/JNK signaling induced a pathway leading to premature loss of ER and nuclei in the central lens fiber cells by autophagy
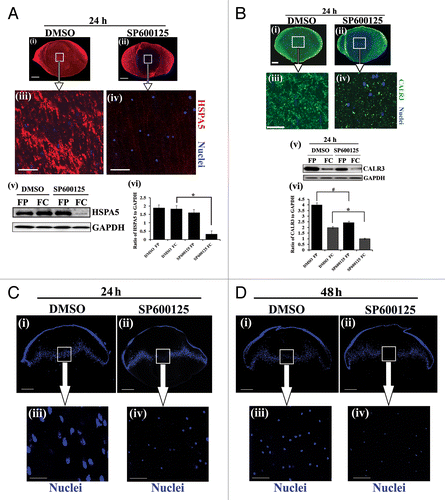
We began our studies of the signaling pathways involved in inducing the autophagic pathway that removes organelles during lens development by investigating the potential role of the signaling protein MAPK/JNK in this process. This avenue of investigation was suggested by our observation that there was a dramatic inhibition of MAPK/JNK signaling in the central region of the lens (FC) coincident with the formation of the OFZ at E15 (). For these studies the activation state of MAPK/JNK was determined by both immunolocalization and western blot analysis for phosphorylation of JUN (p-JUN/p-c-JunSer63/73), the direct downstream target of MAPK/JNK.Citation65 To investigate whether there was a link between the inactivation of MAPK/JNK signaling and the induction of organelle loss in the developing lens, MAPK/JNK activity was blocked in whole lens organ cultures using 2 distinct MAPK/JNK-specific inhibitors, SP600125Citation66 and JNK-IN-8.Citation67 E13 lenses were used for this study because it is a time point in development before there is significant loss of organelles. The lenses were exposed to either SP600125 or JNK-IN-8 for 24 h, which effectively suppressed MAPK/JNK activation in both cortical and central lens fiber cells, as shown by immunoblot analysis for p-JUN (Ser63/73) in differentiation-specific fractions of the lens isolated by microdissection (Figs. S6 and S7A). Both immunostaining and western blot analysis for the ER marker HSPA5 showed that inhibition of MAPK/JNK signaling induced premature loss of ER from cells in the central fiber zone (; Fig. S7C and S7D). Similar studies were performed with the ER marker CALR3. The results showed loss of expression of CALR3 in both cortical and central lens fiber cells following MAPK/JNK inhibition with SP600125 ().
Figure 3. Temporal inactivation of MAPK/JNK in chick embryonic lenses. (A and B) Immunostaining (i and ii) and immunoblotting analysis (iii and iv) of lens cryosections for phospho-(p)-JUN Ser63/73, the direct downstream target of MAPK/JNK, at (A) E13 and (B) E15 are shown. Boxed region in (i) is represented at high magnification in (ii). (i and ii) Images were acquired by confocal microscopy. Analysis of p-JUN expression in E13 (A) and E15 (B) lenses by immunoblotting is shown in (iii), to its right, densitometric analysis of p-JUN expression ratio adjusted to the loading control GAPDH is shown (iv). Error bars represent SE. Results indicate presence of active MAPK/JNK signaling in the central lens fiber cells at E13 and inhibition of the MAPK/JNK signal in this region of the lens at E15, coincident with loss of nuclei and organelles to form the OFZ, suggesting that inactivation of MAPK/JNK may be involved in this process. Images in (i) are projection images of acquired z-stacks, (ii) is a 1-μm thick optical slice. Scale bar, 200 μm (i) and 20 μm (ii). Results are representative of 3 independent studies.
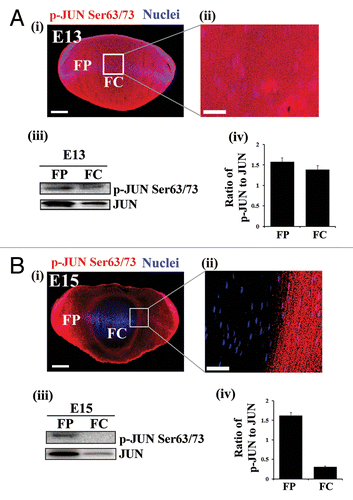
Figure 4. MAPK/JNK inactivation induces premature loss of ER and nuclei from the central lens fiber cells. (A and B, i–iv) Mid-sagittal lens cryosections from whole lens organ cultures, exposed to the vehicle, DMSO, and the MAPK/JNK inhibitor, SP600125, for 24 h, were immunostained for the ER markers HSPA5/Bip (A, i–iv, red), or CALR3 (B, i–iv, green), and colabeled with the nuclear dye TO-PRO-3 (A and B, i–iv, blue). Low magnification confocal images (i and ii) in (A and B) represent projection images of optical slice of 8-μm thickness; scale bar, 200 μm. Boxed areas in (A and B, i and ii) are shown at higher magnification below (iii and iv). Optical slices of 1-μm thickness were selected from an acquired z-stack; scale bar, 20 μm. Data show premature loss of ER in central lens fiber cells is induced when MAPK/JNK activation is suppressed. (A and B, v and vi) Immunoblot studies show that expression of the ER markers (A, v) HSPA5 and (B, v) CALR3 were significantly decreased in the central lens fiber cells (FC) after a 24 h exposure to the MAPK/JNK inhibitor SP00125. Quantification of HSPA5 and CALR3 expression is shown in (A, vi) and (B, vi), respectively. See Figure S7 for immunoblot and immunolocalization results for HSPA5 expression following exposure to the MAPK/JNK inhibitor JNK-IN-8. (C and D) Mid-sagittal lens cryosections obtained from whole lenses that were exposed to the vehicle DMSO, or MAPK/JNK inhibitor, SP600125, for 24 h (C) and 48 h (D) were fixed and labeled with TO-PRO-3 to detect nuclei. Low magnification confocal images (i and ii) in (C and D) represent projections of acquired z-stacks; scale bar, 200 μm. Boxed areas in (i and ii) are shown at higher magnification below (iii and iv). These images were 1-μm optical slices selected from an acquired z-stack; scale bar, 20 μm. Results show pyknosis of nuclei is induced prematurely when MAPK/JNK activation was suppressed for 24 h, followed by nuclear loss after 48 h. See also Figure S8. Results are representative of at least 3 or more independent studies. Error bars represent S.E., */#P ≤ 0.05, t test.
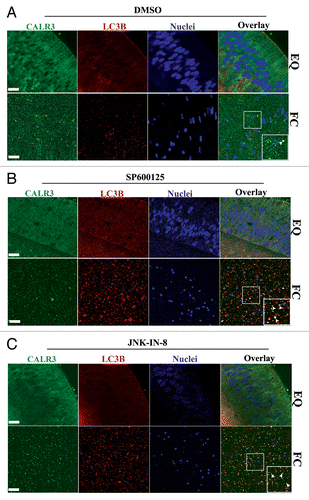
To examine if inhibition of MAPK/JNK induced autophagic loss of organelles from the central lens fiber cells, we performed co-immunostaining analysis for the organelle marker CALR3 with the autophagy proteins LC3B and BECN1. Our results showed increased presence of CALR3 in both LC3B-positive () and BECN1-positive (Fig. S9) vesicles in the FC cells following inactivation of MAPK/JNK with either SP600125 or JNK-IN-8. This result demonstrates that removal of ER following inactivation of MAPK/JNK occurs by an autophagic process. The removal of organelles induced by blocking MAPK/JNK activity was specific to differentiating lens fiber cells as there was no such effect on the organelles of lens cells in the equatorial zone (; Fig. S9). These results strongly support the conclusion that the autophagy process signaled by MAPK/JNK inactivation is regulated in a spatial-temporal manner to form the OFZ.
Figure 5. Suppressing MAPK/JNK activation induced removal of lens organelles from the central lens fiber cell zone by autophagy. Immunofluorescence analysis of mid-sagittal lens cryosections co-labeled for CALR3 (green), LC3B (red), and nuclei (TO-PRO-3, blue) prepared from lenses grown in organ culture for 24 h in the presence of (A) the vehicle DMSO, (B) the MAPK/JNK inhibitors SP600125, or (C) the MAPK/JNK inhibitor JNK-IN-8. (A–C, upper panels) equatorial zone, EQ; (A–C, lower panels) central fiber zone, FC. Overlay of CALR3 (green) and LC3B (red) shown in panels on the right. Boxed areas in the FC overlay images are shown at higher magnification in the bottom right corner. Examples of the presence of ER (green) in LC3B-positive autophagic vesicles (red) are indicated by white arrowheads. Represented optical slices were taken from an acquired z-stack. Each optical slice is 0.2-μm thick. Data shows that inhibiting MAPK/JNK activation induced removal of ER in autophagic vesicles in central lens fiber cells. Little colocalization of ER in LC3B-labeled autophagic vesicles was observed in the EQ zone; scale bar, 20 μm. Results are representative of 3 independent studies. See also Figure S9 for similar studies with BECN1.
