
Abstract
Pharmacological activation of autophagy is becoming an attractive strategy to induce the selective degradation of aggregate-prone proteins. Recent evidence also suggests that autophagy impairment may underlie the pathogenesis of several neurodegenerative diseases. Mutations in the gene encoding SOD1 (superoxide disumutase 1) trigger familial amyotrophic lateral sclerosis (ALS), inducing its misfolding and aggregation and the progressive loss of motoneurons. It is still under debate whether autophagy has a protective or detrimental role in ALS. Here we evaluate the impact of BECN1/Beclin 1, an essential autophagy regulator, in ALS. BECN1 levels were upregulated in both cells and animals expressing mutant SOD1. To evaluate the impact of BECN1 to the pathogenesis of ALS in vivo, we generated mutant SOD1 transgenic mice heterozygous for Becn1. We observed an unexpected increase in life span of mutant SOD1 transgenic mice haploinsufficient for Becn1 compared with littermate control animals. These effects were accompanied by enhanced accumulation of SQSTM1/p62 and reduced levels of LC3-II, and an altered equilibrium between monomeric and oligomeric mutant SOD1 species in the spinal cord. At the molecular level, we detected an abnormal interaction of mutant SOD1 with the BECN1-BCL2L1 complex that may impact autophagy stimulation. Our data support a dual role of BECN1 in ALS and depict a complex scenario in terms of predicting the effects of manipulating autophagy in a disease context.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common fatal paralytic disease affecting the adult population involving the selective degeneration of upper and lower motoneurons from the spinal cord, cortex, and brainstem. The loss of motoneurons results in progressive muscle weakness, atrophy, paralysis, and generally an early death of the patient.Citation1,Citation2 Alterations in more than 15 genes have been identified in familial ALS (fALS) (reviewed in ref. Citation3), including mutations in SOD1 (superoxide dismutase 1, soluble),Citation4 TARDBP/TDP43 (TAR DNA binding protein),Citation5,Citation6 FUS (fused in sarcoma),Citation7,Citation8 and the recently defined hexanucleotide repeat expansions in the intronic region of C9orf72.Citation9 Mutations in the genes encoding SOD1 together with C9orf72 account for at least 50% of all fALS cases.Citation9-Citation11 Since the discovery of the association between SOD1 and fALS, more than 160 point mutations have been reported in the SOD1 gene to cause fALS.Citation12 In addition, misfolding of wild-type SOD1 has recently been reported in spinal cord motoneurons of sporadic ALS cases.Citation12-Citation14
Although the pathogenic mechanism underlying ALS is not fully understood, several cellular perturbations have been identified, including mitochondrial dysfunction, altered axonal transport, neuroinflammation, exitotoxicity, endoplasmic reticulum (ER) stress, and accumulation of ubiquitinated protein inclusions (reviewed in refs. Citation11 and Citation12). The presence of mutant SOD1 aggregates in spinal cord tissue is a common characteristic of all mutant SOD1 transgenic mice generated so far, and this is thought to be a fundamental event triggering its harmful effects.Citation2,Citation15
Macroautophagy (referred to hereafter as autophagy) is the main catabolic mechanism of the cell involved in the turnover of organelles and long-lived proteins. Autophagy is characterized by the formation of autophagosomes, double-membrane vesicles that sequester cargo material that is then degraded and recycled after fusion to lysosomes.Citation16,Citation17 A general consensus in the field indicates that mutant proteins involved in proteinopathies are efficient substrates for autophagy-mediated degradation, as demonstrated, for example, for mutant SOD1 and TARDBP.Citation18-Citation20 Alterations in the levels of autophagy markers have been extensively described in most cellular and animal models of ALS (examples in refs. Citation18, Citation19, and Citation21–Citation24), in addition to human postmortem samples derived from patients.Citation23,Citation25 Recent reports suggest that the pharmacological stimulation of autophagy may have protective effects on ALS. For example, we have recently reported that activation of MTOR-independent autophagy delays experimental ALS in mutant SOD1 transgenic mice.Citation26 In contrast, inhibition of MTOR with rapamycin treatment has no effectsCitation27 or even detrimental consequencesCitation28 on ALS progression in a similar model. Further in contrast, stimulation of both MTOR-dependent and independent autophagy correlates with neuroprotection in TARDBP transgenic mice.Citation18,Citation29,Citation30 However, none of these studies have directly demonstrated that autophagy mediated the effects on ALS progression in vivo because the drugs used target many cellular processes in addition to autophagy. Direct manipulation of essential autophagy regulators is needed to address the functional involvement of the pathway in ALS.
Growing evidence suggests that autophagy impairment may contribute to neurodegeneration as proposed, for example, in Alzheimer (AD)Citation31,Citation32 and Huntington (HD)Citation33 disease (reviewed in refs. Citation34 and Citation35). Genetic inactivation of specific autophagy genes, Atg5 and Atg7, in the central nervous system results in spontaneous neurodegeneration, associated with accumulation of protein aggregates in the brain, motor dysfunction, and extensive neuronal loss.Citation36-Citation38 Interestingly, mutations in some genes associated with ALS, Chmp2b (charged multivesicular body protein 2B) and the lipid phosphatase FIG4, trigger modifications in the autophagy pathway.Citation39,Citation40 Moreover, recent genetic screenings in ALS patients also have identified alterations in SQSTM1/p62 and UBQLN2, proteins that participate in the regulation of the autophagy pathway.Citation41-Citation43 These data suggest that perturbation in the proteostasis network may contribute to the etiology of ALS.
BECN1 is one of the first proteins described to participate in the mammalian autophagy pathwayCitation44 and is an essential regulator of the class III phosphatidylinositol 3-kinase (henceforth PtdIns3K, whose catalytic subunit is encoded by PIK3C3) in a complex involved in the nucleationCitation45 and expansion steps during autophagosome formation.Citation46,Citation47 Recently, it has been shown that autophagy activation is stimulated by a direct phosphorylation of BECN1 by ULK1.Citation48 BECN1 is also negatively regulated by a physical association with BCL2L1/BCLXL or BCL2.Citation49 Besides, BECN1 may also regulate ATG5-ATG7-independent autophagy,Citation50 placing BECN1 as an integrator of different stimuli engaging autophagy. Cellular levels of BECN1 have been correlated with autophagy activity and its decreased expression and/or availability is associated with normal aging,Citation51 increased vulnerability to cancer,Citation52 and to some neurodegenerative disease such as ADCitation53 and HD.Citation51 In ALS, however, the role of BECN1 has not been investigated. Although Becn1 knockout mice are embryonic lethalCitation54,Citation55 Becn1 haploinsufficient animals have a reduction in autophagy activity and have been employed to demonstrate a functional contribution of autophagy in models of AD,Citation53 hypoxia,Citation56 exercise and glucose metabolism,Citation57 cardiac diseases,Citation58 and cancer.Citation54,Citation55,Citation59 Based on this evidence, here we investigated the impact of manipulating BECN1 levels in the context of ALS in cellular and animal models of the disease. Our results revealed an unexpected scenario where, although Becn1 haploinsufficiency had an impact upon autophagy and SOD1 levels, it actually led to protection against experimental ALS, with a significant delay in the onset of the disease and prolonged life span. At the molecular level, we identified a novel interaction between mutant SOD1 and the BECN1-BCL2L1 complex that may alter the threshold for autophagy induction. Our results uncover a complex involvement of BECN1 on ALS where it may have distinct effects on specific diseases.
Results
Upregulation of BECN1 expression in an animal model of ALS
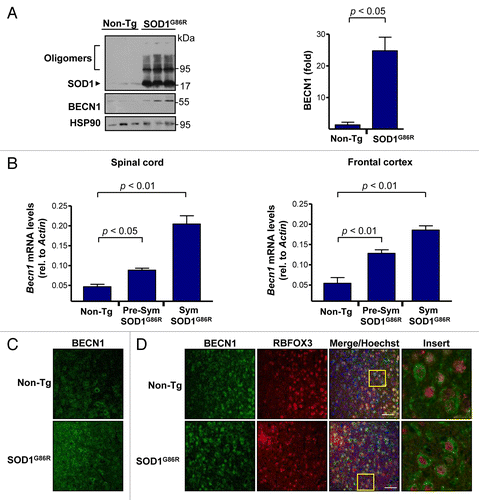
To study the possible contribution of BECN1 to ALS pathogenesis, we first measured the expression levels of BECN1 in mutant SOD1 transgenic mice. We employed the SOD1G86R transgenic mice (the equivalent of human SOD1G85R) because this model drives its expression by the endogenous promoter, and encodes an enzyme with minimal SOD1 activity.Citation23,Citation60 These mice begin the symptomatic phase at approximately 100 d old in a pure C57BL/6 background as we recently described,Citation61 and the disease progresses until almost complete paralysis at 130 to 180 d old. Western blot analysis of spinal cord extracts confirmed the presence of mutant SOD1 oligomers in the model at the symptomatic stage of the disease (). In the same extracts, we observed a significant induction of BECN1 levels in SOD1G86R mice when compared with littermate control nontransgenic animals (, right panel). These effects were also recapitulated when we quantified Becn1 mRNA levels by real-time PCR in total cDNA obtained from spinal cord tissue of animals at both the presymptomatic and late-symptomatic stages of the disease (, left panel). Similar changes were observed in the frontal cortex of the same animals (, right panel). We also determined the expression pattern of BECN1 using histological analysis of brain cortex. A global upregulation of BECN1 levels was detected in the ALS mice using immunofluorescence (), showing a characteristic morphological pattern resembling neurons. To confirm these results, we costained neurons with RBFOX3/NeuN (neuronal nuclei) and BECN1 and observed a high degree of coexpression of this marker and BECN1 (). Staining of astrocytes with anti-GFAP did not reveal significant coexpression with BECN1 in mutant SOD1 transgenic mice (data not shown). Thus, BECN1 levels are induced in neurons of our ALS mouse model.
