
Abstract
Ionizing radiation induces cellular senescence to suppress cancer cell proliferation. However, it also induces deleterious bystander effects in the unirradiated neighboring cells through the release of senescence-associated secretory phenotypes (SASPs) that promote tumor progression. Although autophagy has been reported to promote senescence, its role is still unclear. We previously showed that radiation induces senescence in PTTG1-depleted cancer cells. In this study, we found that autophagy was required for the radiation-induced senescence in PTTG1-depleted breast cancer cells. Inhibition of autophagy caused the cells to switch from radiation-induced senescence to apoptosis. Senescent cancer cells exerted bystander effects by promoting the invasion and migration of unirradiated cells through the release of CSF2 and the subsequently activation of the JAK2-STAT3 and AKT pathways. However, the radiation-induced bystander effects were correlated with the inhibition of endogenous autophagy in bystander cells, which also resulted from the activation of the CSF2-JAK2 pathway. The induction of autophagy by rapamycin reduced the radiation-induced bystander effects. This study reveals, for the first time, the dual role of autophagy in radiation-induced senescence and bystander effects.
Introduction
Radiotherapy is one of the best therapeutic choices for cancer treatment. Cellular responses to ionizing radiation produce a spectrum of effects ranging from growth arrest to apoptotic cell death and senescence.Citation1 Cellular senescence is a phenomenon of prolonged cell cycle arrest. Thus, it is considered as an important tumor suppressive mechanism.Citation2,Citation3 However, senescent cells are still metabolically active and ultimately develop senescence-associated secretory phenotypes, which participate in tumor suppression, tumor promotion, aging, and tissue repair.Citation4 SASPs composed of soluble proteins (cytokines, chemokines, and growth factors), secreted proteases and secreted insoluble proteins/extracellular matrix components may influence the fate of unirradiated neighboring cells, which is known as the bystander effect.Citation5,Citation6
Autophagy is a genetically regulated process characterized by the formation of double-membrane cytosolic vesicles (autophagosomes) that sequester cytoplasmic content and deliver it to the lysosomes for degradation.Citation7 Basal autophagy is essential to maintain cellular homeostasis and genomic integrity by degrading the aged or malfunctioning organelles and the damaged or misfolded proteins.Citation7 It has been shown that autophagy is frequently upregulated in tumors in response to therapy and may protect the tumors against therapy-induced apoptosis.Citation8 However, the role of autophagy in cancer is still controversial as it may suppress tumors during cancer development but promote cell survival during cancer progression.Citation7
PTTG1/securin is an oncogene that is highly expressed in various tumors and has been correlated with tumor invasiveness and poor prognosis.Citation9,Citation10 Recent studies have shown that inhibition of PTTG1 suppresses tumor growth and metastasis in vitro and in vivo.Citation11 Our previous studies show that PTTG1-depleted and -low expressing breast cancer cells senesce after irradiation.Citation12,Citation13 In the present study, we further investigated the role of autophagy in radiation-induced senescence and bystander effects. We found that radiation-induced autophagy in PTTG1-depleted breast cancer cells promoted senescence. Inhibition of autophagy resulted in a switch from radiation-induced senescence to apoptosis. In addition, senescent cancer cells induced the release of CSF2 and the activation of JAK2-STAT3 and AKT pathways, which led to radiation-induced bystander effects that promoted the invasion and migration of unirradiated cancer cells and human umbilical vein endothelial cells (HUVECs). However, radiation-induced bystander effects were correlated with the inhibition of endogenous autophagy in bystander cells, which also resulted from the CSF2-JAK2 pathway. Induction of autophagy by rapamycin treatment reduced the radiation-induced bystander effects. Our study provides new insight into the role of autophagy in radiation-induced senescence and bystander effects in human breast cancer cells.
Results
Radiation induces autophagy in PTTG1-depleted human breast cancer cells
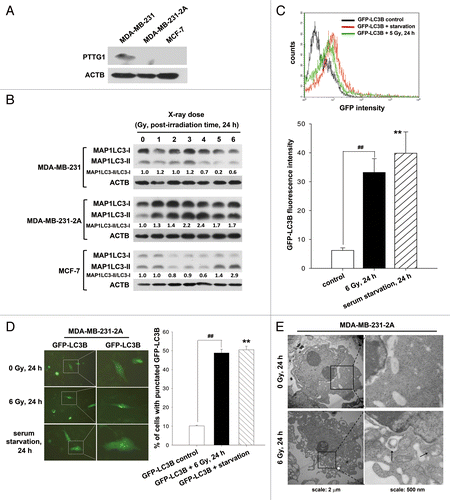
Our previous studies show that radiation induces senescence in PTTG1-depleted human breast cancer cells.Citation12,Citation13 Autophagy has been reported to be an effector of senescence.Citation14-Citation16 To investigate whether radiation induced autophagy in human breast cancer cells expressing different levels of PTTG1 (), MAP1LC3 expression was examined by western blot analysis. Two forms of MAP1LC3 exist, including cytosolic MAP1LC3-I (autophagy-inactive) and processed MAP1LC3-II (autophagy-active). MAP1LC3-II is localized in the autophagosome membranes, and the level of MAP1LC3-II expression is proportional to the number of autophagic vacuoles.Citation17 We found that radiation increased the ratio of MAP1LC3-II/MAP1LC3-I in MDA-MB-231-2A cells (PTTG1-knockdown MDA-MB-231 cells) and MCF-7 cells, but not in the parental MDA-MB-231 cells (), suggesting that radiation induced autophagy in PTTG1-depleted cancer cells. To confirm that radiation induced autophagy, a plasmid encoding green fluorescent EGFP-MAP1LC3 was transfected into MDA-MB-231-2A cells. Flow cytometry analysis showed that radiation increased the fluorescence of EGFP-MAP1LC3 (). Serum-starved cells were used as a positive control (). Because the recruitment of MAP1LC3-II to the autophagosomes is characterized by a punctate pattern of its subcellular localization,Citation18 we next examined the formation of EGFP-MAP1LC3 puncta by fluorescence microscopy. Approximately 50% of the MDA-MB-231-2A cells developed punctate patterns of EGFP-MAP1LC3 in the cytoplasm after irradiation, as did the serum-starved cells (). In addition, electron microscopy analysis showed more autophagosome-like vacuoles in the cytoplasm of the irradiated MDA-MB-231-2A cells ().
Figure 1. Radiation induced autophagy in MDA-MB-231-2A cells. (A) The levels of PTTG1 in MDA-MB-231, MDA-MB-231-2A, and MCF-7 cells were examined by western blot analysis. (B) MDA-MB-231-2A cells were exposed to different doses of radiation followed by 24 h of recovery time. The ratio of MAP1LC3-II/MAP1LC3-I was analyzed by western blot analysis. (C) EGFP-MAP1LC3-transfected MDA-MB-231-2A cells were exposed to 6-Gy radiation or serum starvation. EGFP intensity was measured by flow cytometry. (D) EGFP-MAP1LC3-transfected MDA-MB-231-2A cells were exposed to 6-Gy radiation or serum starvation. EGFP-MAP1LC3 puncta were measured using fluorescence microscopy. Scale bar: 500 µm. In (C and D), ** indicates significant differences (P < 0.01) between the control and serum starved cells. ## indicates significant differences (P < 0.01) between the control and irradiated cells. (E) MDA-MB-231-2A cells were exposed to 6-Gy radiation, and autophagosome-like structures (arrows) were observed by TEM.