The formation of the OFZ also involves the loss of nuclei, with lens nuclei typically becoming rounded and pyknotic (condensed), prior to their disappearance from the central lens fiber cells.Citation46 Associated with this process, DNASE2B/DNase II-like acid DNase localizes with lysosomes in lens fiber cells at the border of the OFZ where it accumulates around the nuclei just prior to their elimination.Citation68 We examined the status of nuclei at 24 and 48 h in ex vivo culture following exposure of E13 lenses to the MAPK/JNK inhibitor SP600125 (). Inhibiting MAPK/JNK activity induced premature nuclear condensation after just 24 h (), which was followed by nuclear loss at 48 h (). In the control group, nuclei became condensed after 2 d in organ culture, exactly patterning the timing of this process in vivo (E13-E15), evidence that the control lenses continued their normal differentiation pathway in ex vivo culture. Similarly, the MAPK/JNK inhibitor JNK-IN-8 induced premature condensation and nuclear loss (Fig. S7C, iii and iv). Electron microscopy analysis confirmed the effect of MAPK/JNK inhibition on nuclear morphology, even in the cortical lens fiber zone where the appearance of nuclei was suggestive of early stages of nuclear fragmentation (Fig. S8). Together, the studies in this section show that MAPK/JNK inactivation initiated the signaling events that induced the loss of organelles and nuclei from the central lens fiber cells, as is required for formation of the OFZ during lens development.
To determine if induction of autophagy due to MAPK/JNK inhibition was specific to differentiating lens cells or is a phenomenon that can be observed in other cell types as well, we blocked endogenous MAPK/JNK activity in the breast cancer cell line MCF-7 with both MAPK/JNK inhibitors, SP600125 and JNK-IN-8. Here, endogenous refers to the MAPK/JNK signaling pathway activated during normal cell growth, not MAPK/JNK activity that is a transducer of a stress pathway, as in response to starvation. This example was chosen specifically, as blocking activation of MAPK/JNK in response to a cellular stress using the MAPK/JNK inhibitor SP600125Citation40,Citation41,Citation69,Citation70 prevents induction of autophagy in MCF-7 cells. Our results showed that both SP600125 (25 μM) and JNK-IN-8 (1 μM) effectively blocked MAPK/JNK activation (phosphorylation of JUN/c-Jun) in MCF-7 cells (Fig. S10A). In contrast, the MAPK/JNK activator anisomycin induced MAPK/JNK activity, while the MTOR inhibitor rapamycin had only a small effect on the activation state of MAPK/JNK. Blocking endogenous MAPK/JNK signaling in the MCF-7 cells with both MAPK/JNK inhibitors led to significant conversion of LC3B-I to LC3B-II, a measure of autophagy induction, whereas activation of MAPK/JNK with anisomycin prevented induction of both LC3B-I and LC3B-II (Fig. S10B). Note that the MCF-7 cells have a basal level of LC3B-II expression (Fig. S10B), as these cells use autophagy as a mechanism to derive energy and surviveCitation71 Similar to the MAPK/JNK inhibitor results, exposure of MCF-7 cells to rapamycin induced increased LC3B-II expression (Fig. S10B). To further demonstrate the connection between MAPK/JNK inactivation and the induction of autophagy in MCF-7 cells, we exposed the cells to the live autophagy dye CYTO-ID, which labels autophagic vesicles, following inactivation of MAPK/JNK with both SP600125 and JNK-IN-8. Significant vesicular localization of the CYTO-ID autophagic vesicle marker was observed following MAPK/JNK inactivation (Fig. S10C).Citation72,Citation73 To further examine the effect of inhibition of endogenous MAPK/JNK activity on MCF-7 cells, we co-labeled with the live dyes ER-Tracker and LysoTracker to detect ER and lysosomes, respectively, in real-time (Fig. S10D), as well as co-immunostained for the autophagy protein ATG14 and the ER marker CALR3 (Fig. S10E). The results showed increased colocalization of ER-Tracker and LysoTracker in vesicular structures, as well as for ATG14 and CALR3, following MAPK/JNK inactivation. These data parallel our results with the developing lens, suggesting that the induction of organelle loss by autophagy following inactivation of endogenous MAPK/JNK signaling pathways may be a common phenomenon. This pathway represents a distinct mechanism from that involved in the prevention of autophagy induction when MAPK/JNK activation is blocked in cells exposed to stress.
MTOR signaling implicated in removal of nuclei and organelles during lens development
In forming the MTORC1 complex the autophagy regulator MTOR kinase binds to and phosphorylates the scaffolding protein RPTOR, which then recruits downstream targets of MTOR such as RPS6KB (ribosomal protein S6 kinase, 70kDa), making them accessible to MTOR phosphorylation, and preventing activation of the autophagy pathway.Citation74,Citation75 Together immunolocalization () and immunoblot (, WCL) analyses demonstrated that MTOR was expressed throughout the E15 lens and that its levels dropped in the central lens fiber cells where OFZ formation is induced. While the level of RPTOR expression was more similar in cortical and central lens fiber cells, the association of RPTOR with MTOR was dramatically decreased in the central fiber region of the lens (), evidence that the MTORC1 complex had become disassembled. Disassembly of this complex would shut off the MTOR signal that prevents autophagy induction, which could lead to the autophagic removal of organelles in this region of the developing lens. In support of this conclusion control E13 lenses at 24 h in organ culture exhibited a lower level of phosphorylation of the MTOR target RPS6KB (Thr389) in the central fiber cells than in cortical fiber cells ().
Figure 6. Premature loss of nuclei and organelles from central lens fiber cells involve inactivation of MTORC1. (A) Cryosection from E15 lens immunostained for MTOR (red). Image is represented as a projection of acquired z-stacks, obtained by confocal microscopy; scale bar, 200 μm. Results show diminished expression of MTOR in the central lens fiber cells (FC). (B) Immunoblot analysis of whole cell lysates (WCL) showed relative expression of MTOR and RPTOR in cortical (FP) and central (FC) fiber zones of the E15 lenses. Co-immunoprecipitation analysis in fiber cell zones isolated by microdissection of E15 lenses demonstrated that the association between MTOR and RPTOR (a measure of the MTORC1 complex) was diminished in the central lens fiber cell zone (FC), consistent with autophagy induction for the formation of the OFZ. This result was quantified as the ratio of RPTOR to MTOR in FP and FC. These results imply disassembly of the MTORC1 complex occurs with lens fiber cell maturation. Error bars represent S.E., *P ≤ 0.05, t test. (C) Cryosections cut from E13 lenses exposed for 24 h to (i and iii) the vehicle, DMSO, or (ii and iv) the MTOR inhibitor rapamycin were immunostained for the ER marker HSPA5/Bip (red) and co-labeled with the nuclear dye TO-PRO-3 (blue). Images were obtained by confocal microscopy. (i and ii) are projection images shown at low magnification; scale bar, 200 μm. Boxed areas in (i and ii) are shown at higher magnification on the right (iii and iv). Each optical slice is 1 μm and was selected from an acquired z-stack; scale bar, 20 μm. (v) Immunoblot analysis of expression of the ER marker HSPA5 in FP and FC lens fiber zones from E13 lenses grown for 24 h in organ culture in the presence of either the vehicle DMSO or the MTOR inhibitor rapamycin. Results indicate premature loss of ER from the central lens fiber cells when the MTOR signal was inhibited with rapamycin. (D) Mid-sagittal cryosections were cut from E13 lenses exposed for 24 h in organ culture to (i and iii) the vehicle, DMSO or (ii and iv) the MTOR inhibitor rapamycin, labeled with the nuclear dye, TO-PRO-3 (blue). (i and ii) are representative projection images at low magnification obtained by confocal microscopy. Boxed regions are shown at higher magnification to the right (iii and iv). Images were selected from acquired z-stacks; each optical slice is 1 μm. Scale bars denote 200 μm (i and ii) and 20 μm (iii and iv). Results show pyknosis and premature loss of nuclei specifically in the central lens fiber cells when the MTOR signal is blocked. Results are representative of 3 independent studies.