Figure 1. Increased levels of BECN1 in the spinal cord and frontal cortex of transgenic SOD1G86R mice. (A) SOD1 oligomers, BECN1, and HSP90 expression were determined in spinal cord protein extracts from symptomatic SOD1G86R transgenic (end point of disease) or non-transgenic litter mate control mice (Non-Tg) by western blot. Each lane represents an independent mouse. HSP90 was monitored as a loading control. Right panel: quantification of BECN1 expression levels in spinal cord of transgenic SOD1G86R mice and non-transgenic age-matched controls. Mean and standard error are shown for 3 independent animals per group. (B) In parallel, Becn1 mRNA levels were measured by real-time PCR in the SOD1G86R transgenic mice at the presymptomatic stage (Presym; 90 d after birth) or end point of disease (Sym) in spinal cord (left panel) and in frontal cortex (right panel). Mean and standard error are shown for the analysis of 3 animals per group. (C) Immunofluorescence assay of BECN1 and RBFOX3 staining were performed in frontal cortex tissue derived from SOD1G86R and non-transgenic litter-mate control mice (Non-Tg). A merged image of the triple staining is presented. (D) Using samples described in (C), the expression of BECN1 and the neuronal marker RBFOX3 were analyzed by indirect immunofluorescence followed by confocal microscopy analysis. The right panel shows a magnification of the area marked with a yellow square. A representative image is presented of the analysis of at least 3 independent animals for genotype. Scale bars: 50 μm.
Generation of mutant SOD1G86R transgenic mice heterozygous for Becn1
We then investigated the possible impact of manipulating BECN1 levels in vivo in the context of ALS. As mentioned, becn1 knockout mice are embryonic lethal;Citation54,Citation55 however, Becn1 heterozygous animals have been employed to manipulate autophagy levels in different settings. We generated SOD1G86R mice heterozygous for BECN1 by cross-breeding SOD1G86R transgenic mice and Becn1 heterozygotes. From this crossing strategy, we studied 4 different genotypes: wild-type animals (Becn1+/+), BECN1 heterozygotes (Becn1+/−), SOD1G86R transgenic mice (Becn1+/+ SOD1G86R), and SOD1G86R transgenic mice that are heterozygous for BECN1 (Becn1+/− SOD1G86R). We confirmed the decrease in BECN1 levels in Becn1+/− mice (Fig. S1A and S1B). Using this approach we obtained 140 animals and the rate of generation of Becn1+/− SOD1G86R mice was almost normal ().
Table 1. Generation of mutant SOD1 transgenic mice that are heretozygous for Becn1
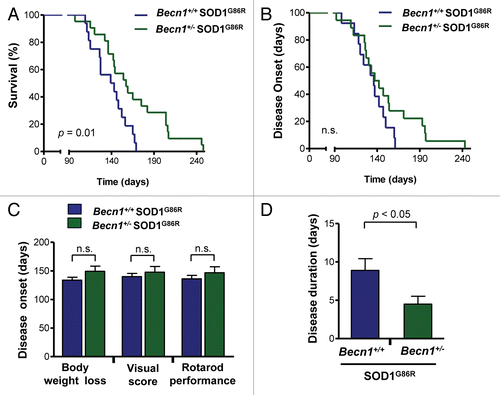
Since we were able to generate viable Becn1+/− SOD1G86R mice, we monitored the progression of experimental ALS by analyzing life span and disease onset. Based on previous findings we expected that targeting Becn1 should exacerbate the development of ALS. Contrary to our prediction, we observed that Becn1 haploinsufficiency significantly prolonged the survival of SOD1G86R mice in a large group of animals (, P = 0.01). Mutant transgenic SOD1 mice showed a survival curve with low variability and with an average life span of 141 d, whereas Becn1+/− SOD1G86R mice had an average survival of 154.5 d with individual animals living from 60 to 104 d longer than the average Becn1+/+ SOD1G86R mice. To monitor disease onset of mutant SOD1 transgenic mice, we analyzed the disease progression by measuring body weight loss, decrease in motor performance by the rotarod assay, and visual appearance of a variety of disease signs (visual symptoms) (see details in Materials and Methods). Mice were monitored once a week starting at 60 d old, and then after 90 d tested every 2 d until paralysis of the hind limbs was observed (end-stage of disease), when the animals were sacrificed. The positive effects of Becn1 heterozygosity in the survival curve were associated with a tendency toward delayed disease onset as measured by body weight loss (; Fig. S1C). Although calculation of the average disease onset did not show significant differences between genotypes using various tests (), analysis of the duration of the symptomatic phase of the disease indicated a shortened symptomatic phase in Becn1+/− SOD1G86R mice as measured with the rotarod assay (). Taken together, these results uncover an unanticipated impact of BECN1 in ALS, where its expression may have detrimental effects on disease progression.
Figure 2.Becn1 haploinsufficiency prolongs life span of mutant SOD1 transgenic mice. (A) Life-span curves in SOD1 transgenic mice (SOD1G86R) on a Becn1 wild-type (Becn1+/+ SOD1G86R, n = 16) or heterozygote (Becn1+/− SOD1G86R, n = 21) background (P = 0.01 obtained by Kaplan-Meier statistics). (B) In the same animals presented in A, disease onset was measured as the loss of 5% of animal total body weight. (C) Average disease onset was calculated by 3 different parameters as described in Materials and Methods: rotarod performance, visual observation of disease signs, and the loss of 5% of animal total body weight. The Student t test analysis indicated no differences between groups. Average and standard error are presented. (D) The duration of the disease was calculated using the measurements of rotarod performance and the survival curve. Average and standard error is presented. P value was determined by the Student t test.
Becn1 haploinsufficiency alters mutant SOD1 oligomerization
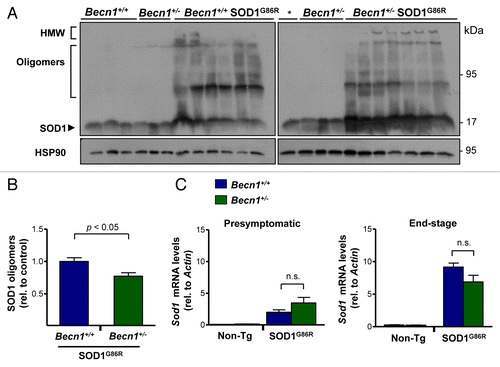
We then evaluated the accumulation of mutant SOD1 aggregates in spinal cord extracts from mutant SOD1 transgenic mice. Western blot analysis of protein extracts prepared in 1% NP40 in the absence of a reducing agent resolved different SOD1 species including monomers, oligomers, and high-molecular weight (HMW; stacking gel) aggregates (). Unexpectedly, we observed a distinct effect of targeting BECN1 on SOD1 aggregation. Becn1+/− SOD1G86R animals showed enhanced accumulation of monomeric forms of mutant SOD1 (), whereas the oligomeric species were reduced (). As predicted, the levels of HMW species were also increased in Becn1+/− SOD1G86R mice (). As a control, we confirmed that the mRNA expression of the Sod1 transgene was not altered in Becn1+/− SOD1G86R mice in presymptomatic or end-stage of the disease, as measured by real-time PCR (). Interestingly, increased levels of Sod1 mRNA were observed in mutant SOD1 transgenic mice as the disease progresses.
