
Abstract
Preclinical studies indicate autophagy inhibition with hydroxychloroquine (HCQ) can augment the efficacy of DNA-damaging therapy. The primary objective of this trial was to determine the maximum tolerated dose (MTD) and efficacy of HCQ in combination with radiation therapy (RT) and temozolomide (TMZ) for newly diagnosed glioblastoma (GB). A 3 + 3 phase I trial design followed by a noncomparative phase II study was conducted in GB patients after initial resection. Patients received HCQ (200 to 800 mg oral daily) with RT and concurrent and adjuvant TMZ. Quantitative electron microscopy and immunoblotting were used to assess changes in autophagic vacuoles (AVs) in peripheral blood mononuclear cells (PBMC). Population pharmacokinetic (PK) modeling enabled PK-pharmacodynamic correlations. Sixteen phase I subjects were evaluable for dose-limiting toxicities. At 800 mg HCQ/d, 3/3 subjects experienced Grade 3 and 4 neutropenia and thrombocytopenia, 1 with sepsis. HCQ 600 mg/d was found to be the MTD in this combination. The phase II cohort (n = 76) had a median survival of 15.6 mos with survival rates at 12, 18, and 24 mo of 70%, 36%, and 25%. PK analysis indicated dose-proportional exposure for HCQ. Significant therapy-associated increases in AV and LC3-II were observed in PBMC and correlated with higher HCQ exposure. These data establish that autophagy inhibition is achievable with HCQ, but dose-limiting toxicity prevented escalation to higher doses of HCQ. At HCQ 600 mg/d, autophagy inhibition was not consistently achieved in patients treated with this regimen, and no significant improvement in overall survival was observed. Therefore, a definitive test of the role of autophagy inhibition in the adjuvant setting for glioma patients awaits the development of lower-toxicity compounds that can achieve more consistent inhibition of autophagy than HCQ.
Keywords: :
Introduction
Concomitant radiation therapy with temozolomide followed by maintenance TMZ has been established as the standard of care for patients with newly diagnosed glioblastoma.Citation1 Despite this advance, GB remains a uniformly lethal malignancy. Autophagy is a therapeutic target in cancer due to its ability to promote the survival of cancer cells in the face of metabolic and therapeutic stress.Citation2 In glioma cells autophagy has been demonstrated to be an adaptive response to both radiation and TMZ,Citation3-Citation7 suggesting that inhibition of autophagy could have therapeutic benefit.
Chloroquine (CQ) and its derivative HCQ are able to inhibit autophagy. Chloroquine has been used for 60 y in humans for malaria prophylaxis and treatment, rheumatoid arthritis, and human immunodeficiency virus.Citation8-Citation10 It is an inexpensive orally available drug that has central nervous system penetration. In vitro studies indicated that higher concentrations of CQ derivatives than traditionally used for malaria and rheumatic disorders may be necessary to effectively block tumor cell autophagy and elicit tumor cell death. HCQ has less cumulative retinal toxicityCitation11 and is likely safer to dose-escalate than CQ.Citation2
We hypothesized that the addition of HCQ to standard chemoradiation and adjuvant chemotherapy would be safe, and evidence of autophagy inhibition and preliminary antitumor activity could be seen in adults with newly diagnosed GB. To test this hypothesis we designed a phase I/II clinical trial that incorporated a traditional phase I dose-escalation component, novel methods to assess the pharmacokinetic-pharmacodynamic (PK-PD) relationship for HCQ dose and autophagy inhibition, and assessment of overall survival in the phase II portion of the trial.
Results
Patient characteristics
A total of 92 patients (58 men) were enrolled between September 2007 and August 2010; 16 to the phase I portion and 76 to the phase II. One of the 16 subjects in the phase I portion enrolled but never received any drugs. The median age of all subjects was 58 y (range 20 to 83) with a median Karnofsky performance status (KPS) of 90 (range 60 to 100). Other baseline characteristics and demographics are shown in .