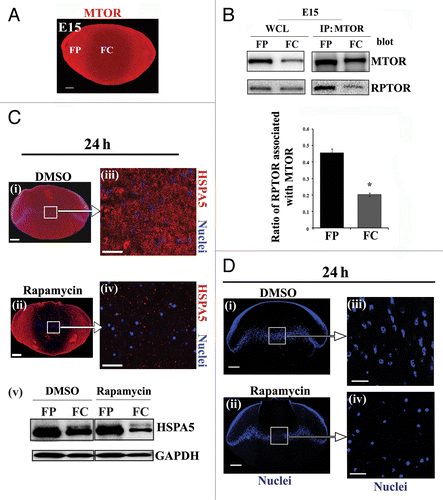
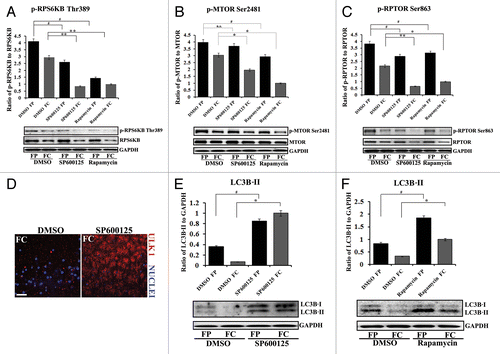
Figure 7. Suppressing the MAPK/JNK-MTOR-RPTOR signaling pathway led to premature loss of nuclei and organelles in lens fiber cells by autophagy. (A–C) Immunoblot analysis for (A) phospho-(p)-RPS6KB Thr389, RPS6KB, (B) phospho-(p) MTORSer2481, MTOR, and (C) phospho-(p)-RPTORSer863, RPTOR, in cortical (FP) and central (FC) fiber cell zones that were microdissected from lenses exposed to the MAPK/JNK inhibitor, SP600125, the MTOR inhibitor, rapamycin, or the vehicle DMSO. Both the MAPK/JNK and MTOR inhibitors suppressed activation of (A) p-RPS6KB Thr389, (B) p-MTORSer2481 and (C) p-RPTOR Ser863 in both cortical and central lens fiber cells. Immunoblot for GAPDH was included as a loading control. Total expression of RPS6KB and RPTOR were also diminished by the inhibitors in the FC zone. Quantification of the results is represented as the ratio of the phosphorylated (activated) form to the total expression level of the protein. Results show that in the presence of the MAPK/JNK inhibitor phosphorylation of RPTOR on Ser863, activation of MTOR and its downstream target RPS6KB were suppressed. Similar results were observed for rapamycin. (D) Immunostaining in cryosections cut from lenses grown for 24 h in organ culture in the presence of the MAPK/JNK inhibitor, SP600125, or its vehicle DMSO, showed upregulated perinuclear expression of the autophagy marker, ULK1 (red) in central lens fiber cells (FC) when MAPK/JNK activation was suppressed. Sections were colabeled for nuclei with TO-PRO-3 (blue). Images were obtained by confocal microscopy. Each optical slice is 0.2 μm and was selected from an acquired z-stack; scale bar, 20 μm. (E and F) Immunoblot analysis for LC3B-I and its lipid modified, autophagosome-associated form LC3B-II in FP and FC fiber cell zones microdissected from lenses that were exposed for 24 h to (E) the MAPK/JNK inhibitor SP600125, (F) rapamycin, or (E and F) the vehicle DMSO. Immunoblot for GAPDH was included as a loading control. Quantification of results is represented as the ratio of LC3B-II/GAPDH. The results reveal a significant increase in the expression of LC3B-II in both FP and FC following inactivation of either MAPK/JNK or MTOR signaling pathways. Results are representative of 3 or more independent studies. Error bars represent S.E., */#P ≤ 0.05 and **P ≤ 0.01, t test; n.s., nonsignificant.
Since a drop in MTOR expression and activation is often correlated with induction of autophagy,Citation76,Citation77 and our results showed decreased expression and activation of MTOR in the central lens fiber cells, we examined whether MTOR inactivation would be sufficient to induce premature formation of the OFZ. For this study MTOR activation was suppressed with the pharmacological inhibitor rapamycin, which blocks formation of the MTORC1 complex, to induce autophagy.Citation78 Exposure of E13 lenses to rapamycin for 24 h in organ culture effectively suppressed phosphorylation of RPS6KB on Thr389 in both cortical and central lens fiber cells (). Suppressing MTOR activity in these lenses induced the premature loss of organelles from lens fiber cells, as shown both by immunostaining and immunoblotting for the ER marker HSPA5 in the central fiber zone (). MTOR inactivation with rapamycin also induced nuclear condensation and loss (), similar to that observed in the MAPK/JNK inhibitor studies. Therefore, inactivation of MTOR is sufficient to induce the premature formation of the OFZ in the developing lens.
MAPK/JNK signals upstream of MTOR to induce autophagy in the developing lens
MAPK/JNK has been shown to target phosphorylation of RPTOR under conditions of osmotic stress.Citation30 However, the possibility of a link between MAPK/JNK inactivation and MTOR function with respect to autophagy induction has not been examined previously in the context of development. Since RPTOR phosphorylation on Ser863 is necessary for MTORC1 signaling,Citation36 we examined the effect of blocking MAPK/JNK activation on the phosphorylation of both RPTOR and MTOR. Immunoblot analysis was performed with activation (phospho)-specific antibodies on both cortical and central fiber cells isolated from E13 lenses that had been exposed in organ culture for 24 h to the MAPK/JNK inhibitor SP600125 or its vehicle dimethyl sulfoxide (DMSO; ). The results showed that suppressing MAPK/JNK activity caused significant inhibition of the MTOR signaling pathway in both these lens fiber cell zones. MAPK/JNK inhibition blocked the phosphorylation (activation) of MTOR (Ser2481, ), RPTOR (Ser863, ), and the direct downstream target of MTOR, RPS6KB (). In parallel studies, we examined the effect of rapamycin on MTOR signaling and compared the results to those obtained with the MAPK/JNK inhibitor. Both had similar effects on MTOR signaling in lens fiber cells that included inactivation of S6K, MTOR and RPTOR in cortical and central lens fiber cells (). However, the inhibition of RPTOR phosphorylation was greater with the MAPK/JNK inhibitor than with rapamycin. These results support a role for MAPK/JNK activation upstream of MTOR signaling in the pathway that blocks induction of autophagy in the developing lens, and suggested that it is inactivation of MAPK/JNK that was the inducing signal for formation of the OFZ.
To examine the link between the inactivation of the MAPK/JNK-MTOR signaling pathway and the induction of the autophagic process in the lens fiber cells, MAPK/JNK and MTOR signaling were blocked independently in lens organ cultures (E13) using SP600125 and rapamycin, respectively. Lenses were examined after a 24 h treatment period for expression of ULK1, a marker of phagophore formation, the expression and cleavage of the autophagosome marker LC3B, and both the presence and number of autophagic vesicles, determined by transmission electron microscopy analysis. Immunofluorescence studies showed that inactivation of MAPK/JNK induced expression of ULK1 and its association with vesicular structures in the central fiber cells of the lens (). Interestingly, the localization of ULK1 in these cells was perinuclear, a common feature of autophagy.Citation79 Both MAPK/JNK (; Fig. S7B) and MTOR () inhibitors induced high levels of LC3B-II expression in cortical and central lens fiber cells.
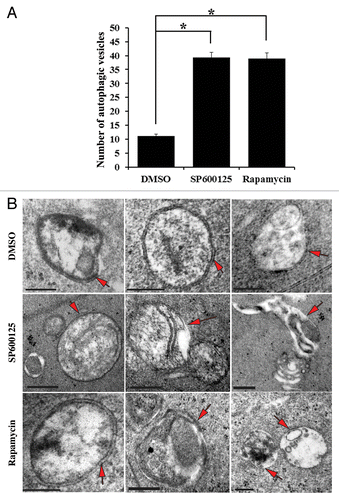
While the data presented above provide strong evidence of the induction of a molecular pathway associated with executing the autophagic process in lens fiber cells following inhibition of MAPK/JNK and MTOR signaling pathways, a standard of proof of autophagy induction remains observation of autophagic vesicles at the ultrastructural level by electron microscopy imaging. For these studies (E13) lenses were grown for 24 h in organ culture in the presence of the MAPK/JNK inhibitor SP600125, rapamycin, or the vehicle control, DMSO. EM analysis in a region of the cortical fiber zone close to the region of formation of the OFZ revealed a significant increase in the number of autophagic vesicles (both autophagosomes and autolysosomes) in response to inactivation of either MAPK/JNK or MTOR (). In control lenses an average of one autophagic vesicle was observed per unit area compared with 3 per unit area following inactivation of MAPK/JNK (SP600125) and 3 per unit area that form after exposure to the MTOR inhibitor rapamycin. Examples of autophagic vesicles commonly observed under each of these conditions are shown in . These include both double-membrane autophagic vesicles surrounding cytoplasmic organelles and cellular debris, and autolysosomes, single-walled autophagic vesicles (translucent interior) that result from the fusion of autophagosomes with hydrolytic-enzyme containing lysosomes. These results support a pathway in which the inactivation of MAPK/JNK-MTOR signaling was responsible for the induction of autophagy in lens fiber cells in order to create the OFZ during lens development.
Figure 8. Inhibition of either MAPK/JNK or MTOR signaling induced formation of autophagic vesicles in lens fiber cells. (A) Quantification of the number of autophagic vesicles in lenses grown in organ culture for 24 h in the presence of the vehicle (DMSO), the MAPK/JNK inhibitor (SP600125, 25 μM) or the MTOR inhibitor (rapamycin, 100 nM) showed a significant increase in the number of autophagic vesicles (including autophagosomes and autolysosomes) when MAPK/JNK or MTOR signaling was inhibited. These results provide ultrastructural evidence that autophagy is induced to remove lens organelles when MAPK/JNK and MTOR signals were suppressed. (B) Representative electron micrographs of autophagic vesicles (red arrows) observed in lenses that were exposed to the vehicle (DMSO), the MAPK/JNK inhibitor (SP600125) or the MTOR inhibitor (rapamycin). Autophagic vesicles shown include both autophagosomes and autolysosomes. Double-membraned vesicles are characteristic of autophagosomes, and are shown containing cellular organelles, cellular debris and/or translucent material. Single-membranes with translucent interiors containing cellular debris and degrading organelles are typical of autolysosomes. The images were acquired from an area in the fiber cells at the border of organelle-free zone formation; scale bar, 500 nm. Results are representative of 4 independent studies. Error bars represent SE *P ≤ 0.05, t test.