Figure 3. Targeting Becn1 alters SOD1 aggregation in ALS transgenic mice. (A) SOD1 aggregation was determined in spinal cord protein extracts derived from Becn1+/+ SOD1G86R and Becn1+/− SOD1G86R mice at the symptomatic stage using western blot analysis. Each well represents an independent animal ordered by crescent genotype and crescent life span. Of note SOD1 oligomers and high molecular (HMW) aggregates are detected in this analysis. “*” represents the same sample from the first lane of Becn1+/+ SOD1G86R mice presented in the left western blot used as a normalization marker to select equivalent film exposure for proper comparison. (B) Mutant SOD1 oligomers were quantified from experiments presented in (A). For the analysis, the average signal of control transgenic mice (*) was normalized to 1 to compare both gels. (C) In parallel, Sod1 mRNA levels were measured by real-time PCR in samples from presymptomatic and end-of- disease stages (right panel). Actin levels were monitored for normalization of gene expression. In (B) and (C), mean and standard error are presented for the analysis of independent animals (at least n = 6 per group). P values were calculated with the Student t test. n.s., nonsignificant differences.
Altered autophagy levels in Becn1+/− SOD1G86R mice
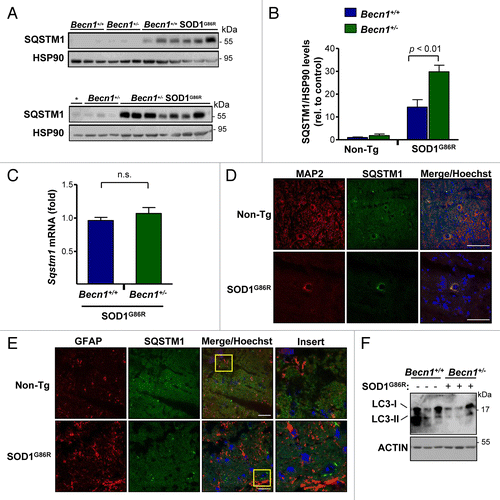
To confirm the impact of Becn1 haploinsufficiency on autophagy levels in our mouse model, we measured the expression of SQSTM1 (also known as p62).Citation62,Citation63 SQSTM1 is a well-described autophagy substrate, and operates as an adaptor protein that acts as a bridge between aggregates and autophagy clearance, and its levels are increased when autophagy is impaired. Analysis of SQSTM1 levels in spinal cord extracts from symptomatic SOD1G86R mice indicated a significant accumulation when compared with non-transgenic animals as described before.Citation21,Citation26,Citation64 Remarkably, SQSTM1 levels were further increased in Becn1+/− SOD1G86R animals (), showing a 2-fold enhancement that was statistically significant (). As a control, we monitored Sqstm1 mRNA levels using real-time PCR, which showed no differences in the expression levels in Becn1+/+ SOD1G86R compared with Becn1+/− SOD1G86R animals (). We then defined the expression pattern of SQSTM1 using histological analysis. SQSTM1 expression was in general restricted to neurons as assessed after costaining with the neuronal marker MAP2 (), compared with astrocytes (). To complement these studies, the levels of MAP1LC3B (hereafter LC3; microtubule-associated protein 1 light chain 3 β) were also measured by western blot. We observed a clear reduction of the lipidated LC3-II form of near 80% on average in Becn1+/− SOD1G86R mice when compared with SOD1G86R control animals (). Finally, we also monitored the levels of BCL2L1 and BCL2, 2 negative regulators of BECN1, in our experimental groups and did not detect any changes between groups (Fig. S1D and S1E).
Figure 4. Altered levels of autophagy markers in transgenic SOD1 mice heterozygous for Becn1. (A) Levels of SQSTM1 were monitored in spinal cord extracts of symptomatic SOD1G86R transgenic mice by western blot and quantified by densitometry. Each lane represents a single mouse, ordered by genotype and then by life span (incremented values). “*” represents the same sample from the first lane of a Becn1+/+ SOD1G86R mouse shown in the left gel used as a normalization sample. (B) SQSTM1 levels were quantified from samples described in (A). Statistical analysis was performed using a one-way ANOVA test; data represent mean and standard error of at least 4 independent animals per genotype. (C) Sqstm1 mRNA levels were measured by real-time PCR in samples presented in (A). Mean and standard error is shown. n.s., nonsignificant differences. (D) Immunofluorescence analysis of SQSTM1 (green) and MAP2 staining (neuronal marker, red) or (E) SQSTM1 and GFAP staining (astrocytes, red) was performed in spinal cord tissue derived from SOD1G86R and non-transgenic littermate control mice (Non-Tg). Hoechst staining was also performed (blue). A merged image of the triple staining is presented together with a zoom of the selected area (yellow square). A representative image is presented of the analysis of 3 independent animals for genotype. Scale bars: 10 µm. (F) Levels of LC3-I and II were evaluated by western blot in spinal cord samples from Becn1+/+ SOD1G86R and Becn1+/− SOD1G86R mice. The western blot for LC3-I and II was performed in a 17% SDS-PAGE. Each well represents an independent animal analyzed.
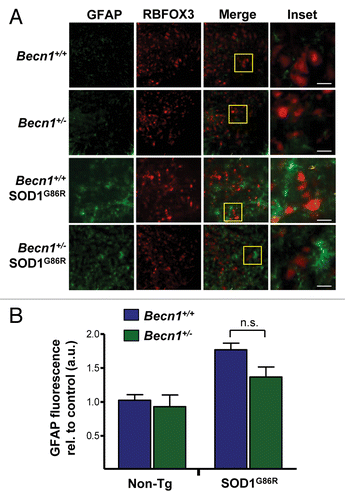
We also performed histological characterization of the spinal cord of mutant SOD1 transgenic mice to visualize glial activation and motoneuron survival. We determined the levels of astrocyte activation after GFAP staining and microscopy in both Becn1+/− SOD1G86R and control animals at the late stage of the disease (). There was only a trend of protection in Becn1+/− SOD1G86R mice after quantifying GFAP signal (). RBFOX3 staining was performed to visualize neuronal survival in the ventral horn of the spinal cord. No differences in the neuronal content of Becn1+/+ SOD1G86R and Becn1+/− SOD1G86R mice were observed in late-symptomatic animals (). In addition, we evaluated the levels of GFAP and RBFOX3 in spinal cord of SOD1G86R mice in presymptomatic stage of the disease in animals of the same age (Fig. S2). We did not observe any differences between Becn1+/+ SOD1G86R and Becn1+/− SOD1G86R animals.
Figure 5. Astrocyte and neuronal content in SOD1G86R mice. (A) Immunofluorescence assay of GFAP (astrocyte marker) and RBFOX3 (neuronal marker) staining was performed in spinal cord tissue derived from Becn1+/+,Becn1+/−, Becn1+/+ SOD1G86R, and Becn1+/− SOD1G86R at the late stage of disease. Hoechst staining was also performed. A merged image of the triple staining is presented together with a zoom of the selected area (yellow square). A representative image is presented of the analysis of 3 independent animals for genotype. Scale bars: 10 µm. (B) Quantification of the GFAP signal intensity is presented for 3 animals from each genotype. Mean and standard deviation are presented. P value was calculated with the Student t test. n.s., nonsignificant differences.
BECN1 expression decreases the aggregation of mutant SOD1 in NSC34 motoneuron cells
We then investigated the impact of BECN1 in a cell-culture model of ALS. To monitor the levels of BECN1 in cells expressing mutant SOD1, we transiently expressed human wild-type (SOD1WT) or mutant SOD1G85R as EGFP fusion proteins in the motoneuron cell line NSC34. As shown in after 72 h of transfection, accumulation of SOD1G85R aggregates was detected in NSC34 cells, correlating with increased levels of LC3-II form. On the other hand, the expression of SOD1WT did not increase LC3-II levels in this experimental setting. As a control to measure LC3-II generation, NSC34 cells were treated with a cocktail of lysosome inhibitors including bafilomycin A1 and pepstatin (). In these experiments, a clear upregulation of BECN1 was observed in cells expressing SOD1G85R (). We then wanted to confirm the previously reported role of autophagy on mutant SOD1 levels.Citation18,Citation19,Citation23 For this purpose, cells expressing SOD1G85R-EGFP were treated with the PtdIns3K inhibitor 3-methyladenine (3-MA). We observed a marked accumulation of mutant SOD1 species, including monomers and aggregates (Fig. S3A). Similar results were observed when lysosomal activity was blocked with bafilomycin A1 and pepstatin (not shown).