Table 1. Demographics
Dose escalation and toxicities
In the phase I part of the study no dose-limiting toxicities (DLTs) were encountered at daily doses of HCQ 200 mg/d and HCQ 400 mg/d. At the HCQ 800 mg/d dose level, 3/3 patients experienced grade 4 thrombocytopenia and 2/3 patients experienced grade 4 neutropenia. One patient experienced grade 4 neutropenia and sepsis. These toxicities occurred in all 3 subjects during chemoradiation. This early and severe myelosuppression (Fig. S1) was viewed as a DLT and per protocol the HCQ 400 mg/d cohort was expanded by 3 patients; no further DLTs were observed. There were no DLTs in 3/3 patients at HCQ 600 mg/d, which was therefore determined to be the MTD. In addition to dose-limiting myelosuppression in the HCQ 800 mg/d dose cohort, other grade 2 toxicities common to all dose levels included nausea, fatigue, constipation, and diarrhea. The grades 1 to 4 toxicities that were possibly, probably or definitely associated with HCQ, TMZ, or both in the phase I study at each dose level are shown in Tables S1–S4. The grade 3 and 4 toxicities with a possible, probable, or definite relationship to HCQ, TMZ or both in the phase II part of the trial are shown in .
Table 2. Phase II adverse events related to study drug
Efficacy
As of 03/04/2013, the time of final analysis, 75 patients had died and 17 were living. Median onset of protocol treatment from initial surgery was 4.9 wk. The median time on treatment was 21.6 wk (range 0.1 to 91.1). The average number of adjuvant treatment cycles of concurrent TMZ and HCQ was 5.1. Ninety patients (98%) were off treatment, 66% due to disease progression or death, 3% due to toxicity, 12% due to treatment delay of more than 14 d, 9% due to patient refusal of further treatment, or 3 due to investigator withdrawal for noncompliance. The estimated hazard rate was 0.59 (95% CI: 0.46 to 0.76) for the phase II portion of the study. The median OS was 15.6 mo (95% CI: 13 to 17.0). For patients ages 18 to 70 y old (n = 63), median OS was 16.5 (95%CI: 13.0–18.0). These results are compared with the European Organization for Research and Treatment of Cancer EORTC phase III study in and Figure S2. Overall, 19 of the 76 patients (25%) lived beyond 24 mo.
Table 3. Overall survival
Pharmacodynamic and population pharmacokinetic analysis
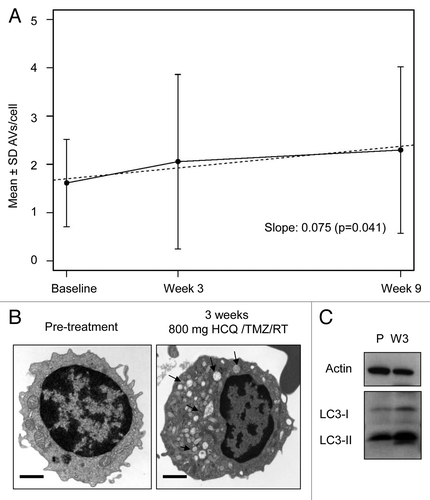
To determine if autophagy inhibition was achieved in patients, electron microscopy (EM)-based scoring for number of autophagic vacuoles was conducted on serially collected peripheral blood mononuclear cells for any patient that had at least 2 PBMC samples amenable to EM (40/92 patients). A mixed-effects model revealed a significant linear trend in mean AV, with mean vacuole count increasing by 0.075 per wk (P = 0.041) in all patients (). A striking accumulation of AV of varying shapes and sizes was observed in electron micrographs from a patient in the HCQ 800 mg/d cohort (). Quantification of morphological parameters of AV in one patient treated in the 800 mg HCQ cohort demonstrated a treatment-associated 9-fold increase in the percent of cytoplasm involved by AV, a 10-fold increase in AV perimeter, and a 2.5-fold increase in AV diameter (Fig. S3A). Accumulation of AV and a treatment-associated increase in the LC3-II/LC3-I ratio indicative of an accumulation of AV were present in PBMC from patients treated at lower nontoxic dose levels (; Fig. S3B).
Figure 1. Pharmacodynamic evidence of autophagy inhibition in patients treated with temozolomide (TMZ), radiation (RT), and hydroxychloroquine (HCQ). (A) Mixed-effects model of mean ± SD autophagic vacuoles (AVs)/cell. Dotted line: regression line. (B) Representative electron micrographs from a patient treated with chemoradiation and HCQ 800 mg/d for 3 wk. Arrows, AV; scale bar: 2 µm. (C) Immunoblotting against LC3 in the lysates of PBMC obtained from the same patient in (B). P, pretreatment; W3, 3 wk of treatment.