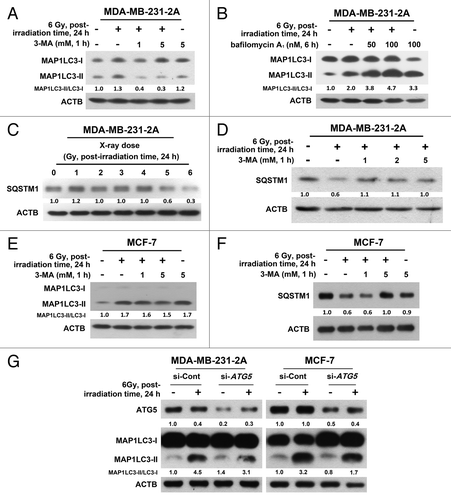
Increased autophagosome formation or impaired autophagosome-lysosome fusion can result in MAP1LC3-II accumulation. To discriminate between these 2 possibilities, MDA-MB-231 cells were treated with a 3-methyladenine (3-MA), a class III phosphatidylinositol 3-kinase (PtdIns3K) inhibitor, to block autophagosome formation, or bafilomycin A1, a vacuolar-type H+-ATPase inhibitor, to block autophagosome-lysosome fusion. As shown in , radiation-induced MAP1LC3-II accumulation was reduced by treatment with 3-MA. However, radiation still enhanced MAP1LC3-II accumulation in the presence of bafilomycin A1 (), suggesting that radiation-induced MAP1LC3-II accumulation was not due to the inhibition of autophagic degradation. SQSTM1/p62 is degraded by autophagy.Citation19 A decrease in SQSTM1 was consistently observed after irradiation (), and this effect can also be blocked by 3-MA (). Similar phenomena were also observed in MCF-7 cells (), although radiation-induced MAP1LC3-II accumulation was only slightly inhibited by 3-MA (). To confirm the effect of 3-MA, an siRNA against ATG5, which is known to initiate the formation of autophagosome,Citation20 was transfected into both MDA-MB-231-2A and MCF-7 cells. Inhibition of ATG5 expression could attenuate radiation-induced MAP1LC3-II accumulation ().
Figure 2. Effect of the autophagic inhibition on radiation-induced autophagy in MDA-MB-231-2A and MCF-7 cells. (A) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. The MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis. (B) MDA-MB-231-2A cells were recovered for 18 h after exposure to 6-Gy radiation and were treated with or without bafilomycin A1 for 6 h. The MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis. (C) MDA-MB-231-2A cells were exposed to different doses of radiation followed by 24 h of recovery time. SQSTM1 expression was measured by western blot analysis. (D) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. After 24 h of recovery time, SQSTM1 expression was measured by western blot analysis. (E and F) MCF-7 cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. After 24 h of recovery time, the MAP1LC3-II/MAP1LC3-I ratio (E) and SQSTM1 expression (F) were measured by western blot analysis. (G) MDA-MB-231-2A and MCF-7 cells were transfected with ATG5 or non-targeting siRNAs for 48 h before exposure to 6-Gy radiation. After 24 h of recovery time, the MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis.
Inhibition of autophagy switches radiation-induced senescence to apoptosis
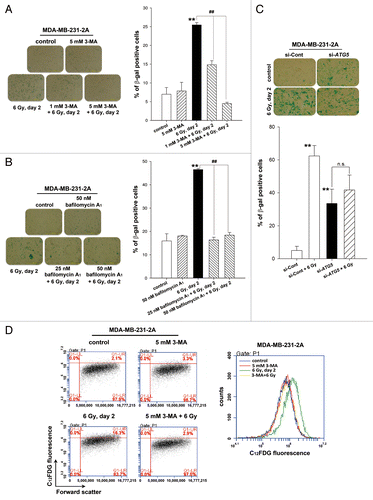
To investigate whether autophagy is necessary for radiation-induced senescence, MDA-MB-231-2A cells were treated with 3-MA and bafilomycin A1 to inhibit autophagy prior to irradiation, and senescence was examined by β-galactosidase staining. As shown in , 25 to 45% of the irradiated cells were undergoing senescence compared with 7% to 16% of the control cells. Radiation-induced senescence was blocked by 3-MA and bafilomycin A1 (). Depletion of ATG5 by transfecting siRNA also inhibited radiation-induced senescence (). Intriguingly, ATG5 knockdown alone increased β-galactosidase activity (). This phenomenon was similar to a previous study reporting that autophagy impairment induces premature senescence in primary human fibroblasts.Citation21 These results were further confirmed by flow cytometry analysis using the fluorescent substrate 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG), which is cleaved by β-galactosidase (). Rapamycin, an autophagy inducer, was also found to induce senescence in MDA-MB-231-2A cells (Fig. S1). These results suggest that the activation of autophagy promotes senescence in MDA-MB-231-2A cells.
Figure 3. Autophagy promoted radiation-induced senescence in MDA-MB-231-2A cells. (A) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. Bright-field microscopy observation and SA-β-gal staining were performed 2 d after irradiation. (B) MDA-MB-231-2A cells were recovered for 18 h after irradiation and were treated with or without bafilomycin A1. The cells were then observed by bright-field microscopy and stained with SA-β-gal 2 d after irradiation. ** indicates significant differences (P < 0.01) between the control and irradiated cells. ## indicates significant differences (P < 0.01) between the inhibitor-treated and untreated cells. (C) MDA-MB-231-2A cells were transfected with ATG5 or nontargeting siRNAs for 48 h before exposure to 6-Gy radiation. Bright-field microscopy observation and SA-β-gal staining were performed 2 d after irradiation. ** indicates significant differences (P < 0.01) compared with si-Cont cells. n.s. indicates no significant differences between the unirradiated and irradiated si-ATG5 cells. (D) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. Cells were stained with C12FDG, and the fluorescence was analyzed by flow cytometry.