Figure 6. BECN1 expression reduces SOD1 levels in a motoneuron cell line. (A) NSC34 cells were transiently transfected with expression vector for human wild-type and mutant SOD1G85R fused to EGFP. After 72 h, SOD1 levels were monitored in cell extracts prepared in 1% Triton X-100 buffer by western blot. LC3-II levels were also measured in the same samples. As a control, NSC34 cells were treated with 200 nM bafilomycin A1 (Bafilo.) and 10 mg/ml pepstatin (Peps.) lysosome inhibitors for 16 h to induce LC3-II accumulation. (B) Levels of BECN1 and LC3-II were evaluated in cells after transient transfection of NSC34 cells with the mutant SOD1G85R–EGFP vector. HSP90 levels were monitored as a loading control. (C) NSC34 cells were cotransfected with SOD1G85R-EGFP, FLAG-BECN1 in the presence or absence of FLAG-BCL2L1 expression vectors. After 72 h, SOD1 protein oligomers were measured in cell extracts prepared in 1% Triton by western Blot. HSP90 levels were monitored as loading control. Right panel: Quantification of relative SOD1 levels from 3 independent experiments performed and normalized to the values obtained in control cells only expressing mutant SOD1. Mean and standard error are presented. P value was calculated with the Student t test. (D) NSC34 cells were cotransfected with SOD1G85R-EGFP, in the presence or not of BECN1WT, BECN1delta, and FLAG-BCL2L1 -tagged vectors. After 72 h, SOD1-EGFP protein oligomers were measured in cell extracts prepared in 1% Triton X-100 by western Blot. HSP90 levels were measured as loading control. Right panel: As control, BECN1 expression was verified. HSP90 levels were measured as loading control.
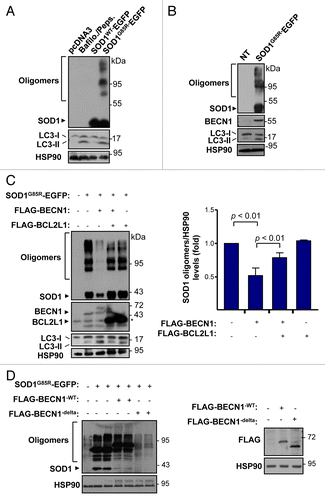
To further study the impact of BECN1 on mutant SOD1 levels, we transiently transfected NSC34 cells with an expression vector for a FLAG-tagged version of BECN1 together with a SOD1G85R-EFGP vector. Expression of BECN1WT dramatically reduced the levels of mutant SOD1 (). Quantification of several experiments revealed a 50% reduction in the total levels of mutant SOD1 aggregates (), but also monomers (Fig. S3B). In agreement with this result, we recently reported that stimulation of autophagy with trehalose induces the clearance of aggregated and monomeric forms of mutant SOD1.Citation26 BCL2 and BCL2L1 are negative regulators of autophagy by a direct interaction with BECN1.Citation49 As expected, transient expression of FLAG-BCL2L1 partially reduced the effects of BECN1 on mutant SOD1 aggregation (). Consistent with these findings, expression of an active form of BECN1 that lacks the BCL2 and BCL2L1 binding site (BECN1delta)Citation49 has a strong effect in reducing mutant SOD1 levels both at the level of monomers and aggregates (). In addition, we inhibited autophagy with 3-MA in NSC34 cells overexpressing SOD1 and BECN1, showing increased accumulation of SOD1 aggregates (Fig. S3C). Taken together, these results indicate a functional role of BECN1 in the clearance of mutant SOD1.
An abnormal interaction of mutant SOD1 with BECN1 through BCL2L1
Our results suggest that BECN1 expression may have an unfavorable effect on ALS progression in mouse models. Studies in Parkinson disease (PD),Citation65 Huntington disease,Citation51 and ADCitation66 models indicate that an abnormal interaction of disease-related proteins with BECN1 may alter its biological function, contributing to neurodegeneration. Because BCL2 and BCL2L1 are negative regulators of BECN1, and mutant SOD1 has an altered association with BCL2,Citation67,Citation68 we decided to explore the possible effects of mutant SOD1 on the BECN1-BCL2L1 complex. Analysis of the subcellular distribution of SOD1-EFGP and FLAG-BECN1 in NSC34 cells using immunofluorescence and confocal microscopy analysis indicated an enhanced colocalization of mutant SOD1 with BECN1 when compared with SOD1WT (). We then tested the possible formation of a protein complex between mutant SOD1 and BECN1. We cotransfected expression vectors for SOD1G93A-MYC, FLAG-BCL2L1, and FLAG-BECN1WT into 293T HEK cells and then performed immunoprecipitation (IP) of MYC followed by western blot analysis. Remarkably, BECN1 was efficiently precipitated with mutant SOD1, suggesting the formation of a protein complex (). As expected, BCL2L1 was also coprecipitated in these experiments (). Virtually identical results were obtained when another ALS-linked mutant, SOD1G85R-MYC, was tested (). Using the same experimental system, no interactions between SOD1WT-MYC and BECN1 was detected when compared with mutant SOD1, whereas BCL2L1 was still precipitated with SOD1WT ().
Figure 7. Mutant SOD1 interacts with the BECN1-BCL2L1 complex. (A) Colocalization analysis between FLAG- BECN1 and wild-type or mutant SOD1G85R-EGFP in NSC34 cells. NSC34 cells were transiently cotransfected with expression vectors for human SOD1WT or SOD1G85R-EGFP fusion protein and FLAG- BECN1. After 24 h, SOD1 distribution was visualized by confocal fluorescent microscopy (green) and FLAG- BECN1 detected by indirect immunofluorescence (red). Merge images are provided. Right panel: SOD1G85R-EGFP and FLAG-BECN1 colocalization index was calculated using the Manders coefficient. Mean and standard error is presented. P value was calculated with the Student t test. (B) SOD1G93A-MYC, (C) SOD1G85R-MYC or (D) SOD1WT-MYC or SOD1G93A-MYC (control) were expressed in 293T cells together with BCL2L1 and BECN1-FLAG tagged versions. Immunoprecipitation (IP) assays were performed using anti-MYC antibody-agarose complexes followed by western blot analysis to assess the interaction with BCL2L1 and BECN1. Total protein extracts are shown as control. Results are representative of 3 independent experiments. (E) SOD1G93A-MYC IP was performed in NSC34 cells cotransfected with mutant FLAG-BECN1WT or BECN1delta, and FLAG-BCL2L1 followed by western blot analysis. (F) IP was performed with anti-MYC antibody-agarose complexes in NSC34 cells cotransfected with BECN1-MYC, FLAG-BCL2L1 and mutant SOD1 (both EGFP-tagged). This figure is representative of 3 independent experiments.
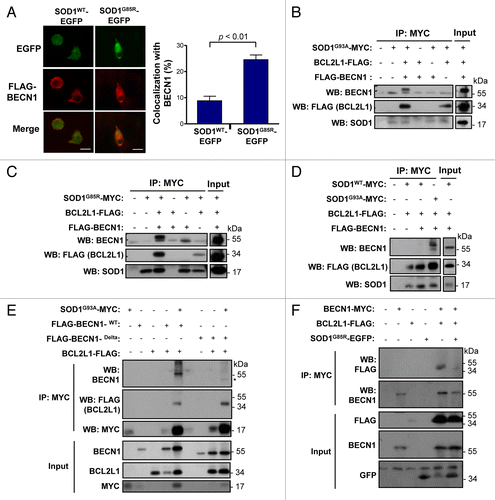
We then repeated these experiments on the motoneuron cell line NSC34. IP of SOD1G93A-MYC confirmed the formation of a protein complex with FLAG-BECN1 and FLAG-BCL2L1 (). Remarkably, deletion of the BCL2L1 interaction site in BECN1 reduced the association between BECN1 and mutant SOD1 (), suggesting the need for an interaction between BCL2L1 and BECN1 for the association with mutant SOD1. Finally, we addressed the functional impact of mutant SOD1 on the stability of the BECN1-BCL2L1 complex. For this purpose, we immunoprecipitated BECN1-MYC and addressed its association with BCL2L1 in the presence or absence of mutant SOD1. As shown in , expression of SOD1 significantly decreased the stability of the interaction between BECN1 and BCL2L1. In summary, our results suggest that ALS-linked mutant SOD1 alters the BECN1-BCL2L1 complex possibly through an abnormal physical association.