The population PK analysis was conducted with 262 nonbaseline blood HCQ concentration observations from 72 patients collected over a period of up to 276 ds. The median number of samples per patient was 4 (range, 1 to 6). The population model accounted for differences in dosing schedule (once vs. twice daily) and unequal doses (400 mg and 200 mg in divided doses over the 24 h dosing interval) in the 600 mg cohort. The population model PK parameters do not specifically represent steady-state values, as they were determined from multiple repeated single doses taken by individual patients during their period of participation in the study. To obtain steady-state PK parameters, individual estimates of PK parameters were simulated from the population model (see Patients and Methods). shows the individual predicted concentrations vs. the observed concentrations using a 2-compartment model with first-order absorption and a lag time. Inter-individual variabilities for the population typical values for lag time (tlag), apparent oral clearance from the central compartment (Cl/F), inter-compartmental clearance (Q/F), apparent volume of distribution of the central compartment (V/F), apparent distribution volume of peripheral compartment (V2/F), and first-order absorption rate constant (Ka), were consistent with the published literature (). Individual PK parameter estimates derived from the population model were most variable for clearance and distribution volumes (Table S5). The mean tlag was 1.06 h (range, 0.97 to 1.25 h), with mean Ka 0.51 h (range, 0.42 to 0.82), mean Ci 11.85 L/hr (range, 6.41 to 28.18), and mean V/F 483.96 L (range, 26 to 5483 L) with V2/F of 963 L (range, 558 to 2208 L).
Figure 2. Population pharmacokinetic-pharmacodynamic analysis. (A) Individual predicted HCQ whole blood concentrations vs. observed concentrations using a 2-compartment population pharmacokinetic model. (B) Observed HCQ concentrations in whole blood by dose cohort. (C) CART analysis. (D) Median AV change in patients with estimated HCQ Cmax above or below 1785 ng/mL.
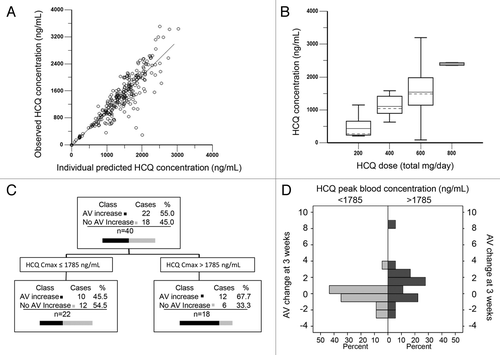
Table 4. Hydroxychloroquine population pharmacokinetic parameter estimates
The range of observed individual measurements of HCQ concentration followed a dose-proportional relationship (). Despite polypharmacy in this patient population, which included coadministration of corticosteroids, trimethoprim-sulfamethoxazole, and anti-emetics, the HCQ concentrations in whole blood from groups of patients receiving the same dose did not show a significant time-dependent trend (Fig. S4A). Individual PK parameter estimates derived from the population analysis were used to simulate individual blood concentration vs. time curves, assuming a 2-compartment model with first-order absorption and a lag time. Simulated PK parameters for patients who had complete sets of PD samples (Table S5), allowed for investigation of the PK-PD relationship between HCQ and AV accumulation in PBMC.
The PK-PD relationship between HCQ and AV accumulation in PBMC at 3 wk was first investigated by using an exploratory classification and regression trees (CART; Salford Predictive Modeler Builder v6.6), which identified a threshold effect of the HCQ peak blood concentration (). The CART analysis was conducted using PK and PD data of 40 patients, with the target variable defined as any positive change in number of AV from baseline to 3 wk and Cmin, Cmax, and area under the curve (AUC) as predictors.
The effect of the threshold value for Cmax on the change in AV from baseline to 3 wk was then investigated using the Kruskal-Wallis test for comparing median values and the Kolmogorov-Smirnov 2-sample test to identify any significant shift in the distribution. Patients with Cmax below 1785 ng/mL (n = 22) produced a median AV change of −0.02 (IQR: −0.52, 0.46), while patients with Cmax above 1785 ng/mL (n = 18) produced a median AV change of 1.01 (interquartile range: −0.21, 2.10) (; Kruskal-Wallis two-sided P = 0.0342; Kolmogorov-Smirnov P = 0.0156). A similar PK-PD correlation in PBMC at 9 wk was not significant (Fig. S4B).