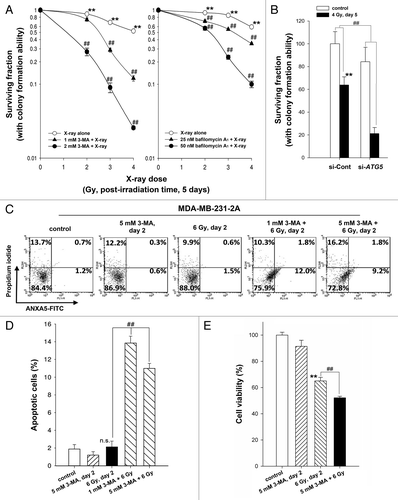
To investigate the role of autophagy in radiosensitivity, cell viability was assessed using a clonogenic survival assay. As shown in , inhibition of autophagy by 3-MA and bafilomycin A1 enhanced cell death in irradiated MDA-MB-231-2A cells. The radiosensitizing effect of pharmacological inhibition of autophagy was confirmed by ATG5 knockdown (). Furthermore, Annexin V (ANXA5)-PI double staining showed that the rate of apoptosis increased to 10% to 13% in the irradiated cells when autophagy was inhibited by 3-MA (). Consistently, the reduction of cell viability by radiation was also enhanced by 3-MA (). These results suggest that autophagy promotes cell survival and plays a decisive role in choosing between radiation-induced senescence and apoptosis. Because the increase of apoptosis induced by autophagy inhibition seems not to be sufficient to explain the extent of sensitization to radiation, other events, such as cell cycle arrest or necrosis, may also occur in parallel. Interestingly, the inhibition of autophagy by 3-MA blocked radiation-induced senescence but did not initiate apoptosis in irradiated MCF-7 cells (Fig. S2). This phenomenon may be due to the lack of CASP3/caspase 3 activity in MCF-7 cells.Citation22
Figure 4. Inhibition of autophagy enhanced the radiosensitivity of MDA-MB-231-2A cells. (A) Left part: MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. Right part: MDA-MB-231-2A cells were recovered for 18 h after irradiation and treated with bafilomycin A1 for 6 h. Then, the cells were washed with PBS, and clonogenic survival assays were performed. ** indicates significant differences (P < 0.01) between the control and irradiated cells. ## indicates significant differences (P < 0.01) between the inhibitor-treated and untreated cells. (B) MDA-MB-231-2A cells were transfected with ATG5 or non-targeting siRNAs for 48 h before exposure to 4-Gy radiation. Then, clonogenic survival assays were performed. ** indicates significant differences (P < 0.01) between the si-Cont and irradiated si-Cont cells. ## indicates significant differences (P < 0.01) between the si-Cont and si-ATG5 cells. (C) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. Apoptotic cell death was measured by ANXA5-PI double staining 2 d after irradiation. ANXA5+ and PI-, as well as ANXA5+ and PI+ cells were quantified in (D). ## indicates significant differences (P < 0.01) between the inhibitor-treated and untreated cells. n.s. indicates no significant differences between the control and irradiated cells. (E) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation. Cell viability was measured by the MTT assay 2 d after irradiation. ** indicates significant differences (P < 0.01) between the control and irradiated cells. ## indicates significant differences (P < 0.01) between the inhibitor-treated and untreated cells.
CSF2 contributes to radiation-induced bystander effects via the JAK2-STAT3 and AKT pathways
Our recent study shows that senescent MDA-MB-231-2A cells exhibited radiation-induced bystander effects through the promotion of invasion and migration of unirradiated MDA-MB-231 cells and HUVECs.Citation13 Cytokine array analysis indicated that radiation-induced senescent MDA-MB-231-2A cells secreted high levels of CSF2/GM-CSF.Citation13 Although CSF2 was originally identified as a hematopoietic growth factor,Citation23 it has been shown to stimulate tumor growth and migration through an autocrine or paracrine mechanism.Citation24-Citation26 A recent study demonstrates that CSF2 is secreted by senescent fibroblasts.Citation27 ELISA analysis was employed to measure the concentration of CSF2 in the conditioned media (CM) from irradiated MDA-MB-231-2A cells, and the results showed that CSF2 is release by senescent MDA-MB-231-2A cells (). However, irradiated MDA-MB-231-2A cells pretreated with 3-MA lose their ability to secrete CSF2 (). Boyden chamber and wound healing assays were used to evaluate invasion and migration, respectively. As shown in , CM from irradiated MDA-MB-231-2A cells (2A-CM) promoted the invasion and migration of unirradiated MDA-MB-231 cells. The 2A-CM-induced bystander effects were blocked when MDA-MB-231-2A cells were pretreated with 3-MA (), which is consistent with the inhibition of senescence by 3-MA (). It is possible that the inhibition of the bystander effects by 3-MA resulted from the induction of apoptosis (). To evaluate this possibility, CM from irradiated MCF-7 cells (MCF-7-CM) with or without 3-MA pretreatment was used because, in these cells, apoptosis was not observed when radiation-induced senescence was inhibited by 3-MA (Fig. S2B). However, as shown in Figure S3, the MCF-7-CM-induced bystander effects were blocked when MCF-7 cells were pretreated with 3-MA. Therefore, based on the results shown in , we investigated whether CSF2 was required for the CM-induced bystander effects using a neutralized antibody to deplete CSF2 from the 2A-CM. As shown in and , CSF2-depleted 2A-CM did not promote the invasion and migration of unirradiated MDA-MB-231 cells. However, addition of the CSF2-neutralized antibody did not have an effect on the 10% serum-stimulated cell invasion and migration (). In addition, an isotype IgG that was used as a negative control did not affect the 2A-CM-induced bystander effects (Fig. S4).
Figure 5. CSF2 contributed to the radiation-induced bystander effects by promoting the invasion and migration of MDA-MB-231 cells. (A) MDA-MB-231-2A cells were pretreated with or without 3-MA before exposure to 6-Gy radiation, and the levels of CSF2 in 2A-CM were measured by ELISA. (B and C), MDA-MB-231 cells were treated with serum-free, 10% serum, 2A-CM, or 2A-CM from MDA-MB-231-2A cells pretreated with 3-MA before exposure to irradiation. In (D and E), CSF2-neutralizing antibody (5 µg/mL) was added to the 2A-CM for 1 h and then incubated with MDA-MB-231 cells. The invasion and migration of MDA-MB-231 cancer cells were measured using a Boyden chamber and a wound-healing assay, respectively. The numbers of the invaded and migrated cells were quantified. In (B and C), ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates significant differences (P < 0.01) between the CM-treated and CM (3-MA)-treated cells. In (D and E), ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates significant differences (P < 0.01) between the CSF2 antibody-treated and untreated cells.
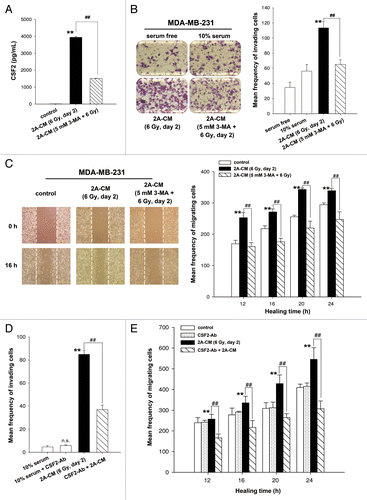
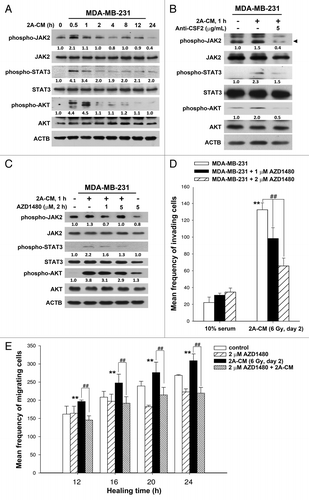
Recent reports show that JAK-STAT pathway is the major signaling cascade downstream from cytokines, chemokines, and growth factor receptors, consequently leading to the migration and invasion of cancer cells.Citation26 In addition, activation of the AKT pathway, which occurs in multiple cancers, promotes cell survival, and is also associated with the enhancement of tumor cell invasion.Citation27 CM from irradiated MDA-MB-231-2A cells was found to induce the phosphorylation of JAK2-STAT3 and AKT in bystander MDA-MB-231 cells (), and this phosphorylation was inhibited by addition of the CSF2 antibody to the CM (). These results also suggest CSF2 in the CM transiently activates the JAK2-STAT3 and AKT pathways, which is similar to the kinetic analysis showing transient JAK2 phosphorylation in CSF2-stimulated peripheral blood polymorphonuclear leukocytes.Citation28 However, the transient activation of JAK2 is enough to phosphorylate downstream proteins and then rapidly modulate gene expression.Citation28 To investigate the role of CSF2 downstream signaling in cell invasion and migration, AZD1480, a small-molecule inhibitor of JAK2, was used to inhibit the phosphorylation of JAK2, STAT3, and AKT in MDA-MB-231 cells (). In addition, the CM-induced cell invasion and migration were inhibited by AZD1480 (). Therefore, the radiation-induced bystander effects resulted from the CSF2-mediated activation of the JAK2-STAT3 and AKT signaling pathways.