Inactivation of MAPK/JNK and MTOR in a primary, differentiating lens culture system induces autophagic-dependent loss of nuclei and organelles
To further investigate the relationship between MAPK/JNK and MTOR signaling and their inactivation in triggering the autophagic removal of nuclei and organelles from mature lens fiber cells, we conducted studies with a primary lens cell culture model that mimics lens cell differentiation as it occurs in vivo. In this culture system primary lens epithelial cells differentiate to form mini lens-like structures called lentoids (Fig. S11).Citation47 With this culture system it was possible to map the molecular components of the autophagy-inducing signaling pathway by blocking their activation with inhibitors. As was found for the lens organ culture system, 24 h exposure of lentoid-containing cultures to the MAPK/JNK inhibitor SP600125 effectively blocked the phosphorylation of JUN, a direct downstream target of MAPK/JNK (). In contrast, the MTOR inhibitor rapamycin had no effect on JUN phosphorylation. This result confirms the linearity of the MAPK/MTOR signaling axis, showing that MAPK/JNK activates MTOR in these cells, but not the reverse. Both the MAPK/JNK inhibitor and rapamycin suppressed phosphorylation (activation) of MTOR, RPTOR, and RPS6KB (). Again, inhibition of MAPK/JNK signaling had a much more potent inhibitory effect of the phosphorylation of RPTOR than did rapamycin (). This result suggested that when MAPK/JNK signaling was inhibited, the assembly of the MTORC1 complex was impaired, removing the block on the autophagy signal.
Figure 9. MAPK/JNK is upstream to MTORC1 in signaling the premature loss of nuclei and organelles in the lens fiber cells. (A–J) Primary lens cell cultures containing mature lentoid structures (multilayered, multicellular, differentiated mini-lens-like structures, shown in Fig. S11) were exposed for 24 h to either the MAPK/JNK inhibitor SP600125, the MTOR inhibitor, rapamycin or their vehicle DMSO. (A–D) Extracts were immunoblotted for (A) phospho-(p)-JUN Ser63/73, JUN, (B) phospho-(p)-pRPS6KB Thr389, RPS6KB, (C) phospho-(p)-MTOR Ser2481, MTOR, or (D) phospho-(p)-RPTOR Ser863, RPTOR. GAPDH was included as a loading control. Quantification is represented as a ratio of phosphorylated (activated) form to total expression levels of the protein. Results show that the MAPK/JNK inhibitor SP600125 effectively blocked activation of JUN, a direct downstream target of JNK, while rapamycin had no significant effect on JUN phosphorylation. Both SP600125 and rapamycin suppressed activation of (B) RPS6KB, (C) MTOR and (D) RPTOR, with MAPK/JNK inhibition causing a greater block of RPTOR phosphorylation than MTOR inhibition. (E) Extracts were immunoblotted for CALR3 and GAPDH. Quantification is represented as a ratio of CALR3 to GAPDH. Loss of the ER marker CALR3 was induced when either MAPK/JNK or MTOR signals were inhibited. (F and G) Cultures were immunostained for (F, i–iii) the Golgi marker, FTCD (F) or (G, i–iii) the mitochondrial marker, TOMM20 (red), colabeled for nuclei with TO-PRO-3 (blue) and examined by confocal microscopy. Images display both a single optical plane and orthogonal (z) sections (above and to the right). Results showed significant loss of Golgi, mitochondria and nuclei in lentoids when either MAPK/JNK or MTOR signals were blocked. (H and I) Extracts were immunoblotted for (H) BECN1 or (I) LC3B, and the loading control GAPDH. Quantification of the results was represented as (H) the ratio of BECN1 to GAPDH and (I) the ratio of LC3B-II to GAPDH. The results demonstrated significant upregulation of BECN1 and LC3B-II following exposure to SP600125 or rapamycin, showing induction of autophagy markers upon inhibiting the MAPK/JNK-MTOR pathway. (J) Live confocal image analysis on lentoid containing cultures exposed for 24 h to (ii and v) the MAPK/JNK inhibitor SP600125 or (iii and vi) the MTOR inhibitor rapamycin labeled with the fluorescent Cyto-ID dye (green) that specifically labels autophagic vesicles. Results showed that both inhibitors induced formation of autophagic vesicles. Lower panels show an overlay of the Cyto-ID fluorescent image with a DIC image. DMSO-exposed lentoids showed little or no labeling with Cyto-ID (i and iv). Images are 1-μm thick optical slices, acquired as z-stacks using confocal microscopy; scale bar, 20 μm. All graphs were plotted as a ratio of the phosphorylated protein to total protein expression or to loading control, GAPDH. Results are representative of at least 3 independent experiments. Error bars represent SE *P ≤ 0.05, t test.
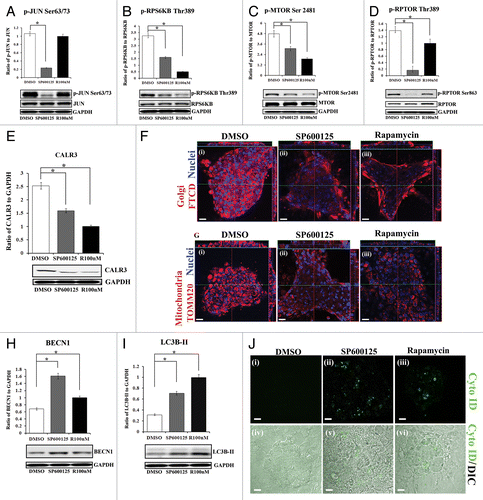
The link of the loss of nuclei and organelles to disinhibition of the autophagy pathway that occurs following suppression of either MAPK/JNK or MTOR signaling was examined in lentoid-containing primary lens cultures. Blocking either MAPK/JNK or MTOR signaling for 24 h induced premature loss of ER (CALR3; ), Golgi (FTCD; ) and mitochondria (TOMM20; ). This premature loss of organelles occurred concurrently with the induction of molecules intrinsically associated with the autophagy process, including BECN1 () and LC3B-II (). Studies with this culture system using the live autophagy marker Cyto ID, which specifically labels autophagic vesicles, demonstrated the induction of autophagy in real-time in response to suppression of either MAPK/JNK or MTOR signaling (). In contrast, exposure of primary lens cell cultures to anisomycin (0.5 μM), a known inducer of MAPK/JNK activation, increased MAPK/JNK activity (phosphorylation of JUN), had little effect on the activation of MTOR, prevented conversion of LC3B-I to LC3B-II, and downregulated BECN1 (Fig. S12). This result suggested that, in the lens, increasing activation of MAPK/JNK suppresses autophagy induction. Together, the primary lens cell culture studies led to the conclusion that inactivation of MAPK/JNK leads to autophagy-dependent loss of organelles in differentiating lens fiber cells through suppression of the MTOR signal.
Discussion
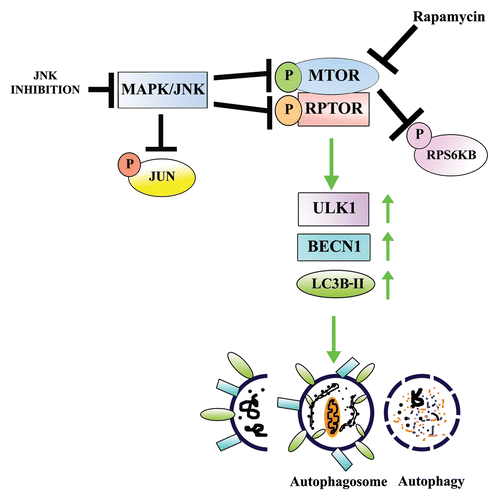
Autophagy is a homeostatic process by which almost all cells recycle and remodel their own intracellular components to maintain cellular homeostasis.Citation17,Citation80 Under conditions of stress such as hypoxia or in response to the need for energy when starved, cells will activate autophagy at a high level literally to eat their own organelles in order to survive.Citation5,Citation81-Citation83 Even under these extreme conditions the autophagic process must be a highly regulated signaling event in order to balance the cells’ momentary needs without leading to the cells’ death. Similarly, when organelles must be removed during cell differentiation for the specific developmental program of a tissue, this process must be regulated by signaling molecules able to turn on, and off, autophagic signaling pathways, which is sometimes required to have great spatial-temporal precision. The MTORC1 signaling pathway has long been known to be the main gatekeeper of autophagy in the cell.Citation27,Citation84 When active, the downstream signaling effectors of MTOR suppress autophagy induction.Citation25,Citation28 Finding systems best suited to understand the upstream regulators of this signaling pathway for normal cell behavior has remained a significant challenge. In our studies of the development of the lens we were able to identify MAPK/JNK as a crucial upstream signaling regulator of MTOR/MTORC1 signaling, the inactivation of MAPK/JNK leading to suppression of MTORC1 signaling, releasing the autophagy signal to execute the coordinated loss of nuclei and organelles that are one of the hallmarks of lens development ().