Discussion
ALS is a progressive and disabling neurodegenerative disease, characterized by paralysis and premature death of the patient. There is no definitive treatment for this illness, and the only currently approved drug by the FDA is riluzole, an anti-glutamatergic compound that has only modest benefits in ameliorating disease signs.Citation69 Clarifying the common mechanisms shared by sporadic and familial cases of ALS should bring insights for the development of effective therapies for both disease forms. Although the participation of protein aggregation in the pathogenesis of neurodegenerative diseases is still a subject of considerable debate, it is clear that abnormal folding of SOD1 and other ALS-linked proteins such as TARDBP and FUS, directly correlates with neuronal toxicity and motor dysfunction in the disease. This is why strategies to reduce the load of misfolded proteins are emerging as an attractive approach for disease intervention in neurodegenerative diseases, including ALS.Citation70-Citation72
BECN1 is an essential regulator of autophagy with validated biological functions in several neurodegenerative diseases, cancer, and ischemia, among other pathological conditions.Citation70-Citation72 Interestingly, in normal human brain, there is a progressive decrease in Becn1 mRNA and protein levels during aging.Citation51 Moreover, a decrease in BECN1 levels is observed in AD brains and also mouse models of the disease.Citation53,Citation73 In agreement with this idea, several studies have demonstrated that autophagy impairment is a relevant pathological event in AD models.Citation32,Citation74 Functional studies using Becn1 heterozygous mice and also gain of function approaches with gene therapy demonstrated that stimulation of BECN1-dependent autophagy has a positive effect in reducing amyloid deposits in ADCitation53 and Machado-Joseph disease models.Citation75 Similar results are reported in an α-Synuclein (Snca) transgenic mouse, a model of PD and Lewy body disease.Citation73,Citation76 In other pathological conditions, BECN1 expression is upregulated including models of brain ischemia,Citation77 HD,Citation78 traumatic brain injury,Citation79 PD,Citation80 and lipid storage disorders,Citation81,Citation82 in addition to the brain of patients infected with HIV.Citation83 Here we report the upregulation of BECN1 in a cellular and in a mouse model of ALS, confirming our previous findings in human postmortem tissue derived from sporadic ALS and fALS cases.Citation23
Despite initial expectations that the reduction of BECN1 levels would accelerate ALS disease progression, it actually has a significant protective effect. These results were associated with a complex signature of SOD1 oligomeric species, where monomeric and high molecular weight aggregates were accumulated in the spinal cord of Becn1+/− SOD1G86R mice. We speculate that the reduction in oligomeric forms may actually contribute to protection against ALS. Recent reports suggest that large SOD1 protein inclusions may have a protective effect on ALS, and also in HD, because they could trap neurotoxic oligomeric species that are highly diffusible and more abundant in terms of specific concentrations.Citation84,Citation85 In agreement with this concept, we have recently reported that targeting the ER stress transcription factor ATF4 protects against ALS and also enhances mutant SOD1 aggregation.Citation86 We have also reported that the ablation of another ER stress transcription factor termed XBP1 in ALS mouse models provides neuroprotection through a shift in the protein homeostasis network toward the upregulation of autophagy in motoneurons.Citation23 Based on these observations, we monitored ER stress levels in mutant SOD1 mice on a wild-type or Becn1+/− background and did not detect any significant differences on the levels of the classical stress markers Hspa5/Bip and Ddit3/Chop (Fig. S4).
Treatment of mutant SOD1 mice with trehalose protects against ALS, correlating with the stimulation of MTOR-independent autophagy,Citation26 which induces autophagy flux in the nervous system.Citation87 In contrast, treatment of SOD1 mice with rapamycin has detrimentalCitation28 or no effectsCitation27 on disease progression and animal survival.Citation27,Citation28 In these experiments, although changes on LC3 levels are demonstrated upon rapamycin treatment, no effects on mutant SOD1 levels are reported.Citation28 These results are surprising since rapamycin treatment has been shown to be beneficial in diverse neurodegenerative disease models.Citation88 In contrast, treatment of a TARDBP transgenic mouse model with rapamycin, spermidine, carbamazepine, or tamoxifen delays experimental ALS, correlating with the upregulation of autophagy markers.Citation89 Although that study has great therapeutic potential, the association with autophagy as a mechanism of action of the compounds tested was only correlative. Besides, it is well known that most of the drugs tested to stimulate autophagy have many other cellular targets.Citation90 Our current study using Becn1 heterozygous mice is the first direct demonstration for the possible involvement of autophagy on ALS, providing unexpected results.
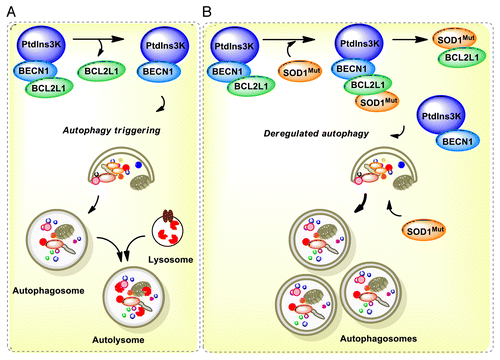
We speculate that autophagy impairment may contribute to ALS through alterations of BECN1 function due to abnormal protein-protein interactions as reported in other diseases.Citation35 Then, reducing BECN1 levels may bypass this specific molecular alteration on the pathway as suggested here by reducing the levels of autophagy (see model in ). Supporting this concept, an early report shows that mutant huntingtin inclusions sequester BECN1, which may impair its function.Citation51 This observation is confirmed in cell-culture and animal models of the disease, in addition to human postmortem tissue from HD patients.Citation51 Similarly, PINK1, a gene also involved in PD and mitophagy, binds to BECN1, altering the stimulation of autophagy. PINK1 has also been shown to interact with BCL2L1, a negative regulator of BECN1.Citation91 A recent report also indicates that the PARK2/Parkin-BECN1 complex is altered in mouse models of AD.Citation66 Since mutant SOD1 interacts with BCL2,Citation67 here we explored if the BECN1 complex is altered in a cellular model fALS. Our results uncover an abnormal association of mutant SOD1 with the BECN1-BCL2L1 complex that was not observed with SOD1WT. This interaction was associated with a reduced stability of the complex, suggesting that mutant SOD1 may alter the autophagy machinery through BECN1. Thus, BECN1 may have a dual participation in the disease, both promoting the degradation of misfolded proteins (downstream effect) and the modulation of the threshold for autophagy activation due to an abnormal interaction with mutant SOD1 (upstream alteration) (). This study identifies for the first time a protective role of Becn1 haploinsufficiency in neurodegenerative diseases, indicating a specific contribution of the pathway to ALS that contrasts with its known role in other pathologies. Complementary mechanisms of protection of Becn1 deficiency remain to be explored. Since we only used one particular mutant SOD1 mice, our findings need to be confirmed in other ALS mouse models to assess the actual contribution of BECN1 to the disease process. Interestingly, the first characterization of Becn1+/− mice demonstrates that loss of one copy of Becn1 is inductive in long-term cancer.Citation54,Citation55 Autophagy impairment in cancer has been proposed to trigger genomic instability possibly due to the occurrence of oxidative stress, promoting cancer cell survival.Citation62,Citation63 Although markers of the unfolded protein response were not altered in Becn1+/− mice, we cannot exclude the possibility that global adaptive changes in the proteostasis network may contribute to the protective effects observed in these animals as we describe in other models.Citation23,Citation92 This report highlights the need for a better understanding of the actual contribution of autophagy to ALS to move forward developing therapeutic strategies to treat this devastating disease.
Figure 8. Working model. (A) In a normal condition, BECN1 is inhibited by a direct interaction with BCL2 or BCL2L1 anti-apoptotic proteins. Once autophagy is triggered, BCL2 or BCL2L1 are released from BECN1 and the PtdIns3K (whose catalytic subunit is PIK3C3) is activated, resulting in formation of autophagosomes and downstream autophagy-mediated degradation of cargoes. (B) In ALS, the expression of mutant SOD1 leads to 2 possibilities: a formation of a triple complex between SOD1 and the BECN1-BCL2L1 complex or to an abnormal association with the BECN1 and BCL2L1 complex, destabilizing the interaction leading to abnormal autophagy levels. These pathological effects may be attenuated by the reduction of BECN1 levels in BECN1 heterozygous animals, which may normalize autophagy activity. In addition, mutant SOD1 aggregates are substrates of autophagy-mediated degradation.
Material and Methods
Plasmids and reagents
Pepstatin and E64D were purchased (P4265 and E3132, respectively) from Sigma-Aldrich. Bafilomycin A1 was purchased from Calbiochem (196000). Cell media and antibiotics were obtained from Life Technologies. Fetal calf serum was obtained from Atlanta Biologicals (S11050H). Hoechst was purchased from Molecular Probes (51731). All transfections were performed using the Effectene reagent (Qiagen, 301425). DNA were purified with Qiagen plasmid midi kits (Qiagen, 12143).
SOD1-EGFP expression vectors were described before.Citation93 For BECN1 cell overexpression, we employed FLAG or MYC epitope-tagged vectors cloned into pCR3.1. The constructs of interest are wild-type BECN1 (FLAG-BECN1wt or BECN1wt-MYC) and BCL2 binding defective mutants of BECN1 (FLAG-BECN1delta), which carries a deletion in the BH3 domain.Citation49 For IPs, we also employed the pcDNA3.1/MYCHis-SOD1 vectors (SOD1wt, SOD1G93A, and SOD1G85R-MYC)Citation94 and FLAG-BCL2L1.