Figure 6. CSF2 contributed to the radiation-induced bystander effects via the JAK2-STAT3 and AKT pathways. (A) MDA-MB-231 cells were treated with 2A-CM for the indicated time intervals. The levels of phospho-JAK2, phospho-STAT3, and phospho-AKT were examined by western blot analysis. (B) CSF2-neutralizing antibody (5 µg/mL) was added to the 2A-CM for 1 h and then incubated with MDA-MB-231 cells. phospho-JAK2, phospho-STAT3, and phospho-AKT were examined by western blot analysis. (C–E) MDA-MB-231 cells were treated with or without the JAK2 inhibitor AZD1480 before incubation with 2A-CM. The levels of phospho-JAK2, phospho-STAT3, and phospho-AKT were examined by western blot analysis (C). The invasion (D) and migration (E) of cells were measured using Boyden chamber and wound healing assays, respectively. In (D and E), ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates significant differences (P < 0.01) between the inhibitor-treated and untreated cells.
Inhibition of endogenous autophagy in bystander cells via the CSF2-JAK2 pathway promotes their invasion and migration
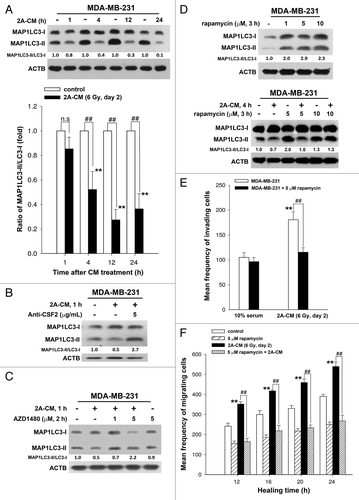
To investigate the role of endogenous autophagy in the radiation-induced bystander effect on unirradiated MDA-MB-231 cells, the MAP1LC3-II/MAP1LC3-I ratio was examined using western blot analysis in 2A-CM-treated MDA-MB-231 cells. As shown in , 2A-CM reduced the MAP1LC3-II/MAP1LC3-I ratio, indicating that endogenous autophagy in bystander cells was downregulated. This phenomenon was blocked when CSF2 in 2A-CM was depleted with an antibody (). The JAK2 inhibitor AZD1480 also rescued the decrease in the MAP1LC3-II/MAP1LC3-I ratio in unirradiated MDA-MB-231 cells (). Rapamycin was used to induce autophagy in unirradiated MDA-MB-231 cells (, upper part). It was found that rapamycin compensated for the loss of MAP1LC3-II/MAP1LC3-I ratio by 2A-CM (, lower part), and inhibited the 2A-CM-induced cell invasion and migration (), indicating that the induction of endogenous autophagy in bystander cells counteracted the radiation-induced bystander effects.
Figure 7. Endogenous autophagy inhibited the radiation-induced bystander effects by promoting the invasion and migration of unirradiated MDA-MB-231 cells. (A) MDA-MB-231 cells were exposed to 2A-CM for the indicated time intervals, and the MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis and quantified at each time point. ** indicates significant differences (P < 0.01) compared with the CM-treated cells at day 1. ## indicates significant differences (P < 0.01) between the control and CM-treated cells at each time points. n.s. indicates no significant differences between the control and CM-treated cells. (B) CSF2-neutralizing antibody (5 µg/mL) was added to the 2A-CM for 1 h and then incubated with MDA-MB-231 cells for 1 h. The MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis. (C) MDA-MB-231 cells were treated with or without AZD1480 before incubation with 2A-CM. The MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis. (D) Upper part: MDA-MB-231 cells were treated with rapamycin for 3 h. Lower part: MDA-MB-231 cells were exposed to 2A-CM with or without rapamycin for 4 h. Western blot analysis was performed to examine the MAP1LC3-II/MAP1LC3-I ratio. (E and F), MDA-MB-231 cells were pretreated with or without rapamycin for 3 h before exposure to 2A-CM. The invasion (E) and the migration (F) of the cells were measured using a Boyden chamber and wound healing assays, respectively. ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates significant differences (P < 0.01) between the rapamycin-treated and untreated cells.
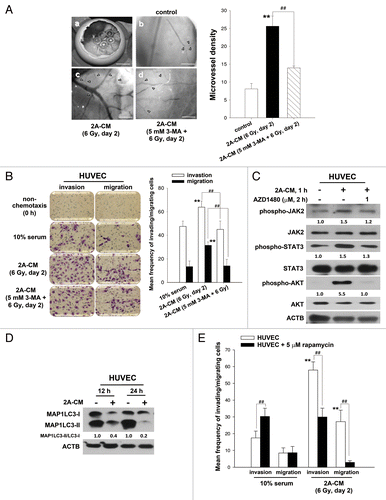
It has been reported that new blood vessels, which consist of endothelial cells, are essential for the delivery of nutrients and oxygen to the tumor microenvironment and for providing a route for angiogenesis and metastasis.Citation29 To investigate whether radiation-induced bystander effects promote angiogenesis, the chicken chorioallantoic membrane (CAM) assay was performed. The results showed that CM induced angiogenesis (). Endothelial cells play a major role in angiogenesis, and it has been demonstrated that HUVECs are able to form capillary-like structures and maintain the phenotype of endothelial cells in vivo, including angiogenesis.Citation29-Citation31 To investigate whether CM increased the invasive or migratory ability of endothelial cells, HUVECs were incubated with CM. We found that CM induced the invasion and migration of HUVECs (). However, CM from irradiated MDA-MB-231-2A cells pretreated with 3-MA was no longer able to promote the invasion and migration of HUVECs () or angiogenesis (). The phosphorylation of JAK2-STAT3 and AKT was also found in CM-treated HUVECs, and this phosphorylation could be blocked by AZD1480 (). Furthermore, endogenous autophagy in HUVECs was downregulated by CM, and activation of autophagy by rapamycin reduced the CM-induced invasion and migration of HUVECs (). Therefore, endogenous autophagy plays an inhibitory role in radiation-induced bystander effects toward unirradiated cancer and endothelial cells.