Figure 10. Proposed model for the role of MAPK/JNK in regulating MTOR-dependent autophagy for the removal of nuclei and organelles to form the OFZ during lens development. Our results showed that both MAPK/JNK and MTOR are critical components of the machinery that is involved in loss of nuclei and organelles during lens development. Active MAPK/JNK phosphorylates both MTOR and RPTOR in the MTORC1 complex to activate MTORC1 and block autophagy. Conditions that lead to inactivation of MAPK/JNK prevent phosphorylation of RPTOR and activation of MTOR, both essential components of MTORC1, causing disinhibition of the autophagy signal and inducing loss of nuclei and organelles in a tightly regulated manner from the central lens fiber cells to create the organelle-free zone.
It is, in fact, this very unique developmental process that is required for normal lens function that made this discovery possible. Transparency of the lens is essential to its role of focusing incoming images exactly on the site of the retina that processes these images for the brain. In addition to the remarkable geometric precision of its cellular organization, one of the incredible properties of lens cells that allows this tissue to belie its own cellularity and appear almost as if it was made of glass, is the developmental loss of nuclei and organelles from cells in the light path.Citation43,Citation85 The removal of these organelles during lens development, referred to as formation of the “organelle-free zone,”Citation45,Citation86 is highly regulated both spatially and temporally, beginning simultaneously over a wide area in the center of the lens. Some previous studies have established the need for specific molecules for nuclear degradation in the lens,Citation43,Citation68,Citation87,Citation88 however prior to this study the signals that induced formation of the OFZ remained uninvestigated.
Our studies with 2 different MAPK/JNK-specific inhibitors (SP600125, JNK-IN-8) revealed that inhibition of the MAPK/JNK signaling pathway was sufficient to induce premature formation of the OFZ in embryonic lenses. While autophagy seems an obvious mechanism for OFZ formation, until the discovery of Brennan et al.Citation52 that human lenses express a full complement of genes responsible for the autophagic process, and the revelation that a mutation in a gene associated with autophagy was linked to congenital cataracts,Citation89 autophagy was not considered to be required for normal lens development. More recently, Wignes et al.Citation90 demonstrated that congenital cataract in the αBR120G crystalline mutant mouse is associated with maintained expression of SQSTM1 and inhibition of autophagy, and Costello et al. provided extensive electron microscopy evidence of the presence of autophagic vesicles in the lens.Citation91 The role of autophagy in the formation of OFZ in the lens has long been a subject of debate as selective deletion of autophagy regulators, including Atg5 and Pik3c3, fail to abolish OFZ formation in the lens.Citation19 However, recent evidence shows that ablation of either of these molecular players is not alone sufficient to block the autophagy process.Citation20,Citation21 Such discrepancies could be explained by differences in gene targeting approach (choice of exon deletion), cell type (context) dependence, influence of genetic background, and time of development examined.Citation21 Thus, there may be specificity or redundancy in the function of autophagy molecules in different tissue contexts and, in the embryonic lens, distinct pathways responsible for autophagy-dependent removal of organelles during development from those involved in maintaining homeostasis.
Our studies with the developing lens confirm that autophagy is at least one mechanism that is responsible for the formation of the OFZ, and place MAPK/JNK signaling as the molecular switch that regulates the developmental timing of autophagy induction in the lens differentiation program. Similar premature induction of autophagy in the central region of the embryonic lens and formation of the OFZ could be induced by rapamycin, which directly targets MTOR to block its signaling pathway, releasing the autophagic process. Our studies show that MAPK/JNK is upstream of MTOR (modeled in ). Blocking MAPK/JNK activity suppresses MTOR signaling associated with MTORC1 to activate autophagy and induce premature formation of the OFZ. Both MTOR and MAPK/JNK target essential elements of the MTORC1 complex, most importantly the scaffolding protein RPTOR.Citation29,Citation74,Citation75 Phosphorylation of RPTOR is essential to its scaffold function in bringing together the MTORC1 complex and suppressing autophagy in the cell.Citation36 Interestingly, inhibition of MAPK/JNK signaling has a much greater impact on blocking RPTOR phosphorylation than rapamycin, suggesting that this target is a key element in the role of MAPK/JNK in regulating the timing of autophagy induction during lens development. The role of MAPK/JNK in this pathway is multifaceted, as blocking MAPK/JNK signaling also suppresses activation of MTOR, not just MTOR’s downstream signaling effectors. And studies with differentiating lens cells suggest that this pathway is not a circular one, as blocking MTOR activation with rapamycin has no measurable effect on the activity of MAPK/JNK. In contrast, activation of MAPK/JNK with anisomycin maintained MTOR activation and prevented autophagy induction, further support that the role of MAPK/JNK signaling in development involves its function in a pathway that prevents the induction of autophagy.
Immunostaining and immunoblotting for phospho-JUN, a molecular readout for MAPK/JNK activity, revealed that a dramatic, spatially coordinated shutdown of MAPK/JNK signaling occurs in the central region of the embryonic lens with the precise timing required for its central role in regulating formation of the OFZ. While our studies have revealed the downstream effectors of MAPK/JNK in autophagy-dependent removal of lens organelles, the regulators of the activity state of the MAPK/JNK signaling cascade still remain to be determined. Such an investigation promises to be challenging, as MAPK/JNK activation state is dependent on a balance between the activity of the upstream kinases and phosphatases that have been identified as its regulators, among which there are many candidates. Importantly, most of these known regulators of MAPK/JNK activity themselves are sensors of extracellular cues, changes in the cells’ environment, even changes in cell shape. It is likely that there are many types of regulation at play here. Prior to formation of the OFZ, there are the signals that must keep MAPK/JNK active for its role in maintaining activity of MTOR and the MTORC1 complex, preventing loss of organelles. Then at the exact time in lens development when organelles need to be removed from the central lens fiber cells, other regulators must be responsible for turning off MAPK/JNK activation to inactivate the MTORC1 complex and induce autophagy. We suggest that these regulatory events are likely to converge at the plasma membrane, specifically on scaffolds assembled at integrin adhesion junctions. This is a particularly attractive scenario in light of the discovery by Pereira et al.Citation92 that the level of MAPK/JNK activity can be altered by the specific matrix microenvironment in which cells find themselves. These investigators go on to show that at focal adhesions both integrin receptors and TLN/talin are key factors in suppressing MAPK/JNK activity, while a static stretch of cells leads to integrin-dependent MAPK/JNK activation. These results are particularly exciting for our own studies as they suggest that alterations in the way cells interact with their matrix, or each other, can regulate MAPK/JNK signaling activity. This possibility would intricately tie the morphogenesis of the lens itself during its development to the signaling changes in MAPK/JNK activity that lead to induction of autophagy and formation of the OFZ, and therefore warrants further investigation.
In summary, our studies demonstrate that inactivation of MAPK/JNK in the ocular lens induces upregulation of autophagy proteins by suppressing the MTOR-RPTOR signaling axis, leading to the spatio-temporal removal of organelles from the central lens fiber cells to form the organelle-free zone in the lens.
Materials and Methods
Lens microdissection
Lenses were isolated from chicken embryos (B&E Eggs, York Springs, PA) at different stages of development and microdissected into the central anterior epithelium (EC), equatorial epithelium (EQ), cortical fiber (FP), and nuclear fiber (FC) zones as previously described.Citation93
Preparation of ex vivo whole lens organ cultures
E13 lenses were placed in Complete Medium (Medium 199 [Life Technologies, 11150-059] with 10% fetal bovine serum, 1% penicillin and 1% streptomycin) at 37 °C. Lenses free of opacities after 2 h in culture were treated for 24 h or 48 h, as indicated, with either the specific MAPK/JNK inhibitor SP600125 (25 µM; Enzo Life Sciences, BML-EI305-0010),Citation66 the specific and irreversible MAPK/JNK inhibitor JNK-IN-8Citation67 (1 μM; EMD Millipore, 420150), the MTOR kinase inhibitor/autophagy inducer, rapamycin (100 nM; Calbiochem/EMD Millipore, 553211), or the vehicle control DMSO (0.1% Me2SO; Sigma, 276855). Lenses were microdissected for immunoprecipitation or direct western blot analysis, fixed, and cryosectioned for immunostaining studies, or fixed and prepared for electron microscopic studies.
Preparation of primary lens cell cultures
Primary lens cell cultures were prepared as described previouslyCitation47 using lenses isolated from E9 quail lenses. Briefly, cells were isolated by trypsinization and agitation and plated on laminin, and cultured in Complete Medium. After the cells had differentiated and formed mature lentoid structures (D10) they were treated for 24 h with MAPK/JNK inhibitor, SP600125 (25 µM), the MTOR inhibitor, rapamycin (100 nM), the MAPK/JNK activator anisomycin (Tocris Bioscience, 1290; 0.5 μM) or the vehicle control DMSO. Details on preparation of the SP600125 inhibitor have been previously described.Citation66 For treatment of embryonic lenses with inhibitors in ex vivo organ culture or in primary lens cell cultures complete medium was replaced with medium containing SP600125 (25 μM), JNK-IN-8 (1 μM), rapamycin (100 nM) or anisomycin (0.5 μM).