Discussion
Autophagy is an intracellular process in which damaged organelles are sequestered in AVs. After fusion with lysosomes, the contents of the AV undergo acid-dependent degradation and are then recycled.Citation12 In eukaryotic cells autophagy serves at least 2 functions: 1) as an intracellular mechanism for the disposal of damaged organelles and proteins, and 2) to catabolize substrates during cellular stress in order to generate energy for cell survival.Citation13 Therapeutic activation of stress response genes such as TP53/p53 (Trp53 in mouse models) can induce autophagy in addition to apoptosis.Citation14 While persistent autophagy by itself can result in cancer cell death, there is mounting evidence that autophagy represents a key component of the damage response that cancer cells use to avoid death when exposed to metabolic and therapeutic stresses. However, when autophagy is inhibited, a cell that is reliant on autophagy will undergo either apoptotic or nonapoptotic cell death.Citation15,Citation16
CQ and its derivative HCQ are weak bases that are thought to deacidify lysosomes and thus inhibit the last step in autophagy. Our previous work demonstrated that autophagy inhibition with CQ augmented the efficacy of alkylating chemotherapy in a mouse tumor model.Citation14 In patients with newly diagnosed GB, a previous trial of radiation and carmustine with or without CQ found a trend toward increased median overall survival.Citation17 Preclinical studies indicate that micromolar concentrations of CQ and its derivatives are required to inhibit autophagy.Citation14 Since low-dose CQ prescribed for malaria prophylaxis achieves low nanomolar blood concentrations, dose escalation would likely be necessary to achieve effective autophagy inhibition in a clinical setting.Citation8 As studies demonstrate that CQ and HCQ are equipotent autophagy inhibitors, and based on its safer toxicity profile, we chose HCQ for a dose-escalation study.Citation18,Citation19 We cannot, however, exclude the possibility that TMZ, RT, and CQ would have proven to be a more active combination.
Surprisingly, the dose escalation in this study generated dose-limiting myelosuppression at HCQ 800 mg daily in combination with concomitant low-dose continuous TMZ and RT. All 3 patients treated at this dose experienced early, severe and sustained neutropenia and/or thrombocytopenia that were incompatible with further treatment on protocol. While it is possible that this is a reflection of TMZ-induced myelosuppression that can occur during chemoradiation,Citation20 it is in striking contrast to other clinical trials involving HCQ such as a phase I trial of TMZ (150 mg/m2 po daily 7 d on/7 d off) in combination with 1200 mg daily HCQ, in which no myelosuppression was observed (Rangwala et al., this issueCitation24). Other HCQ combination trials also reached HCQ 1200 mg/d dosing (Vogl et al., this issueCitation25; Rangwala et al., this issueCitation26). This suggests that continuous dosing of TMZ or the combination of both during chemoradiation produce more sustained autophagy than intermittent TMZ in the hematopoietic compartment and therefore sensitizes these host cells to HCQ-induced cytotoxicity. If so, these results demonstrate that the cytotoxic regimen that is paired with autophagy inhibitors can produce significant variability in toxicity, and also raise concern over an increasingly common practice of adding high doses of HCQ to various cytotoxic cancer regimens outside the context of a clinical trial. The considerable variability in HCQ pharmacokinetics underscores the importance of testing this approach in clinical trials.
The observed concentrations of HCQ in glioma patients in this study were consistent with those reported for the same doses given to patients with rheumatoid arthritis.Citation21 The population PK model developed for our PK-PD analysis was necessitated by the PK properties of HCQ and the outpatient visit schedule of adjuvant therapy for GB patients. Although the pharmacokinetics of HCQ have been represented by 1-, 2-, and 3-compartment models, our data supported a 2-compartment model, due to the greatest number of samples having been obtained near the beginning and end of the dosing interval.Citation21-Citation23 As with previous studies of HCQ pharmacokinetics, there was wide inter-and intra-patient variability. Unlike other investigators who have fixed Ka and tlag to published values, we chose to estimate these parameters because the disposition of HCQ in patients with advanced cancer has not been reported, nor have the effects of polypharmacy and gastrointestinal side effects been described. These estimates may have influenced the accuracy of V/F, which showed considerable variability. The population PK parameters indicated that similar to rheumatoid arthritis patients taking only single-agent HCQ, GB patients taking HCQ in combination with TMZ and radiation, and the many supportive care medications, demonstrated dose-proportional and sustained exposure to HCQ.