Figure 8. Endogenous autophagy inhibited the radiation-induced bystander effects by promoting the invasion and migration of HUVECs. (A) Observation window of the fertilized chicken eggs was prepared (a) and incubated with control medium (b), 2A-CM (c) or CM from MDA-MB-231-2A cells pretreated with 3-MA before irradiation (d). Microvessels were observed under a dissecting scope (arrowhead). The method for counting the numbers of microvessels was described in Materials and Methods. Scale bars: (a) 5 mm; (b–d) 2 mm. (B) HUVECs were incubated with CM from MDA-MB-231-2A cells pretreated with or without 3-MA before irradiation. The effects on cell invasion and migration were examined using a Boyden chamber and wound healing assays, respectively. ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates a significant differences (P < 0.01) between the CM- and CM (3-MA)-treated cells. (C) MDA-MB-231-2A cells were pretreated with 3-MA before irradiation. Then, the HUVECs were treated with 2A-CM. The levels of phospho-JAK2, phospho-STAT3 and phospho-AKT were examined by western blot analysis. (D) HUVECs were exposed to 2A-CM, and the MAP1LC3-II/MAP1LC3-I ratio was measured by western blot analysis and quantified at each time point. (E) HUVECs were pretreated with or without rapamycin (5 µM) for 3 h and incubated with 2A-CM. The effects on cell invasion and migration were examined using a Boyden chamber and wound healing assays, respectively. ** indicates significant differences (P < 0.01) between the control (10% serum) and CM-treated cells. ## indicates significant differences (P < 0.01) between the rapamycin-treated and untreated cells.
Discussion
Radiotherapy is effective in various human malignancies by inducing irreparable DNA strand breaks that leads to apoptosis or senescence.Citation32 Our previous studies show that MDA-MB-231-2A (PTTG1-knockdown) and MCF-7 (low PTTG1 expression) breast cancer cells senesce after irradiation, which enhances the radiosensitivity of the cells.Citation12,Citation13 Although senescence definitely accounts for the radiation-induced phenotype, additional pathways, such as autophagy, may be of equal importance.Citation33 In this study, we found that autophagy played an important role in promoting radiation-induced senescence in MDA-MB-231-2A cells. Inhibition of autophagy resulted in the cells undergoing apoptosis instead of radiation-induced senescence; cells undergoing apoptosis could have enhanced radiosensitivity. It appears that apoptosis is a more efficient way to trigger cell death, as inhibition of autophagy and senescence by treatment with 3-MA and bafilomycin A1 enhanced cell death. Therefore, our results may provide a molecular basis for the development of alternative strategies, such as combination therapy with autophagy inhibitors to improve the efficacy of radiotherapy in breast cancer patients. Indeed, it has been shown that concurrent inhibition of autophagy may improve the outcomes of cancer therapy.Citation34 For example, hydroxychloroquine, an autophagy inhibitor that is currently in phase I and phase II clinical trials, has been used in combination with several chemo- and radiotherapies.Citation34
Autophagy has recently been identified as an effector of senescence.Citation35 However, inhibition of autophagy only delays, but does not fully abrogate the senescence phenotypes.Citation36,Citation37 The finding suggests that autophagy plays an accelerating role during cellular senescence. Generally, common stimuli can simultaneously induce apoptosis and autophagy in a mutually exclusive manner, which may be a result of variable thresholds for both processes or from a different cellular decision between the 2 responses, thereby linking their mutual inhibition.Citation38 In the present study, the inhibition of autophagy resulted in irradiated MDA-MB-231-2A cells undergoing apoptosis instead of radiation-induced senescence, which enhanced the radiosensitivity of the cells; this result suggests that radiation induced autophagy in order to promote cell survival. Therefore, autophagy may also play a role in the cell’s decision to undergo apoptosis or senescence.
Differential responses of irradiated breast cancer cells are observed. We and others’ studies show that radiation induces apoptosis in MDA-MB-231 cells, but induces senescence in MCF-7 cells.Citation13,Citation39 This differential effect may depend on their TP53 status because MCF-7 cells express wild-type TP53, and MDA-MB-231 express a mutant form. Indeed, MCF-7/E6 cells with attenuated TP53 function fail to senesce after irradiation and instead exhibit a delayed apoptosis.Citation39 In addition, our studies show that PTTG1-knockdown MDA-MB-231-2A, but not parental MDA-MB-231 cells, can enter senescence in response to radiation.Citation12,Citation13 The finding is intriguing because the TP53 status is not altered in MDA-MB-231 and MDA-MB-231-2A cells. Therefore, genetic status of TP53 and PTTG1 per se seems not to determine the cell fate (i.e., apoptosis or senescence). The downstream events regulated by TP53 and PTTG1, such as cell cycle arrest or autophagy, may play an essential role.
Irradiated cancer cells can induce damage in neighboring unirradiated cells by intracellular gap-junction communication or signals that are released outside of the cells.Citation40 Our previous study indicated that radiation-induced senescent MDA-MB-231-2A cells secreted multiple cytokines and chemokines, including CSF2, CXCL1, IL6 and IL8.Citation13 These factors are involved in multiple functions during cancer progression.Citation41-Citation44 Among them, CSF2 was secreted into the CM at the highest levels by the irradiated MDA-MB-231-2A cells.Citation13 Recent reports show that autophagy can influence the protein expression profile of senescent cells.Citation35 In this study, autophagy inhibition in MDA-MB-231-2A cells significantly decreased the release of CSF2, suggesting that autophagy plays an important role in promoting the secretion of SASPs. In support of this notion, it has been reported that inhibition of autophagy delays the secretion of several senescence-associated cytokines such as IL6 and IL8.Citation45
The tumor microenvironment contains many distinct cell types, including cancer cells and endothelial cells, which are the 2 major groups involved in tumor angiogenesis and metastasis.Citation46 Angiogenesis is the process of new blood vessel formation, which is necessary to deliver nutrients and oxygen to the tumor microenvironment and to provide routes for metastasis.Citation47 A novel finding in this study was that the downregulation of endogenous autophagy in bystander cells was correlated with the bystander effect that promoted the invasion and migration of neighboring MDA-MB-231 cells and HUVECs, and this effect could be counteracted by treatment with rapamycin, which activates autophagy. These results indicated that autophagy might inhibit the metastasis of cancer cells and the angiogenic activity of endothelial cells. Indeed, a deficiency in BECN1, an initiator of autophagy, is associated with increased hypoxia-induced angiogenesis.Citation48 In contrast, BECN1 overexpression inhibits the proliferation, invasion, and migration of cervical cancer cells.Citation49 However, how autophagy influences metastasis and angiogenesis remains unclear. Because autophagy is one of the major intracellular protein degradation systems,Citation50 it may participate in the regulation of metastasis- and angiogenesis-related protein stability. For example, the activation of autophagy has been reported to promote the degradation of SNAI1 and TWIST1, 2 master regulators of the epithelial-mesenchymal transition and metastasis.Citation51
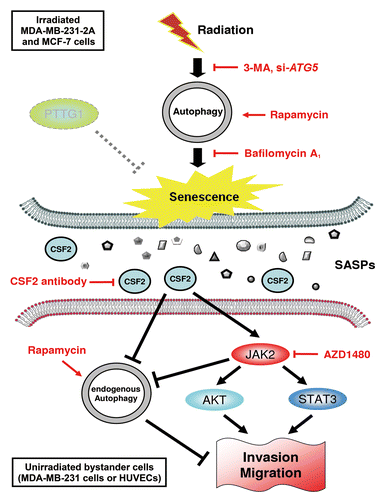
In this study, we provided new insight into the role autophagy plays in radiation-induced senescence and bystander effects. A hypothetic model is proposed in . We found that autophagy in both PTTG1-depleted and -low-expressing human breast cancer cells (MDA-MB-231-2A and MCF-7, respectively) promotes radiation-induced senescence. The radiation-induced senescent cancer cells release CSF2 to promote the invasion and migration of the unirradiated neighboring MDA-MB-231 cells and HUVECs (bystander effects) via the JAK2-dependent AKT and STAT3 pathways. In addition, endogenous autophagy in unirradiated neighboring cells seems to counteract the bystander effects. Therefore, autophagy promotes radiation-induced senescence in irradiated cells but inhibits the bystander effects in human breast cancer cells.