Antibodies
Antibodies to HSPA5/BiP/GRP78 (ER marker, G8918) and FTCD (Golgi marker, G2404) were purchased from Sigma Aldrich. JUN/c-Jun (sc-45), p-JUN/c-JunSer63/73 (sc-16312-R), p-RPS6KB/p70S6K Thr389 (sc-11759), RPS6KB/p70S6K (sc-9027), p-RPTOR/RAPTOR Ser863 (sc-130214), RPTOR/RAPTOR (sc-81537), CALR3/calregulin/calreticulin (sc-166837), ULK1/Atg1 (sc-33182), BECN1/BECLIN1 (for immunostaining, sc-11427), TOMM20 (sc-17764), SQSTM1/p62 (sc-25575) and GAPDH (sc-25778) were purchased from Santa Cruz Biotechnology, Inc. Antibodies to p-MTOR Ser2481 (2974), MTOR (2983) and BECN1/BECLIN1 (for immunoblotting, 3495) were purchased from Cell Signaling Technology. Antibody to LC3B was purchased from Novus Biologicals (NB100-2220). ATG14 (5504) was purchased from Cell Signaling Technology. Antibody to AQP0/Aquaporin-0 was generated as described in a separate study (MP28Citation94).
Immunoblotting
A triton/octylglucoside (OG/T) buffer cocktail composed of 44.4 mM n-octyl β-D-glucopyranoside, 1% Triton X-100, 100 mM NaCl, 1 mM MgCl2, 5 mM EDTA, and 10 mM imidazole, containing 1 mM sodium vanadate, 0.2 mM H2O2, and protease inhibitor cocktail (Sigma Aldrich, P8340) was used to extract the cells as described previously.Citation95 The concentration of protein was determined using the BCA assay (Thermo Sceintific, 23223, 23224). 50 μg of protein extracts were subjected to SDS-PAGE on precast 8–16% tris/glycine gels (Invitrogen, EC6045BOX). Proteins were electrophoretically transferred onto Immobilon-P membranes (Millipore Corp., IPVH00010). Membranes were blocked in 5% skim milk for 1 h and probed for primary antibodies at 4 °C overnight followed by secondary antibodies conjugated to horseradish peroxidase (BIO-RAD, 170-6515, 170-6516). Protein bands were detected using ECL reagent or ECL plus reagent (Thermo Fisher Scientific Inc. Rockford, 32106, 80197) and images were acquired using the FluorChem E & M imager from Protein Simple (FM0418). The FluorChem E & M imager is a digital darkroom technology with an 8.3 megapixel CCD (charge-coupled device) resolution and flat field calibration that corrects for non-uniformities in light gathering.
Immunoprecipitation
For these studies the FP and the FC zones were isolated from 100 E15 lenses by microdissection and extracted with OG/T buffer. To determine association of MTOR with RPTOR the entire FP and FC fractions were immunoprecipitated with primary antibody to MTOR (4 °C overnight), followed by incubation with TrueBlot immunoprecipitation beads from eBiosciences (00-8800-25) for 1 h. The immunoprecipitates and whole cell lysates were subjected to SDS-PAGE, transferred to Immobilon-P membranes and the association of RPTOR with MTOR determined using western blot techniques as described above.Citation95,Citation96
Immunostaining
Freshly isolated lenses, lenses from organ culture, and primary lens cell cultures were fixed in 3.7% formaldehyde (15 min for primary cultures and overnight for whole lenses). For detecting phospho-protein, cultures were pre-treated with pervanadate for 20 min prior to fixation. Whole lenses were cryopreserved in 30% sucrose solution after fixation for 24 h or longer and prepared for cryosectioning. 20-μm thick sections were cut. Samples were then permeabilized with 0.25% Triton X-100 in DPBS buffer (2.7 mM KCl, 1.5 mM KH2PO4, 137.9 mM NaCl, 8.1 mM Na2HPO4–7 H2O [Corning, 21-0310CV]) for 10 min, blocked in blocking buffer for 1 h (5% goat serum, 0.5 g BSA in 50 ml DPBS) and then incubated sequentially in primary antibody either for 3 h at 37 °C or overnight at 4 °C, followed by a fluorescent-conjugated secondary antibody for 2 h (Jackson ImmunoResearch Laboratories, 111-295-144, 115-545-003, 115-295-008). F-actin was localized with Alexa448-conjugated phalloidin (Invitrogen-Molecular Probes, A12379). Nuclei were counterstained with TO-PRO-3 (Invitrogen-Molecular Probes, T3605). All sections were cut serially in the anterior to posterior direction.
Image analysis
Phase contrast microscopy was performed using a Nikon Eclipse TE2000-U microscope (Optical Apparatus), and images were acquired with the Cool Snap HQCitation2 camera (Photometrics) using Image-Pro Plus software (Phase 3 Imaging Systems). Confocal microscopy was performed using the Zeiss LSM510 META confocal microscope. Single optical planes were selected from z-stacks, each 1-μm thick, unless otherwise indicated, using the LSM5 Image Browser. For creating projection images optical slices 8-μm thick were used. Orthogonal views were created using the LSM5 Image Browser. DIC images were acquired using the Zeiss LSM510 confocal microscope.
Transmission electron microscopy
Lenses were fixed in 2% formaldehyde, 2.5% glutaraldehyde for 72 h. Fifty micron-thick serial cross sections were cut using a Vibratome (Technical Product International), and rinsed extensively in 0.1 M phosphate buffer (PB; pH 7.4) and 0.1 M Tris-buffered saline (TBS; pH 7.6). Next, sections were fixed in 2% osmium tetroxide (Electron Microscopy Sciences, 19190) in 0.1 M PB for 1 h and dehydrated in an ascending series of ethanol followed by propylene oxide and flat embedded in Epon 812 (Electron Microscopy Sciences);Citation97 between 2 sheets of Aclar plastic (Electron Microscopy Sciences, 50425-25).
Mid-coronal/crosssections were selected from each lens and 20–25 serial sections of 74-nm thickness were cut from the mid-sagittal vibratome lens section with a diamond knife from each lens (Diatome-US, Fort Washington, PA) using a Leica Ultracut microtome (Leica Microsystems, Wetzlar, Germany) and collected on copper mesh grids ((Electron Microscopy Sciences, G300H-Cu). The grids were then counterstained with 8% uranyl acetate solution for 20 min followed by lead citrate solution for 5 min and examined with an electron microscope (Morgagni, Fei Company, Hillsboro, OR). To examine for the presence of autophagic vacuoles in lens fiber cells, cortical lens fibers close to the zone of organelle-free zone formation were imaged. Five different such regions were examined for the presence of autophagic vesicles. Digital images were captured using the AMT advantage HR/HR-B CCD camera system (AdvanceMicroscopy Techniques Corp., Danvers, MA, USA). Images were assembled and adjusted for brightness and contrast in Adobe Photoshop CS6 software (Adobe Systems, Inc., San Jose, CA).
Electron microscopy quantification
Autophagic vesicles that included both autophagosomes (double-membrane vesicles containing cellular debris or degrading organelles) and autolysosomes (single-membrane vesicles formed after autophagosomes have fused with hydrolytic enzyme-containing lysosomes, giving them a translucent interior) were counted blind for each treatment. For analysis, 4 different areas in the cortical lens fiber zone close to the OFZ were randomly selected. Four neighboring lens fiber cells in cross section were considered as 1 unit area. The total number of autophagic vesicles observed for each treatment group (DMSO, SP600125, rapamycin) was divided by the number of times the experiment was performed (4) to determine the average number of vesicles observed per experiment. The cell number was kept at a constant while counting and the average number of autophagic vesicles observed for each treatment group was quantified and represented graphically.
Live imaging studies
Cyto-ID® Autophagy detection kit (Enzo Life Sciences, ENZO-51031-K200), which specifically labels autophagic vesicles,Citation98 was used according to the manufacturer’s protocol. Briefly, cultured cells were washed with 1× assay buffer prior to incubation with dye solution (3 μl of dye to 1ml of 1× assay buffer for each dish). Samples were shielded from direct light and incubated in the dye for 10–15 min at 37 °C. After the incubation period, cells were imaged live using confocal imaging.
Preparation and treatment of MCF-7 cell cultures
MCF-7 breast cancer cells were obtained from the American Type Culture Collection (ATCC, HTB-22) and maintained in ATCC-formulated Eagle’s Minimum Essential Medium (30-2003), supplemented with 0.1% bovine insulin powder (Gemini Bio-products, 700-112P), 10% FBS and 1% penicillin-streptomycin. After the cells reached 90% confluency they were exposed for 24 h to the MAPK/JNK inhibitors (SP600125, 25 µM; or JNK-IN-8, 1 µM), the MTOR inhibitor rapamycin (100 nM), the MAPK/JNK activator anisomycin (Tocris Biosciences, 1290 0.3 μM) or the vehicle control DMSO. Post-treatment cells either were extracted in OG/T buffer for immunoblot analysis or fixed in 3.7% formaldehyde for immunofluorescence studies as described above. For live imaging of ER and lysosomes in real-time cells were incubated with LysoTracker® Green DND-26 (1 µM; Invitrogen L7526) and ER-Tracker™ Red (2 µM; BODIPY® TR Glibenclamide, Invitrogen, E34250) in Medium 199 (without phenol red, serum or antibiotics) for 20 min at 37 °C. Cells were then washed twice with PBS, transferred to Medium 199 (without phenol red, serum or antibiotics) and imaged live by confocal microscopy. Live imaging of MCF-7 cells with CYTO-ID was performed as described above.