Our finding that patients with higher Cmax and AUC were more likely to demonstrate significant therapy-associated accumulation of AV in PBMC is the first demonstration that autophagy inhibition with HCQ can be achieved in humans. The major limitation of this study is that PBMC cannot fully reflect the complexity of autophagy dynamics that are within the tumor microenvironment. We chose PBMC in this study since serial biopsies of tumor tissue are not feasible in the GB population. Other HCQ studies involving malignancies with more accessible tumors such as multiple myeloma and melanoma have incorporated serial tumor biopsies that can be compared with PBMC to determine the degree of agreement between tumor and surrogate tissue with regard to the EM-based assay (Rangwala, et al., this issueCitation26; Vogl et al., this issueCitation25; Mahalingam et al., this issueCitation27). The current study demonstrates the feasibility of incorporating the EM-based PD assay into clinical trials, and the results suggest that this assay can be used to guide development of other novel autophagy inhibitors. These data also indicate that at HCQ 600 mg/d in combination with TMZ and RT, many patients may not be achieving adequate autophagy inhibition, and therefore the efficacy of the phase II trial of this regimen as measured by prolonged overall survival may be blunted by inconsistent autophagy inhibition by HCQ. Given the tolerability of higher doses of HCQ in combination with intermittent temozolomide (Rangwala et al., this issueCitation24), more consistent and a greater degree of autophagy could likely be achievable in this patient population, if the HCQ dose were maintained at 600 mg/d for the first 6 wk of chemoradiation and then dose-escalated during adjuvant temozolomide. Another approach to consider for future trials is to incorporate an intermittent dose-intense temozolomide and HCQ regimen (as used in Rangwala et al., this issueCitation24) in combination with brain radiation so that higher doses of HCQ might be achieved early in the course of GB therapy. Alternatively, the role of autophagy as a targetable resistance mechanism in front-line GB may best be answered by testing novel autophagy inhibitors that are more potent and tolerable than HCQ.
Patients and Methods
Study design and objectives
This was an open-label, multi-center, phase I/II trial in adults with newly diagnosed GB conducted by the American Brain Tumor Consortium (ABTC) (Clinical Trials.gov NCT00486603). The study was reviewed and approved by the National Cancer Institute and the individual institutional review board of each of the 10 participating institutions (University of Pennsylvania, Johns Hopkins University, Wake Forest University, H Lee Moffitt Cancer Center, Henry Ford Hospital, Emory University, University Hospitals Case Medical Center, Dana Farber Cancer Institute, Massachusetts General Hospital, and University of California San Francisco), and informed consent was obtained from each subject. The primary objective of the phase I study was to determine the maximum tolerated dose of HCQ when administered in conjunction with RT and concomitant and adjuvant TMZ. Upon completion of the phase I trial, patients were enrolled to a single-arm phase II trial and treated with MTD of HCQ in combination with RT and TMZ. The primary objective of the phase II cohort was overall survival. The secondary objectives of this trial were to a) characterize the population PK profile of HCQ; b) determine if PD evidence of autophagy inhibition could be detected in PBMC; and c) correlate PK with PD findings to determine the relationship between HCQ exposure and autophagy inhibition.
Eligibility criteria
Patients ≥ 18 y of age with histologically confirmed supratentorial GB diagnosed < 3 mo prior to registration were eligible. Other criteria included no prior tumor-directed therapy other than surgery, KPS ≥ 60%, normal hematological, adequate renal and liver function (transaminases ≤ 4 times the upper limits of the institutional normal), ability to provide written informed consent, and Mini Mental State Exam score of ≥ 15. Exclusion criteria included pregnancy or breast feeding, carmustine (Gliadel®) wafers, or a history of macular degeneration, diabetic retinopathy, porphyria, or psoriasis. Patients requiring cytochrome P450 enzyme-inducing anticonvulsant drugs were not eligible.