Figure 9. A hypothetical model for the role of autophagy in radiation-induced senescence and bystander effects. Radiation induces autophagy and senescence in PTTG1-depleted breast cancer cells (MDA-MB-231-2A and MCF-7). Inhibition of autophagy by 3-MA and bafilomycin A1 blocks radiation-induced senescence. In contrast, induction of autophagy by rapamycin induces senescence. Conditioned medium (CM) from radiation-induced senescent MDA-MB-231-2A and MCF-7 cells promote the invasion and migration of unirradiated neighboring cancer (MDA-MB-231) and normal endothelial (HUVEC) cells (bystander effects). CSF2 in the CM from the radiation-induced senescent cancer cells acts as senescence-associated secretory phenotype (SASP) to exert the bystander effects. CSF2 promotes cell invasion and migration through either the activation of the JAK2-STAT3 and JAK-AKT pathways or the downregulation of endogenous autophagy in bystander cells.
Materials and Methods
Chemicals and antibodies
3-methyladenine (3-MA; M9281) and bafilomycin A1 (B1793) were purchased from Sigma Chemical. Rapamycin (553210) was purchased from Merck Calbiochem. AZD1480 (S2162) was purchased from Selleck chemicals. MAP1LC3B (2775; 1:1000), SQSTM1 (5114; 1:1000), phospho-JAK2 (Tyr1007/1008, 3771; 1:1000), JAK2 (3230; 1:1000), phospho-STAT3 (Tyr705, 9131; 1:1000), STAT3 (9132; 1:1000), phospho-AKT (Ser473; 9271; 1:1000) and AKT (9272; 1:1000) antibodies were purchased from Cell Signaling Technology. ATG5 (2290254; 1:5000) antibody was purchased from Merck Millipore.
Cell culture
The human breast cancer MDA-MB-231 cell line was grown in DMEM medium (GIBCO, 12800-058) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, 100 μg/mL streptomycin, and 1 mM sodium pyruvate solution. The PTTG1-knockdown MDA-MB-231-2A cells were kindly provided by Dr Ji-Hshiung Chen (Department of Molecular Biology and Human Genetics, Tzu Chi University, Taiwan).Citation12 Human umbilical vein endothelial cells, purchased from Sciencell Research Laboratories (8000), were grown in endothelial cell medium supplemented with 5% FBS, 100 units/mL penicillin and 100 μg/mL streptomycin. The cells were cultured at 37 °C and 5% CO2 in a humidified incubator and were passaged every 2 d.
X-ray irradiation
Radiation was generated using an X-ray machine (RS2000; RAD Source Technologies, Coral Springs, FL) operating at 160 kVp (peak kilovoltage) and 25 mA, as described previously.Citation12 The cells were replenished with fresh medium and immediately subjected to X-ray irradiation. The radiation dosage was chosen for this study according to our previous finding that senescence was induced by various doses of radiation range from 3 to 6 Gy.Citation12
EGFP-MAP1LC3 transfection
MDA-MB-231-2A cells were transfected with 2 µg/mL of PSG5 vector or EGFP-MAP1LC3 plasmid provided by Dr William Jackson (Department of Microbiology and Molecular Genetics, Medical College of Wisconsin, USA) using the FuGENE HD transfection reagent (Roche). Twenty-four hours after transfection, the cells were subjected to irradiation.
siRNA knockdown analyses
ON-TARGET plus SMARTpool human ATG5 siRNA (L-004374-00) and its Non-targeting Pool (D-001810-10-05) were purchased from Thermo Scientific Dharmacon RNAi Technologies. These siRNAs were transiently transfected into cells with Thermo Scientific DharmaFECT 4 siRNA Transfection Reagent (T-2004) according to the manufacturer’s instructions. After 48 h, cells were subjected for other assays.
Flow cytometry and fluorescence microscopy
To observe MAP1LC3′s expression during radiation-induced senescence, EGFP-MAP1LC3-transfected MDA-MB-231-2A cells were exposed to 6-Gy radiation followed by 24 h recovery time. The cells were trypsinized and analyzed by flow cytometry analysis using the Cell Quest software (FACSCalibur, Becton-Dickinson Biosciences). To examine EGFP-MAP1LC3 puncta in irradiated MDA-MB-231-2A cells, the cells were observed using a fluorescence microscope (OLYMPUS IX-71, Olympus, Rungis, France) 24 h after they were irradiated with 6-Gy. For the quantification, cells displaying more than 20 brightly fluorescent EGFP-MAP1LC3 puncta were counted as autophagic cells.
Transmission electron microscopy (TEM)
TEM images were generated by a commercial TEM (Hitachi H07500m, Japan). To prepare the samples, the cells were seeded and allowed to sit for 24 h; the cells were then irradiated with 6-Gy and were allowed to recover for 18 h. After the cells recovered, they were harvested and fixed in 0.1 M phosphate buffer containing 4% formaldehyde for 1 h. The specimens were rinsed in a buffer containing 5% sucrose, post-fixed in 1% osmium solution at 4 °C for 1 h and contrasted with uranyl acetate. The samples were then dehydrated in ethanol followed by acetone and consequently embedded and polymerized using Spurr’s (Emsdiasum). The ready specimens were imaged using the TEM at an operating voltage of 80 kV and with direct magnifications between 25,000× and 100,000×.
Western blot analysis
The preparation of cellular protein extracts and the procedures for western blot analysis were described in our previous study.Citation12
Senescence-associated β-galactosidase (SA-β-gal) and 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG) staining
Senescent cells were analyzed using a β-gal staining kit (Cell Signaling Technology, 9860) in accordance with the manufacturer’s instructions. The percentage of SA-β-gal positive cells was then calculated as described previously.Citation12 In addition, radiation-induced senescence was also observed by C12FDG (Life Technologies, D2893) staining.Citation52 Briefly, MDA-MB-231-2A cells were exposed to 6-Gy radiation followed by a 24 h recovery period. Then, the cells were stained with 33 μM C12FDG for 1 h. The cells were trypsinized and analyzed using an Accuri C6 flow cytometer (Accuri Cytometers, Inc., Ann Arbor, MI) using the CFlow Plus software (Becton-Dickinson Biosciences).
Clonogenic survival assay
MDA-MB-231-2A cells were seeded at a density of 1 × 104 cells per well in 2 mL of culture medium in 6-well plates. After 24 h, the cells were either pretreated with 3-MA before irradiation or treated with bafilomycin A1 for 6 h after radiation and were further incubated for 5 to 7 d. The cells were fixed in a 30% ethanol solution containing 1% crystal violet and visualized using an inverted phase contrast microscope. Twenty random fields were selected, and isolated clusters of more than 30 to 40 cells were counted as a single colony. The relative surviving cell fraction was calculated by dividing the number of colonies from the treated cells by the number of colonies in the control.