Animal experimentation
As an ALS animal model we employed the SOD1G86R transgenic mice (the equivalent of the human SOD1G85R mutation)Citation23,Citation95 that we backcrossed into a C57/b6 background.Citation26,Citation61 The expression of the SOD1 mutant gene is driven by the endogenous SOD1 promoter. In addition, SOD1G86R encodes an enzyme with low SOD activity, and thus expression of the altered enzyme does not significantly affect overall SOD1 cellular activity when added to the genome in the presence of 2 wild-type parental genes. We generated SOD1G86R mice heterozygous for Becn1Citation54 by crossing the SOD1G86R mice with Becn1 heterozygous mice. F1 animals were used in all experiments to maintain the ratio of mix background. The animal care and all animal experiments were performed according to procedures approved by “Guide for the Care and Use of Laboratory Animals” (Commission on Life Sciences, National Research Council. National Academy Press 1996) and approved by the Bioethical Committee of the Faculty of Medicine, University of Chile.
Analysis of disease parameters
To assess disease onset, progression of the disease, and survival of animals, we monitored a group of disease parameters, including loss of motor performance, body weight loss, and qualitative assessment of visual disease signs. For each parameter the day of onset was defined using the following criteria: We determined motor performance using the rotarod test 3 times per wk starting at 60 d old using an acceleration protocol (Panlab SL, Model LE8500, need address for apparatus).Citation26,Citation86 Disease onset was defined as the day before the animals were not able to perform the task. Body weight was measured 3 times per wk beginning at 30 d old. In this case, onset was defined as the day before the animal lost more than 5% of the peak of its body weight. To monitor rotarod performance, measurements started at 60 d old. The onset of the disease was calculated by this parameter considering the day when the animal was not able to maintain itself on the rotor. To determine disease onset by visual disease sign observations, we monitored and scored the appearance of abnormal limb-clasping (c), tremor felt in hind-limbs (t), dirty appearance of skin (s), backbone arching (a), and paralysis (p) (Table S1 and ref. Citation26). We assigned to each visual observation a score 1 when the symptom appeared, 3 when it increased or 5 when the phenotype was severe. As the backbone arching and paralysis frequently appear as a result of a severe neuronal dysfunction, the score for these symptoms started from 3 instead of 1 (Table S1). Using this score disease onset was defined as when the value reached more than 10. The end-stage of disease was determined as the time when an animal was not able to right itself up within 30 s after being placed on its back. The progression of the disease for each parameter was calculated as the difference in d from the end point (sacrifice) and the onset of the disease.
Assays for the detection of mutant SOD1 aggregation
We used the NSC34 (neuroblastoma-spinal cord 34) cell line as a motoneuron-cell like model for the experiments on mutant SOD1 aggregation, inclusions, observation of autophagy markers, and for the production of cell lines with decreased or increased expression of BCN1. NSC34 cells were selected for this study due to valuable characteristics of motoneurons.Citation93,Citation96 For SOD1 expression assays, cells were transiently transfected with the SOD1 ALS-linked expression constructs and autophagy pathways components. The EGFP-SOD1 constructs were employed to visualize and quantify the formation of intracellular SOD1 inclusions in living cells by fluorescent microscopy. Insolubility in nondenaturing detergents of SOD1 oligomers was assessed using biochemical analysis followed by western blot analysis. After 72 h of transfection, total cell extracts were prepared in 1% NP40 buffer (Merck, 301425) in phosphate-buffered saline (PBS; 21600-010) containing a protease inhibitor cocktail (Roche, 4693124001) and sonicated. Protein concentration was determined by micro-BCA assay (Pierce Thermo Scientific, 23235). The following primary antibodies and dilutions were used: anti-GFP 1:2000 (Santa Cruz Biotechnology, sc-9996), anti-HSP90 1:3000 (Santa Cruz Biotechnology, sc-33755), and anti-BECN1 1:10000 (Santa Cruz Biotechnology, sc-11427); anti-SOD1 1:3000 (Calbiochem, 574597); anti-LC3 1:1000 (Cell Signaling, 2775S). The secondary antibodies employed, all at 1:4000 dilutions, were: anti-mouse (Life Technologies, 62-6520), anti-rabbit (Life Technologies, 65-6120), and anti-sheep (Sigma-Aldrich, A3415).
RNA extraction and quantitative real-time PCR
Total RNA was extracted from cell lines directly using TRIzol (Life Technologies, 15596-018). For spinal cord RNA extraction, the tissue was previously homogenized in PBS and 1/3 of the resulting homogenate was separated for TRIzol, and 2/3 were diluted in lysis buffer for western blot assay. cDNA was synthesized with SuperScript III (Life Technologies, 11754250) using random primers p(dN)6 (Roche) and quantitative real-time PCR by standard protocols.Citation97 Actin was used as a housekeeping cell control. Primer sequences for Sqstm1: forward 5′-CGATGACTGG ACACATTTGT CT-3′ and reverse 5′-GTCCTTCCTG TGAGGGGTCT-3′. For Becn1: forward 5′-GAGCCATTTA TTGAAACTCG CCA-3′ and reverse 5′-CCTCCCCGAT CAGAGTGAA-3′. For Sod1: forward 5′-GCCCGCTAAG TGCTGAGTC-3′ and reverse 5′-CCAGAAGGAT AACGGATGCC A-3′. For Bcl2l1: forward 5′- GCTGGGACAC TTTTGTGGAT-3′ and reverse 5′- AAGAGTGAGC CCAGCAGAAC-3′. For Hspa5: forward 5′- TCATCGGACG CACTTGGAA-3′ and reverse 5′- CAACCACCTT GAATGGCAAG A-3′. For Ddit3: forward 5′- CCCTGGCTTT CACCT TGG-3′ and reverse 5′- CCGCTCGTTC TCCTGCTC-3′. For Actb: forward 5′-CTCAGGAGGA GCAATGATCT TGAT-3′ and reverse 5′-TACCACCATG TACCCAGGCA-3′.
Western blot assays of spinal cord tissue
One centimeter lumbar spinal cord tissue was collected and homogenized in RIPA buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% DOC, 0.5% triton X-100) containing a protease inhibitor cocktail (Roche, 4693124001) by sonication. Protein concentration was determined by micro-BCA assay (Pierce Thermo Scientific, 23235). The equivalent of 30–50 μg of total protein was loaded onto 8% or 17% SDS-PAGE minigels depending on the analysis. The following primary antibodies and dilutions were used: anti-BECN1 1:10000 (Santa Cruz Biotechnology, sc-11427), anti-HSP90 1:3000 (Santa Cruz Biotechnology, sc-33755), anti-SOD1 1:3000 (Calbiochem, 574597), anti-LC3 1:1000 anti-LC3 1:1000 (Cell Signaling, 2775S); anti-SQSTM1 1:10000 (Abcam, AB56416). And the secondary antibodies employed, all at 1:4000 dilution, were: anti-mouse (Life Technologies, 62-6520), anti-rabbit (Life Technologies, 65-6120), and anti-sheep (Sigma-Aldrich, A3415).
Tissue analysis
To monitor SOD1 pathogenesis in vivo, animals were euthanized and tissue collected for histology at the end-stage of the disease. Spinal cord tissue was processed for immunohistochemistry using standard procedures as described.Citation98 In brief, mice were perfused transcardially with 4% paraformaldehyde in 0.1 M PBS. The spinal cord was removed and postfixed for 3 h in 4% paraformaldehyde. The spinal tissue was subjected to a sucrose gradient (5%, 10%, and 30% sucrose in PBS), cryoprotected with Optimal Cutting Temperature compound (Tissue Tek, 25608-930), and fast frozen using liquid nitrogen. The tissue was longitudinally sectioned (5-µm thick slices) using a cryostat microtome (Leica). Sections were immunostained using antibodies anti-NeuN (RBFOX3) 1:300 (Millipore, MAB377), anti-GFAP 1:1000 (Dako, N1506), MAP2 1:300 (Millipore, AB5622), and anti-SQTM1 1:1000 (Abcam, AB56416). Secondary antibodies: mouse Alexa 594 1:500 (Molecular Probes, A11007) and anti-rabbit Alexa 488 1:500 (Molecular Probes, A11006). Tissue sections were viewed with an Olympus IX71 microscope and images were captured using a QImaging QICAM Fast 1394 camera.