Treatment plan
Commencement of protocol therapy began as soon as medically appropriate, but, no later than 3 mo after craniotomy. All subjects received conventional external beam RT as utilized by the Radiation Therapy Oncology Group. This consisted of 60 Gy in 30 fractions delivered with megavoltage machines. The initial target treatment volume was defined as the contrast-enhancing lesion plus edema plus a 1 cm margin. The cone-down target volume included the enhancing lesion plus a 1.0 cm margin. Electron, particle, implant, or stereotactic radiosurgery boosts were not allowed. Prophylaxis for pneumocystis jirovecii was mandatory during RT.
Daily oral TMZ (75 mg/m2; Merck) was administered 1 h before each RT session or in the AM on ds without RT. Dose delays or discontinuation occurred for hematological and non-hematological toxicity; once resolved, TMZ was resumed without making up lost doses. Dose reductions were not allowed during RT. The 6 wk of RT and TMZ (chemoradiation) plus a 4-wk rest period was considered the initiation cycle. Maintenance cycles began after the initiation cycle and consisted of TMZ at a dose of 150 mg/m2/d for 5 consecutive d every mo for 6 mo (escalated in subsequent cycles to 200 mg/m2 in the absence of toxicity).
HCQ (obtained by patients through prescriptions at retail pharmacies, and manufactured by numerous generic pharmaceutical manufacturers) administration began on the first d of chemoradiation and continued daily without any planned interruptions including during the 4-wk rest period. Upon completion of 6 maintenance cycles of TMZ + HCQ patients continued with daily HCQ alone in monthly cycles with no limit to the number of cycles.
Dose escalation and definition of maximal tolerated dose
The planned daily doses for HCQ were 200 mg, 400 mg (given as 200 mg twice a day) and 800 mg (given as 400 mg twice a day). After toxicity was observed at 800 mg daily the protocol was amended to include a cohort at 600 mg HCQ daily (given as 400 mg in the morning and 200 mg in the evening). Three patients were treated per dose level. Before accrual to the next dose level began, all patients in a given cohort completed the 10-wk initiation cycle, permitting toxicities to be assessed (DLT period). No dose escalation beyond 800 mg/d was planned.
Treatment-related DLT was defined as any non-hematological grade 3 and 4 toxicities possibly, probably, or definitely HCQ-related (except nausea and vomiting without sufficient antiemetic prophylaxis). Known or reasonably suspected TMZ hematological toxicities were not considered dose-limiting unless the toxicity was felt to be significantly exacerbated by HCQ. Any adverse effect (AE) of ≥ grade 3 attributed to HCQ (nausea, vomiting, diarrhea, rash, and visual field deficit) resulted in the dose being held until the AE had resolved to ≤ grade 1 or baseline. If the AE resolved, reinstitution of treatment occurred at a dose reduced by 200 mg daily with up to 3 dose reductions allowed for subsequent toxicity. During maintenance cycles TMZ dosing was reduced if platelets < 50 × 109/L and/or absolute neutrophil count < 1 × 109/L, by increments of 50 mg/m2, with 100 mg/m2 the lowest allowed treatment dose for continuation on study. Adverse events and toxicities were graded according to Common Terminology Criteria for Adverse Events version 3.0.
Patient evaluation
Evaluations performed within 14 d of beginning therapy included a medical history, physical and neurological examinations, Mini-Mental Status Exam, KPS determination, electrocardiogram, chest radiograph, vital signs, blood count with differential and platelet counts (complete blood count), blood coagulation parameters, serum chemistry profile, urinalysis, and pregnancy test for women of child-bearing potential. During the initiation cycle a complete blood count was performed weekly or more often if grade 3 or 4 myelosuppression was observed. History, physical and neurological examinations, Mini-metal status exam, KPS determination, complete blood count, and serum chemistries were performed within 3 d before every maintenance cycle.
Imaging studies were performed within 14 d of beginning treatment and after every 2 maintenance cycles (of HCQ and TMZ or HCQ alone). If criteria for progression (new lesions on imaging, or neurologic deterioration on stable or increasing corticosteroids without another explanation) were met, the patient would be removed from study. In the absence of progression, treatment with TMZ and HCQ was continued for a maximum of 6 maintenance cycles. This was followed by monthly cycles of HCQ alone until progression or removal from study for other reasons.