MTT assay
Cytotoxicity was measured using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, cells were plated in 24-well plates at a density of 1 × 104 cells/well for 24 h. Cells were then pretreated with 3-MA for 2 h before exposure to 6-Gy radiation. After treatment, the cells were washed once with PBS, and cultured for 2 d. Then, cells were incubated with 0.5 mg/mL of MTT for 3 h. The blue MTT formazan precipitate was dissolved in 100 μL DMSO. The absorbance at 545 nm was measured on a 96-well plate reader (Molecular Dynamics, OPTImax, Sunnyvale, CA). The relative percentage of cell viability was calculated by dividing the absorbance of treated cells by that of the control in each experiment.
Apoptosis detection
Apoptosis induced by autophagy inhibition was determined using the ANXA5-PI double staining assay (BioVision, K101), which stains cells with FITC-conjugated ANXA5 and PI according to the manufacturer’s instructions. MDA-MB-231-2A cells were pretreated with 3-MA then irradiated with 6 Gy. After a post-irradiation period of 2 d, the cells were trypsinized and analyzed by flow cytometry using the Cell Quest software.
Conditioned media and ELISA
To generate the senescent-conditioned media, MDA-MB-231-2A cells were grown in DMEM for 24 h and replenished with fresh medium with or without inhibitors following irradiated with 6-Gy. CM was collected 2 d after radiation and was centrifuged at 2,000 × g for 5 min at 4 °C to remove the cells or cellular debris. An ELISA kit from R&D Systems (DGM00) was used to detect the level of CSF2 in the CM according to the manufacturer’s instructions. Results were obtained from 3 experiments.
Boyden chamber analysis
The ability of the cells to degrade the extracellular matrix is required for their invasive capability.Citation53 Because gelatin can be degraded by the gelatinases that are secreted from invading cells, a gelatin (Sigma, G1393)-coated membrane was used in the Boyden chamber analysis (invasion assay). Cells were grown until they were 80% confluent and were then trypsinized, counted and plated (8000 cells/well) in the upper chambers of the Boyden chambers (Neuro Probe, AP48). The upper chambers contained 1% serum culture media for MDA-MB-231 cells and 5% for the HUVEC. The medium was supplemented with 10% FBS, radiation-induced senescent CM or CM from MDA-MB-231-2A cells pretreated with inhibitors, and this medium was used as the chemoattractant in the lower chamber. After a 16 h incubation, the cells were stained with Giemsa (Merck, 1.09204.0500). In each experiment, the cells were seeded in 6 wells. The cells on the lower surface (invading cells) were counted in one randomly chosen field for each photo image. The mean frequency of the invading cells was averaged from 6 different fields.
Cell migration analysis
Cells were cultured at a density of approximately 8 × 105 cells per well of a 6-well plate for 24 h, resulting in a monolayer that was more than 90% confluent. The monolayer cell was scraped with a 200 μl pipette tip to generate a wound, and the cells were treated with control medium, radiation-induced senescent CM, or CM from MDA-MB-231-2A cells pretreated with inhibitors. Photographs were taken between 12 and 24 h on the same position of the wound. For the migration analysis of the HUVECs, Boyden chamber analysis was performed without coating (transwell migration assay).
The chicken chorioallantoic membrane (CAM) assay
Chicken embryos were purchased from the Animal Health Research Institute (Taiwan, R.O.C.). Briefly, 10-d-old chick embryos were removed from their shells. Sterilized silicone O-rings were placed on the chicken CAM surfaces above the dense subectodermal plexus and 20 μl concentrated CM were concentrated in 10-fold using spin-X UF centrifugal concentrators (Corning). The concentrated CM was then added to the middle of the O-ring and incubated at 37 °C. After 48 h, the angiogenesis of the chicken embryos was examined under a dissecting microscope and photographed. For each sample value, a 0.1 × 0.1-cm grid was added to the digital CAM images, and the average number of vessels in the 7 grids was counted as vascularity. At the end of the experiment, the embryos were sacrificed by opening their air sacs.
Statistical analysis
All data are represented as the mean ± standard error of at least 3 independent experiments. Statistical analysis was performed using a one-way analysis of variance, and further post-hoc testing was performed using the statistical software GraphPad Prism 4 (Graph-Pad Software).
| Abbreviations: | ||
| 3MA | = | 3-methyladenine |
| ACTB | = | actin, beta |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| BECN1 | = | Beclin 1, autophagy-related |
| C12FDG | = | 5-dodecanoylaminofluorescein di-β-D-galactopyranoside |
| CAM | = | chicken chorioallantoic membrane |
| CM | = | conditioned media |
| CSF2/GM-CSF | = | colony stimulating factor 2 (granulocyte-macrophage) |
| CXCL1/GRO-α | = | chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) |
| FBS | = | fetal bovine serum |
| HUVECs | = | human umbilical vein endothelial cells |
| IL6 | = | interleukin 6 (interferon, beta 2) |
| IL8 | = | interleukin 8 |
| JAK2 | = | Janus kinase 2 |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MDA-MB-231-2A | = | PTTG1-knockdown MDA-MB-231 cells |
| PTTG1/securin | = | pituitary tumor-transforming 1 |
| SA-β-gal | = | senescence-associated β-galactosidase |
| SASPs | = | senescence-associated secretory phenotypes |
| SNAI1 | = | snail family zinc finger 1 |
| STAT3 | = | signal transducer and activator of transcription 3 (acute-phase response factor) |
| SQSTM1/p62 | = | sequestosome 1 |
| TEM | = | transmission electron microscopy |
| TWIST1 | = | twist family bHLH transcription factor 1 |
Additional material
Download Zip (2.3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a research grant from the National Science Council of Taiwan (NSC99-2314-B-320-004-MY3 and NSC102-2628-B-320-002-MY3) and Tzu Chi Research grant (TCRPP102005).