Densitometric analysis of immunoblots
The densitometric analysis of immunoblots was performed using both Carestream Health Imaging Software (Carestream Health Inc.) and Alpha View® software (Protein Simple).
Statistical analysis
Statistical analysis was performed using t test on 3 or more independent experiments using the SPSS statistics software. Error bars represent SEM. Differences were considered significant when *P ≤ 0.05, #P ≤ 0.05 and, **P ≤ 0.01.
| Abbreviations: | ||
| BECN1 | = | Beclin 1, autophagy-related |
| DMSO | = | dimethyl sulfoxide |
| EQ | = | equatorial epithelium |
| FC | = | central nuclear fiber cells |
| FP | = | cortical fiber cells |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| OFZ | = | organelle-free zone |
| RPTOR | = | regulatory associated protein of MTOR, complex 1 |
Additional material
Download Zip (1.3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
This work was supported by grant EY010577, EY14258 from the National Institutes of Health (NIH) to ASM and in part, by a grant-in-aid of research from the National Academy of Sciences, administered by Sigma Xi, The Scientific Research Society, to SB. We would like to thank Brigid Bleaken for excellent technical assistance and Iris Wolff for performing the E13 and E15 lens microdissections. We thank Janet Richards for help with the EM studies. We thank Dr Janice Walker for guiding us in the MCF-7 cell studies and for critically reading the manuscript.
References
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology 1973; 7:253 - 66; http://dx.doi.org/10.1002/tera.1420070306; PMID: 4807128
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26:239 - 57; http://dx.doi.org/10.1038/bjc.1972.33; PMID: 4561027
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Neufeld TP. Autophagy and cell growth--the yin and yang of nutrient responses. J Cell Sci 2012; 125:2359 - 68; http://dx.doi.org/10.1242/jcs.103333; PMID: 22649254
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 5; http://dx.doi.org/10.1038/nature07006; PMID: 18454133
- Takano-Ohmuro H, Mukaida M, Kominami E, Morioka K. Autophagy in embryonic erythroid cells: its role in maturation. Eur J Cell Biol 2000; 79:759 - 64; http://dx.doi.org/10.1078/0171-9335-00096; PMID: 11089924
- Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima NN, Iwasaki A, He YW, Swat W, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008; 4:309 - 14; PMID: 18188005
- Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012; 119:2895 - 905; http://dx.doi.org/10.1182/blood-2011-08-372383; PMID: 22223827
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A 2009; 106:19860 - 5; http://dx.doi.org/10.1073/pnas.0906048106; PMID: 19910529
- Zhao Y, Chen G, Zhang W, Xu N, Zhu JY, Jia J, Sun ZJ, Wang YN, Zhao YF. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1α/BNIP3 signaling pathway. J Cell Physiol 2012; 227:639 - 48; http://dx.doi.org/10.1002/jcp.22768; PMID: 21465467
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 2008; 17:3897 - 908; http://dx.doi.org/10.1093/hmg/ddn292; PMID: 18782848
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003; 301:1387 - 91; http://dx.doi.org/10.1126/science.1087782; PMID: 12958363
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/10.1152/physrev.00030.2009; PMID: 20959619
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Juenemann K, Reits EA. Alternative macroautophagic pathways. Int J Cell Biol 2012; 2012:189794; http://dx.doi.org/10.1155/2012/189794; PMID: 22536246
- Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem 2013; 288:11436 - 47; http://dx.doi.org/10.1074/jbc.M112.437103; PMID: 23479732
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P, Di Paolo G. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PLoS One 2013; 8:e76405; http://dx.doi.org/10.1371/journal.pone.0076405; PMID: 24098492
- Martens S, Rusten TE, Kraft C. Autophagy at sea. Autophagy 2013; 9:1286 - 91; http://dx.doi.org/10.4161/auto.25838; PMID: 23917436
- Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc Natl Acad Sci U S A 2010; 107:9424 - 9; http://dx.doi.org/10.1073/pnas.0914725107; PMID: 20439739
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Hall MN. mTOR-what does it do?. Transplant Proc 2008; 40:Suppl S5 - 8; http://dx.doi.org/10.1016/j.transproceed.2008.10.009; PMID: 19100909
- Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010; 6:438 - 48; http://dx.doi.org/10.4161/auto.6.4.12244; PMID: 20484971
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 2000; 103:253 - 62; http://dx.doi.org/10.1016/S0092-8674(00)00117-3; PMID: 11057898
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 95; http://dx.doi.org/10.1016/j.febslet.2010.01.017; PMID: 20083114
- Foster KG, Acosta-Jaquez HA, Romeo Y, Ekim B, Soliman GA, Carriere A, Roux PP, Ballif BA, Fingar DC. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 2010; 285:80 - 94; http://dx.doi.org/10.1074/jbc.M109.029637; PMID: 19864431
- Kwak D, Choi S, Jeong H, Jang JH, Lee Y, Jeon H, Lee MN, Noh J, Cho K, Yoo JS, et al. Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal Kinase (JNK)-mediated Raptor protein phosphorylation. J Biol Chem 2012; 287:18398 - 407; http://dx.doi.org/10.1074/jbc.M111.326538; PMID: 22493283
- Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011; 25:310 - 22; http://dx.doi.org/10.1101/gad.1984311; PMID: 21325132
- Hwang M, Perez CA, Moretti L, Lu B. The mTOR signaling network: insights from its role during embryonic development. Curr Med Chem 2008; 15:1192 - 208; http://dx.doi.org/10.2174/092986708784310459; PMID: 18473813
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949 - 51; PMID: 18769111
- Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 2003; 278:15461 - 4; http://dx.doi.org/10.1074/jbc.C200665200; PMID: 12604610
- Foster KG, Acosta-Jaquez HA, Romeo Y, Ekim B, Soliman GA, Carriere A, Roux PP, Ballif BA, Fingar DC. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 2010; 285:80 - 94; http://dx.doi.org/10.1074/jbc.M109.029637; PMID: 19864431
- Wang L, Lawrence JC Jr., Sturgill TW, Harris TE. Mammalian target of rapamycin complex 1 (mTORC1) activity is associated with phosphorylation of raptor by mTOR. J Biol Chem 2009; 284:14693 - 7; http://dx.doi.org/10.1074/jbc.C109.002907; PMID: 19346248
- Waetzig V, Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci 2005; 26:455 - 61; PMID: 16054242
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678 - 88; http://dx.doi.org/10.1016/j.molcel.2008.06.001; PMID: 18570871
- Kim AD, Kang KA, Kim HS, Kim DH, Choi YH, Lee SJ, Kim HS, Hyun JW. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis 2013; 4:e750; http://dx.doi.org/10.1038/cddis.2013.273; PMID: 23907464
- Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006; 2:39 - 46; PMID: 16874071
- Walker J, Menko AS. Integrins in lens development and disease. Exp Eye Res 2009; 88:216 - 25; http://dx.doi.org/10.1016/j.exer.2008.06.020; PMID: 18671967
- Nishimoto S, Kawane K, Watanabe-Fukunaga R, Fukuyama H, Ohsawa Y, Uchiyama Y, Hashida N, Ohguro N, Tano Y, Morimoto T, et al. Nuclear cataract caused by a lack of DNA degradation in the mouse eye lens. Nature 2003; 424:1071 - 4; http://dx.doi.org/10.1038/nature01895; PMID: 12944971
- Wride MA. Lens fibre cell differentiation and organelle loss: many paths lead to clarity. Philos Trans R Soc Lond B Biol Sci 2011; 366:1219 - 33; http://dx.doi.org/10.1098/rstb.2010.0324; PMID: 21402582
- Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res 2009; 88:133 - 9; http://dx.doi.org/10.1016/j.exer.2008.08.017; PMID: 18840431
- Wride MA. Lens fibre cell differentiation and organelle loss: many paths lead to clarity. Philos Trans R Soc Lond B Biol Sci 2011; 366:1219 - 33; http://dx.doi.org/10.1098/rstb.2010.0324; PMID: 21402582
- Menko AS, Klukas KA, Johnson RG. Chicken embryo lens cultures mimic differentiation in the lens. Dev Biol 1984; 103:129 - 41; http://dx.doi.org/10.1016/0012-1606(84)90014-9; PMID: 6370757
- Bassnett S. The fate of the Golgi apparatus and the endoplasmic reticulum during lens fiber cell differentiation. Invest Ophthalmol Vis Sci 1995; 36:1793 - 803; PMID: 7635654