BECN1 expression was analyzed by indirect immunofluorescence using the following protocol. Brain cortex tissue was prepared as described above for spinal cord tissue. However, before sectioning, tissue was incubated in formalin for 24 h, dehydrated in ethanol series, and embedded in Paraplast Plus (Monoject Scientific, 02847-AB). Ten micrometer sections were then processed for deparaffinization with alcohol series. Sections were rinsed in water and PBS for 10 min and incubated with primary BECN1 antibody at 37 °C for 1 h. Sections were washed 3 times in PBS and incubated with secondary antibody for 1 h at RT. Sections were then washed 3 times in PBS and mounted with Fluoromount G (Electron Microscopy Science, 0100-01). Images were acquired using an Olympus Fluoview FV1000 confocal laser scanning microscope. Sections were immunostained using antibodies anti-BECN1 1:200 (Abcam, AB62472), anti-RBFOX3 1:300 (Millipore, MAB377), secondary anti-mouse Alexa 594 1:500 (Molecular Probes, A11007) and anti-rabbit Alexa 488 1:500 (Molecular Probes, A11006).
Immunoprecipitation experiments and colocalization analysis
IP assays were performed in HEK 293T and in NSC34 cells cotransfected with vectors encoding FLAG-BECN1WT, BECN1WT-MYC, SOD1WT-EGFP, SOD1G85R-EGFP, in addition to those encoding FLAG-BCL2L1, SOD1WT, SOD1G93A-MYC, and SOD1G85R-MYC (pcDNA3.1/MYCHis-SOD1 vectors) by using anti-MYC antibody-agarose complexes (Santa Cruz Technology, sc-40 AC). Protein complexes were eluted by competing with 1.4 mg/ml MYC peptide (Sigma-Aldrich, M2435). HEK293T or NSC34 cells seeded in 6-well plates were cotransfected and 48 h later the cell extracts were collected, centrifuged, and the resulting pellet was resuspended in 500 µL of 0.2% NP40 buffer (100 mM KCl, 50 mM Tris [pH 7.5], 50 mM NaF, 1 mM Na3VO4, 250 mM PMSF, and protease inhibitors). Post nuclear protein extracts were incubated with the anti-MYC antibody-agarose complexes for 4 h at 4 °C in a wheel rotor, and then washed twice in 1 ml of NP40 buffer. A third wash was performed in NP40 buffer with 500 mM NaCl. The IP and coimmunoprecipitation verification were assessed by western blot assays.
Statistical analysis
All data were analyzed by the Student t test, one-way ANOVA followed by Bonferroni posttest, or Kaplan-Meier statistics. The GraphPad Prism 5 software was used for statistical analysis.
| Abbreviations: | ||
| AD | = | Alzheimer disease |
| ALS | = | amyotrophic lateral sclerosis |
| fALS | = | familial ALS |
| IP | = | immunoprecipitation |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MTOR | = | mechanistic target of rapamycin |
| PD | = | Parkinson disease |
| SOD1 | = | superoxide dismutase 1 |
Additional material
Download Zip (263.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by the Muscular Dystrophy Association and ALS Therapy Alliance; Millennium Institute no. P09-015-F, ACT1109; FONDEF D11I1007; FONDECYT no. 1140549, AD Association, CONICYT grant USA2013-0003, ECOS-CONICYT C13S02 (CH), postdoc fellow FONDECYT 3140110 (MN), 3130365 (DRR), and FONDECYT 11121524 (SM). VV received a CONICYT PhD fellowship no. 21120411. This work was also funded by NIH RO1 CA109618 (BL). GK is supported by the Ligue Nationale contre le Cancer, Agence Nationale pour la Recherche, European Commission (ArtForce), European Research Council, Institut National du Cancer (INCa), Cancéropôle Ile-de-France, Fondation Bettencourt-Schueller, AXA Chair for Longevity Research, LabEx Immuno-Oncology, and Paris Alliance of Cancer Research Institutes.
We are grateful to Dr Maria Chiara Maiuri and Dr Alfredo Criollo (INSERM U848 Villejuif, France) for their advice in assessing BECN1-BCL2L1 interaction. We thank Dr Julie Atkin (Florey Neuroscience Institutes, Australia) for providing SOD1-GFP constructs and Dr Thomas V O’Halloran (Northwestern University) for providing SOD1-MYC plasmids. We are thankful to Claudia Duran for advices in tissue deparaffinization protocol, Monica Flores for technical help, and Silke Escobar for mice care.
References
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006; 52:39 - 59; http://dx.doi.org/10.1016/j.neuron.2006.09.018; PMID: 17015226
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006; 7:710 - 23; http://dx.doi.org/10.1038/nrn1971; PMID: 16924260
- Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 2013; 9:617 - 28; http://dx.doi.org/10.1038/nrneurol.2013.203; PMID: 24126629
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:59 - 62; http://dx.doi.org/10.1038/362059a0; PMID: 8446170
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008; 4:e1000193; http://dx.doi.org/10.1371/journal.pgen.1000193; PMID: 18802454
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668 - 72; http://dx.doi.org/10.1126/science.1154584; PMID: 18309045
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323:1205 - 8; http://dx.doi.org/10.1126/science.1166066; PMID: 19251627
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208 - 11; http://dx.doi.org/10.1126/science.1165942; PMID: 19251628
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72:245 - 56; http://dx.doi.org/10.1016/j.neuron.2011.09.011; PMID: 21944778
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al, ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72:257 - 68; http://dx.doi.org/10.1016/j.neuron.2011.09.010; PMID: 21944779
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14:248 - 64; http://dx.doi.org/10.1038/nrn3430; PMID: 23463272
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know?. Nat Rev Neurol 2011; 7:603 - 15; http://dx.doi.org/10.1038/nrneurol.2011.150; PMID: 21989245
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 2010; 13:1396 - 403; http://dx.doi.org/10.1038/nn.2660; PMID: 20953194
- Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brännström T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 2010; 5:e11552; http://dx.doi.org/10.1371/journal.pone.0011552; PMID: 20644736
- Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet 2009; 18:3217 - 26; http://dx.doi.org/10.1093/hmg/ddp260; PMID: 19483195
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 2010; 19:3440 - 56; http://dx.doi.org/10.1093/hmg/ddq257; PMID: 20570967
- Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 2006; 281:30524 - 33; http://dx.doi.org/10.1074/jbc.M603337200; PMID: 16920710
- Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett 2010; 469:112 - 6; http://dx.doi.org/10.1016/j.neulet.2009.11.055; PMID: 19944744
- Gal J, Ström AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 2009; 111:1062 - 73; http://dx.doi.org/10.1111/j.1471-4159.2009.06388.x; PMID: 19765191
- Gomes C, Escrevente C, Costa J. Mutant superoxide dismutase 1 overexpression in NSC-34 cells: effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neurosci Lett 2010; 475:145 - 9; http://dx.doi.org/10.1016/j.neulet.2010.03.065; PMID: 20363292
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009; 23:2294 - 306; http://dx.doi.org/10.1101/gad.1830709; PMID: 19762508
- Tian F, Morimoto N, Liu W, Ohta Y, Deguchi K, Miyazaki K, Abe K. In vivo optical imaging of motor neuron autophagy in a mouse model of amyotrophic lateral sclerosis. Autophagy 2011; 7:985 - 92; http://dx.doi.org/10.4161/auto.7.9.16012; PMID: 21628996
- Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2011; 70:349 - 59; http://dx.doi.org/10.1097/NEN.0b013e3182160690; PMID: 21487309
- Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013; 9:1308 - 20; http://dx.doi.org/10.4161/auto.25188; PMID: 23851366
- Bhattacharya A, Bokov A, Muller FL, Jernigan AL, Maslin K, Diaz V, Richardson A, Van Remmen H. Dietary restriction but not rapamycin extends disease onset and survival of the H46R/H48Q mouse model of ALS. Neurobiol Aging 2012; 33:1829 - 32; http://dx.doi.org/10.1016/j.neurobiolaging.2011.06.002; PMID: 21763036
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011; 7:412 - 25; http://dx.doi.org/10.4161/auto.7.4.14541; PMID: 21193837
- Wang IF, Tsai KJ, Shen CK. Autophagy activation ameliorates neuronal pathogenesis of FTLD-U mice: a new light for treatment of TARDBP/TDP-43 proteinopathies. Autophagy 2013; 9:239 - 40; http://dx.doi.org/10.4161/auto.22526; PMID: 23108236
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956 - 9; http://dx.doi.org/10.1126/science.1225967; PMID: 23112296
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146 - 58; http://dx.doi.org/10.1016/j.cell.2010.05.008; PMID: 20541250
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011; 7:788 - 9; http://dx.doi.org/10.4161/auto.7.7.15596; PMID: 21464620
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 2010; 13:567 - 76; http://dx.doi.org/10.1038/nn.2528; PMID: 20383138
- Menzies FM, Moreau K, Rubinsztein DC. Protein misfolding disorders and macroautophagy. Curr Opin Cell Biol 2011; 23:190 - 7; http://dx.doi.org/10.1016/j.ceb.2010.10.010; PMID: 21087849
- Nassif M, Hetz C. Targeting autophagy in ALS: a complex mission. Autophagy 2011; 7:450 - 3; http://dx.doi.org/10.4161/auto.7.4.14700; PMID: 21252621