References
- Verheij M. Clinical biomarkers and imaging for radiotherapy-induced cell death. Cancer Metastasis Rev 2008; 27:471 - 80; http://dx.doi.org/10.1007/s10555-008-9131-1; PMID: 18470482
- Ren JL, Pan JS, Lu YP, Sun P, Han J. Inflammatory signaling and cellular senescence. Cell Signal 2009; 21:378 - 83; http://dx.doi.org/10.1016/j.cellsig.2008.10.011; PMID: 18992324
- Tschöp K, Conery AR, Litovchick L, Decaprio JA, Settleman J, Harlow E, Dyson N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev 2011; 25:814 - 30; http://dx.doi.org/10.1101/gad.2000211; PMID: 21498571
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547 - 56; http://dx.doi.org/10.1083/jcb.201009094; PMID: 21321098
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99 - 118; http://dx.doi.org/10.1146/annurev-pathol-121808-102144; PMID: 20078217
- Morgan WF, Sowa MB. Non-targeted bystander effects induced by ionizing radiation. Mutat Res 2007; 616:159 - 64; http://dx.doi.org/10.1016/j.mrfmmm.2006.11.009; PMID: 17134726
- Choi KS. Autophagy and cancer. Exp Mol Med 2012; 44:109 - 20; http://dx.doi.org/10.3858/emm.2012.44.2.033; PMID: 22257886
- Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol 2009; 3:366 - 75; http://dx.doi.org/10.1016/j.molonc.2009.05.007; PMID: 19559660
- Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev 2007; 28:165 - 86; http://dx.doi.org/10.1210/er.2006-0042; PMID: 17325339
- Chiu SJ, Chao JI, Lee YJ, Hsu TS. Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells. Toxicol Lett 2008; 179:63 - 70; http://dx.doi.org/10.1016/j.toxlet.2008.04.004; PMID: 18499365
- Panguluri SK, Yeakel C, Kakar SS. PTTG: an important target gene for ovarian cancer therapy. J Ovarian Res 2008; 1:6; http://dx.doi.org/10.1186/1757-2215-1-6; PMID: 19014669
- Chen WS, Yu YC, Lee YJ, Chen JH, Hsu HY, Chiu SJ. Depletion of securin induces senescence after irradiation and enhances radiosensitivity in human cancer cells regardless of functional p53 expression. Int J Radiat Oncol Biol Phys 2010; 77:566 - 74; http://dx.doi.org/10.1016/j.ijrobp.2009.12.013; PMID: 20457353
- Yu YC, Yang PM, Chuah QY, Huang YH, Peng CW, Lee YJ, Chiu SJ. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci Rep 2013; 3:1675; http://dx.doi.org/10.1038/srep01675; PMID: 23591770
- Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol 2010; 22:234 - 40; http://dx.doi.org/10.1016/j.ceb.2009.12.005; PMID: 20045302
- Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy 2009; 5:1046 - 7; http://dx.doi.org/10.4161/auto.5.7.9444; PMID: 19652542
- Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest 2010; 90:835 - 43; http://dx.doi.org/10.1038/labinvest.2010.56; PMID: 20212459
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 2011; 6:e23367; http://dx.doi.org/10.1371/journal.pone.0023367; PMID: 21858089
- Essmann F, Engels IH, Totzke G, Schulze-Osthoff K, Jänicke RU. Apoptosis resistance of MCF-7 breast carcinoma cells to ionizing radiation is independent of p53 and cell cycle control but caused by the lack of caspase-3 and a caffeine-inhibitable event. Cancer Res 2004; 64:7065 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-04-1082; PMID: 15466201
- Williams ME, Quesenberry PJ. Hematopoietic growth factors. Hematol Pathol 1992; 6:105 - 24; PMID: 1429340
- Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol 1999; 155:1557 - 67; http://dx.doi.org/10.1016/S0002-9440(10)65472-7; PMID: 10550313
- Obermueller E, Vosseler S, Fusenig NE, Mueller MM. Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res 2004; 64:7801 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-03-3301; PMID: 15520186
- Pei XH, Nakanishi Y, Takayama K, Bai F, Hara N. Granulocyte, granulocyte-macrophage, and macrophage colony-stimulating factors can stimulate the invasive capacity of human lung cancer cells. Br J Cancer 1999; 79:40 - 6; http://dx.doi.org/10.1038/sj.bjc.6690009; PMID: 10408691
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99 - 118; http://dx.doi.org/10.1146/annurev-pathol-121808-102144; PMID: 20078217
- Brizzi MF, Aronica MG, Rosso A, Bagnara GP, Yarden Y, Pegoraro L. Granulocyte-macrophage colony-stimulating factor stimulates JAK2 signaling pathway and rapidly activates p93fes, STAT1 p91, and STAT3 p92 in polymorphonuclear leukocytes. J Biol Chem 1996; 271:3562 - 7; http://dx.doi.org/10.1074/jbc.271.7.3562; PMID: 8631962
- Zhu W, Fu A, Hu J, Wang T, Luo Y, Peng M, Ma Y, Wei Y, Chen L. 5-Formylhonokiol exerts anti-angiogenesis activity via inactivating the ERK signaling pathway. Exp Mol Med 2011; 43:146 - 52; http://dx.doi.org/10.3858/emm.2011.43.3.017; PMID: 21297378
- Chen Z, Htay A, Dos Santos W, Gillies GT, Fillmore HL, Sholley MM, Broaddus WC. In vitro angiogenesis by human umbilical vein endothelial cells (HUVEC) induced by three-dimensional co-culture with glioblastoma cells. J Neurooncol 2009; 92:121 - 8; http://dx.doi.org/10.1007/s11060-008-9742-y; PMID: 19039523
- Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M. Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J Biol Chem 2004; 279:18600 - 7; http://dx.doi.org/10.1074/jbc.M311643200; PMID: 14985355
- Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells: towards novel cytoprotection and radio-sensitization policies?. Autophagy 2009; 5:442 - 50; http://dx.doi.org/10.4161/auto.5.4.7667; PMID: 19164950
- Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009; 5:1232 - 4; http://dx.doi.org/10.4161/auto.5.8.9896; PMID: 19770583
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol 2010; 22:234 - 40; http://dx.doi.org/10.1016/j.ceb.2009.12.005; PMID: 20045302
- Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, Rodier F, Davalos AR, Gewirtz DA. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep?. J Pharmacol Exp Ther 2012; 343:763 - 78; http://dx.doi.org/10.1124/jpet.112.197590; PMID: 22927544
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798 - 803; http://dx.doi.org/10.1101/gad.519709; PMID: 19279323
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Jones KR, Elmore LW, Jackson-Cook C, Demasters G, Povirk LF, Holt SE, Gewirtz DA. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol 2005; 81:445 - 58; http://dx.doi.org/10.1080/09553000500168549; PMID: 16308915
- Rzeszowska-Wolny J, Przybyszewski WM, Widel M. Ionizing radiation-induced bystander effects, potential targets for modulation of radiotherapy. Eur J Pharmacol 2009; 625:156 - 64; http://dx.doi.org/10.1016/j.ejphar.2009.07.028; PMID: 19835860
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547 - 56; http://dx.doi.org/10.1083/jcb.201009094; PMID: 21321098
- Huang WL, Yeh HH, Lin CC, Lai WW, Chang JY, Chang WT, Su WC. Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol Cancer 2010; 9:309; http://dx.doi.org/10.1186/1476-4598-9-309; PMID: 21122157
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001; 11:S27 - 31; http://dx.doi.org/10.1016/S0962-8924(01)02151-1; PMID: 11684439
- Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 2009; 9:81 - 94; http://dx.doi.org/10.1038/nrc2560; PMID: 19132009
- Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest 2010; 90:835 - 43; http://dx.doi.org/10.1038/labinvest.2010.56; PMID: 20212459
- Lorusso G, Rüegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol 2008; 130:1091 - 103; http://dx.doi.org/10.1007/s00418-008-0530-8; PMID: 18987874
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 2003; 3:401 - 10; http://dx.doi.org/10.1038/nrc1093; PMID: 12778130
- Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011; 7:829 - 39; http://dx.doi.org/10.4161/auto.7.8.15598; PMID: 21685724
- Sun Y, Liu JH, Sui YX, Jin L, Yang Y, Lin SM, Shi H. Beclin1 overexpression inhibitis proliferation, invasion and migration of CaSki cervical cancer cells. Asian Pac J Cancer Prev 2011; 12:1269 - 73; PMID: 21875280
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1 - 32; http://dx.doi.org/10.1007/978-3-642-00302-8_1; PMID: 19802558
- Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu ZW. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res 2012; 72:3238 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-11-3832; PMID: 22719072
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:1798 - 806; http://dx.doi.org/10.1038/nprot.2009.191; PMID: 20010931
- Artym VV, Yamada KM, Mueller SC. ECM degradation assays for analyzing local cell invasion. Methods Mol Biol 2009; 522:211 - 9; http://dx.doi.org/10.1007/978-1-59745-413-1_15; PMID: 19247615