- Modak SP, Perdue SW. Terminal lens cell differentiation. I. Histological and microspectrophotometric analysis of nuclear degeneration. Exp Cell Res 1970; 59:43 - 56; http://dx.doi.org/10.1016/0014-4827(70)90622-1; PMID: 4915191
- Bashour A-M, Bloom GS. 58K, a microtubule-binding Golgi protein, is a formiminotransferase cyclodeaminase. J Biol Chem 1998; 273:19612 - 7; http://dx.doi.org/10.1074/jbc.273.31.19612; PMID: 9677387
- Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J 2011; 434:181 - 8; http://dx.doi.org/10.1042/BJ20101569; PMID: 21309747
- Brennan LA, Kantorow WL, Chauss D, McGreal R, He S, Mattucci L, Wei J, Riazuddin SA, Cvekl A, Hejtmancik JF, et al. Spatial expression patterns of autophagy genes in the eye lens and induction of autophagy in lens cells. Mol Vis 2012; 18:1773 - 86; PMID: 22815631
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008; 27:Suppl 1 S137 - 48; http://dx.doi.org/10.1038/onc.2009.51; PMID: 19641499
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571 - 80; http://dx.doi.org/10.1038/cdd.2010.191; PMID: 21311563
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395 - 8; http://dx.doi.org/10.1038/26506; PMID: 9759731
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488 - 92; http://dx.doi.org/10.1038/35044114; PMID: 11100732
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836 - 41; http://dx.doi.org/10.1038/ncb0910-836; PMID: 20811356
- Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L. Molecular cloning and characterization of rat LC3A and LC3B--two novel markers of autophagosome. Biochem Biophys Res Commun 2006; 339:437 - 42; http://dx.doi.org/10.1016/j.bbrc.2005.10.211; PMID: 16300744
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 27; http://dx.doi.org/10.1016/j.molcel.2009.01.021; PMID: 19250912
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen H-Y, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 2007; 19:142 - 9; http://dx.doi.org/10.1016/j.ceb.2007.02.001; PMID: 17303404
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 2001; 98:13681 - 6; http://dx.doi.org/10.1073/pnas.251194298; PMID: 11717429
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 2012; 19:140 - 54; http://dx.doi.org/10.1016/j.chembiol.2011.11.010; PMID: 22284361
- Nakahara M, Nagasaka A, Koike M, Uchida K, Kawane K, Uchiyama Y, Nagata S. Degradation of nuclear DNA by DNase II-like acid DNase in cortical fiber cells of mouse eye lens. FEBS J 2007; 274:3055 - 64; http://dx.doi.org/10.1111/j.1742-4658.2007.05836.x; PMID: 17509075
- Oh S-H, Lim S-C. Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J Pharmacol Exp Ther 2009; 329:112 - 22; http://dx.doi.org/10.1124/jpet.108.144113; PMID: 19139269
- Li Y, Luo Q, Yuan L, Miao C, Mu X, Xiao W, Li J, Sun T, Ma E. JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol Appl Pharmacol 2012; 263:21 - 31; http://dx.doi.org/10.1016/j.taap.2012.05.018; PMID: 22668848
- Gavilán E, Sánchez-Aguayo I, Daza P, Ruano D. GSK-3β signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis 2013; 4:e572; http://dx.doi.org/10.1038/cddis.2013.95; PMID: 23559006
- Coleman J, Xiang Y, Pande P, Shen D, Gatica D, Patton WF. A live-cell fluorescence microplate assay suitable for monitoring vacuolation arising from drug or toxic agent treatment. J Biomol Screen 2010; 15:398 - 405; http://dx.doi.org/10.1177/1087057110364242; PMID: 20237207
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177 - 89; http://dx.doi.org/10.1016/S0092-8674(02)00833-4; PMID: 12150926
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163 - 75; http://dx.doi.org/10.1016/S0092-8674(02)00808-5; PMID: 12150925
- Bohensky J, Leshinsky S, Srinivas V, Shapiro IM. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol 2010; 25:633 - 42; http://dx.doi.org/10.1007/s00467-009-1310-y; PMID: 19830459
- Kapoor V, Zaharieva MM, Das SN, Berger MR. Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt-mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett 2012; 319:39 - 48; http://dx.doi.org/10.1016/j.canlet.2011.12.032; PMID: 22202640
- Jung CH, Ro S-H, Cao J, Otto NM, Kim D-H. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 95; http://dx.doi.org/10.1016/j.febslet.2010.01.017; PMID: 20083114
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011; 13:453 - 60; http://dx.doi.org/10.1038/ncb2204; PMID: 21394080
- Jia K, Levine B. Autophagy and longevity: lessons from C. elegans. Adv Exp Med Biol 2010; 694:47 - 60; http://dx.doi.org/10.1007/978-1-4419-7002-2_5; PMID: 20886756
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728 - 41; http://dx.doi.org/10.1016/j.cell.2011.10.026; PMID: 22078875
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682 - 95; http://dx.doi.org/10.1016/j.cell.2011.07.030; PMID: 21884931
- Wang RC, Levine B. Autophagy in cellular growth control. FEBS Lett 2010; 584:1417 - 26; http://dx.doi.org/10.1016/j.febslet.2010.01.009; PMID: 20096689
- Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol 2010; 22:157 - 68; http://dx.doi.org/10.1016/j.ceb.2009.11.005; PMID: 20006481
- Pendergrass W, Penn P, Possin D, Wolf N. Accumulation of DNA, nuclear and mitochondrial debris, and ROS at sites of age-related cortical cataract in mice. Invest Ophthalmol Vis Sci 2005; 46:4661 - 70; http://dx.doi.org/10.1167/iovs.05-0808; PMID: 16303963
- Bassnett S. Lens organelle degradation. Exp Eye Res 2002; 74:1 - 6; http://dx.doi.org/10.1006/exer.2001.1111; PMID: 11878813
- Nakamura T, Pichel JG, Williams-Simons L, Westphal H. An apoptotic defect in lens differentiation caused by human p53 is rescued by a mutant allele. Proc Natl Acad Sci U S A 1995; 92:6142 - 6; http://dx.doi.org/10.1073/pnas.92.13.6142; PMID: 7597093
- Sanders EJ, Parker E. The role of mitochondria, cytochrome c and caspase-9 in embryonic lens fibre cell denucleation. J Anat 2002; 201:121 - 35; http://dx.doi.org/10.1046/j.1469-7580.2002.00081.x; PMID: 12220121
- Chen J, Ma Z, Jiao X, Fariss R, Kantorow WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet 2011; 88:827 - 38; http://dx.doi.org/10.1016/j.ajhg.2011.05.008; PMID: 21636066
- Wignes JA, Goldman JW, Weihl CC, Bartley MG, Andley UP. p62 expression and autophagy in αB-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp Eye Res 2013; 115:263 - 73; http://dx.doi.org/10.1016/j.exer.2013.06.026; PMID: 23872361
- Costello MJ, Brennan LA, Basu S, Chauss D, Mohamed A, Gilliland KO, Johnsen S, Menko AS, Kantorow M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res 2013; 116:141 - 50; http://dx.doi.org/10.1016/j.exer.2013.08.017; PMID: 24012988
- Pereira AM, Tudor C, Kanger JS, Subramaniam V, Martin-Blanco E. Integrin-dependent activation of the JNK signaling pathway by mechanical stress. PLoS One 2011; 6:e26182; http://dx.doi.org/10.1371/journal.pone.0026182; PMID: 22180774
- Walker JL, Menko AS. alpha6 Integrin is regulated with lens cell differentiation by linkage to the cytoskeleton and isoform switching. Dev Biol 1999; 210:497 - 511; http://dx.doi.org/10.1006/dbio.1999.9277; PMID: 10357906
- Sas DF, Sas MJ, Johnson KR, Menko AS, Johnson RG. Junctions between lens fiber cells are labeled with a monoclonal antibody shown to be specific for MP26. J Cell Biol 1985; 100:216 - 25; http://dx.doi.org/10.1083/jcb.100.1.216; PMID: 3880752
- Basu S, Rajakaruna S, Menko AS. Insulin-like growth factor receptor-1 and nuclear factor κB are crucial survival signals that regulate caspase-3-mediated lens epithelial cell differentiation initiation. J Biol Chem 2012; 287:8384 - 97; http://dx.doi.org/10.1074/jbc.M112.341586; PMID: 22275359
- Walker JL, Zhang L, Zhou J, Woolkalis MJ, Menko AS. Role for alpha 6 integrin during lens development: Evidence for signaling through IGF-1R and ERK. Dev Dyn 2002; 223:273 - 84; http://dx.doi.org/10.1002/dvdy.10050; PMID: 11836791
- Leranth C, Pickel V. Electron Microscopic Preembedding Double-Immunostaining Methods. In: Heimer L, Záborszky L, eds. Neuroanatomical Tract-Tracing Methods 2: Springer US, 1989:129-72
- Chan LL, Shen D, Wilkinson AR, Patton W, Lai N, Chan E, Kuksin D, Lin B, Qiu J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012; 8:1371 - 82; http://dx.doi.org/10.4161/auto.21028; PMID: 22895056