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 63; http://dx.doi.org/10.1016/j.cell.2007.10.035; PMID: 18083104
- Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, Hollinger HC, Hartley JA, Brockington A, Burness CE, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 2010; 5:e9872; http://dx.doi.org/10.1371/journal.pone.0009872; PMID: 20352044
- Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 2009; 18:4868 - 78; http://dx.doi.org/10.1093/hmg/ddp460; PMID: 19793721
- Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, et al, TODEM Study Group. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012; 79:1556 - 62; http://dx.doi.org/10.1212/WNL.0b013e31826e25df; PMID: 22972638
- Synofzik M, Maetzler W, Grehl T, Prudlo J, Vom Hagen JM, Haack T, Rebassoo P, Munz M, Schöls L, Biskup S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging 2012; 33:e13 - 7; http://dx.doi.org/10.1016/j.neurobiolaging.2012.07.002; PMID: 22892309
- Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G, Sazdovitch V, Cazeneuve C, Meininger V, LeGuern E, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 2013; 125:511 - 22; http://dx.doi.org/10.1007/s00401-013-1090-0; PMID: 23417734
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2:330 - 5; http://dx.doi.org/10.1093/embo-reports/kve061; PMID: 11306555
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385 - 96; http://dx.doi.org/10.1038/ncb1846; PMID: 19270696
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468 - 76; http://dx.doi.org/10.1038/ncb1854; PMID: 19270693
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741 - 50; http://dx.doi.org/10.1038/ncb2757; PMID: 23685627
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 2006; 281:14474 - 85; http://dx.doi.org/10.1074/jbc.M600364200; PMID: 16522639
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190 - 9; PMID: 18497889
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; http://dx.doi.org/10.1172/JCI20039; PMID: 14638851
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077 - 82; http://dx.doi.org/10.1073/pnas.2436255100; PMID: 14657337
- Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011; 7:829 - 39; http://dx.doi.org/10.4161/auto.7.8.15598; PMID: 21685724
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511 - 5; http://dx.doi.org/10.1038/nature10758; PMID: 22258505
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914 - 22; http://dx.doi.org/10.1161/01.RES.0000261924.76669.36; PMID: 17332429
- Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, Choi AM. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011; 7:629 - 42; http://dx.doi.org/10.4161/auto.7.6.15100; PMID: 21460622
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 1995; 92:689 - 93; http://dx.doi.org/10.1073/pnas.92.3.689; PMID: 7846037
- Matus S, Castillo K, Hetz C. Hormesis: protecting neurons against cellular stress in Parkinson disease. Autophagy 2012; 8:997 - 1001; http://dx.doi.org/10.4161/auto.20748; PMID: 22858553
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Gal J, Ström AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem 2007; 282:11068 - 77; http://dx.doi.org/10.1074/jbc.M608787200; PMID: 17296612
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 2010; 17:962 - 74; http://dx.doi.org/10.1038/cdd.2009.200; PMID: 20057503
- Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med 2013; 5:1247 - 62; http://dx.doi.org/10.1002/emmm.201302771; PMID: 23737459
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH Jr.. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004; 43:19 - 30; http://dx.doi.org/10.1016/j.neuron.2004.06.021; PMID: 15233914
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH Jr., Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet 2010; 19:2974 - 86; http://dx.doi.org/10.1093/hmg/ddq202; PMID: 20460269
- Bensimon G, Lacomblez L, Meininger V, ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med 1994; 330:585 - 91; http://dx.doi.org/10.1056/NEJM199403033300901; PMID: 8302340
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8:108 - 17; http://dx.doi.org/10.1038/nrneurol.2011.200; PMID: 22187000
- Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration?. Nat Rev Mol Cell Biol 2005; 6:891 - 8; http://dx.doi.org/10.1038/nrm1742; PMID: 16167052
- Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 2011; 43:17 - 28; http://dx.doi.org/10.1016/j.nbd.2010.08.015; PMID: 20732422
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010; 5:e9313; http://dx.doi.org/10.1371/journal.pone.0009313; PMID: 20174468
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011; 134:258 - 77; http://dx.doi.org/10.1093/brain/awq341; PMID: 21186265
- Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, Dufour N, Colomer Gould VF, Koeppen A, Déglon N, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011; 134:1400 - 15; http://dx.doi.org/10.1093/brain/awr047; PMID: 21478185
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 2009; 29:13578 - 88; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009; PMID: 19864570
- Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis 2008; 29:132 - 41; http://dx.doi.org/10.1016/j.nbd.2007.08.005; PMID: 17936001
- Wu JC, Qi L, Wang Y, Kegel KB, Yoder J, Difiglia M, Qin ZH, Lin F. The regulation of N-terminal Huntingtin (Htt552) accumulation by Beclin1. Acta Pharmacol Sin 2012; 33:743 - 51; http://dx.doi.org/10.1038/aps.2012.14; PMID: 22543707
- Diskin T, Tal-Or P, Erlich S, Mizrachy L, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma 2005; 22:750 - 62; http://dx.doi.org/10.1089/neu.2005.22.750; PMID: 16004578
- Wills J, Credle J, Oaks AW, Duka V, Lee JH, Jones J, Sidhu A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 2012; 7:e30745; http://dx.doi.org/10.1371/journal.pone.0030745; PMID: 22292029
- Pacheco CD, Lieberman AP. Lipid trafficking defects increase Beclin-1 and activate autophagy in Niemann-Pick type C disease. Autophagy 2007; 3:487 - 9; PMID: 17611388
- Takamura A, Higaki K, Kajimaki K, Otsuka S, Ninomiya H, Matsuda J, Ohno K, Suzuki Y, Nanba E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun 2008; 367:616 - 22; http://dx.doi.org/10.1016/j.bbrc.2007.12.187; PMID: 18190792
- Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, et al. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol 2013; 19:89 - 101; http://dx.doi.org/10.1007/s13365-012-0145-7; PMID: 23341224
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431:805 - 10; http://dx.doi.org/10.1038/nature02998; PMID: 15483602
- Brotherton TE, Li Y, Glass JD. Cellular toxicity of mutant SOD1 protein is linked to an easily soluble, non-aggregated form in vitro. Neurobiol Dis 2012; 49C:49 - 56; PMID: 22926189
- Matus S, Lopez E, Valenzuela V, Nassif M, Hetz C. Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 2013; 8:e66672; http://dx.doi.org/10.1371/journal.pone.0066672; PMID: 23874395
- Ludolph AC, Bendotti C, Blaugrund E, Hengerer B, Löffler JP, Martin J, Meininger V, Meyer T, Moussaoui S, Robberecht W, et al, ENMC Group for the Establishment of Guidelines for the Conduct of Preclinical and Proof of Concept Studies in ALS/MND Models. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler 2007; 8:217 - 23; http://dx.doi.org/10.1080/17482960701292837; PMID: 17653919
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009; 16:46 - 56; http://dx.doi.org/10.1038/cdd.2008.110; PMID: 18636076
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 2012; 109:15024 - 9; http://dx.doi.org/10.1073/pnas.1206362109; PMID: 22932872
- Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 2013; 123:980 - 9; http://dx.doi.org/10.1172/JCI64099; PMID: 23454761
- Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, Cilia E, Jonas EA, Valente EM. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ 2013; 20:920 - 30; http://dx.doi.org/10.1038/cdd.2013.19; PMID: 23519076
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 2012; 21:2245 - 62; http://dx.doi.org/10.1093/hmg/dds040; PMID: 22337954
- Turner BJ, Atkin JD, Farg MA, Zang DW, Rembach A, Lopes EC, Patch JD, Hill AF, Cheema SS. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci 2005; 25:108 - 17; http://dx.doi.org/10.1523/JNEUROSCI.4253-04.2005; PMID: 15634772
- Niwa J, Yamada S, Ishigaki S, Sone J, Takahashi M, Katsuno M, Tanaka F, Doyu M, Sobue G. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J Biol Chem 2007; 282:28087 - 95; http://dx.doi.org/10.1074/jbc.M704465200; PMID: 17666395
- Hetz C, Thielen P, Fisher J, Pasinelli P, Brown RH, Korsmeyer S, Glimcher L. The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ 2007; 14:1386 - 9; http://dx.doi.org/10.1038/sj.cdd.4402166; PMID: 17510659
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 1992; 194:209 - 21; http://dx.doi.org/10.1002/aja.1001940306; PMID: 1467557
- Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 2009; 33:679 - 91; http://dx.doi.org/10.1016/j.molcel.2009.02.017; PMID: 19328063
- Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis 2012; 3:e272; http://dx.doi.org/10.1038/cddis.2012.8; PMID: 22337234