Statistical considerations and data analyses
The conventional 3 + 3 design with 4 pre-specified dose levels was used in the phase I portion of the study with a target DLT rate of ≤ 33%. The MTD was defined as (a) the dose producing DLT in 2 out of 6 patients, or (b) the dose level below the dose which produced DLT in ≥ 2 out of 3 patients, or in ≥ 3 out of 6 patients. No intra-patient dose escalation was permitted. Patients were evaluable for the cohort if they completed 90% of their expected dose of HCQ for the initiation cycle. Patients who experienced a DLT were evaluable for the cohort after at least one dose of HCQ.
For assessing the efficacy of the regimen in term of overall survival in the phase II portion of the study, the overall failure rate was estimated and compared with the failure rate of 0.6 per person-year of follow-up found in a phase III trial performed in the same patient population treated with RT plus concomitant and adjuvant TMZ.Citation1 The primary endpoint was death due to any cause and survival time was defined from time of histological diagnosis to death. The overall failure rate was expressed as hazard of failure per person-year of follow-up. The trial was designed to have 90% power to detect a hazard ratio of 0.70, a 30% reduction in hazard rate of death from a hazard rate of 0.6 in the phase III trial at an α level of 0.1 (one-sided). A minimum of 55 death events was required at the time of analysis. Central review of pathology and neuroimaging was mandated for all patients with a documented complete or partial response. Survival probability and median time of survival were calculated using the Kaplan-Meier method. All P values reported are 2-sided unless specified. All analyses were performed with SAS software (version 9.2; SAS Institute). For PD analysis of AV a mixed-effects model was fit to the mean autophagic vacuoles for all patients (n = 40) and phase II patients only (n = 20). The model included a fixed effect of continuous measurement time (baseline, week 3, week 9) and a random subject effect.
Pharmacodynamic assessment of autophagy
Sixteen milliliters of venous blood was collected in BD CPT vacutainer tubes (BD, Inc., 37253) from patients prior to treatment, 3 wk after starting protocol therapy (during chemoradiation and HCQ) and 9 wk after starting treatment (while receiving HCQ alone). Manufacturer’s instructions were followed to collect PBMC in 2 cell pellets. Cells obtained from PBMC pellet one were immediately fixed with 2% glutaraldehyde and stored at 4 °C until embedding. Embedding and image capture were performed as previously described.Citation14 For quantification of AV in PBMC using electron microscopy, high-powered micrographs (10,000–12,000×) of 20 to 25 mononuclear cells from multiple distinct low-powered fields in each sample were obtained. Autophagic vacuoles were scored by 2 independent investigators who were blinded to treatment time points. Morphological criteria for AV included 1) circularity, 2) contrast with structures that were white or lighter than the cytoplasm, 3) vacuoles with contents, 4) vacuoles > 200 nm in size and, 5) vacuoles > 200 nm interior to the plasma membrane. Vesicular structures with cristae characteristic of mitochondria in cross section were excluded. The average of 2 investigators counts are presented as mean ± standard error of the mean. PBMC pellet 2 was immediately frozen. For immunoblotting, after thawing on ice, the pellet was lysed with RIPA buffer (prepared according to ref. Citation14). Protein from cell lysates were resolved by gel electrophoresis and transferred to nitrocellulose membrane. Immunoblotting was performed using anti-MAP1LC3B (LC3, 1:500; QCB Biologicals, antibodies raised in rabbits using rat LC3B N terminus peptide) and anti-ACTB (actin, 1:5000; Sigma, A1978) antibodies as previously described.Citation14
Pharmacokinetic analysis of HCQ
Collection of PK samples
Whole blood was collected from each patient at the following time points: 1) within 14 d prior to starting protocol therapy (baseline); 2) wk 3–4; 3) wk 9–10; wk 4 of maintenance cycles 1, 2, 3, and 6. Blood was collected in green top tubes containing sodium heparin (BD, 367884), and stored at ≤ 70 °C until analysis. Pharmacokinetic logs were maintained to document the timing of last meal and most recent HCQ ingestion, timing of blood draw, and concurrent medications.
Determination of HCQ in blood
Study samples were thawed at room temperature and mixed before removing a 100-µL aliquot for analysis. After spiking with 5 µL of internal standard (IS) working solution (10 µg/mL chloroquine [Sigma-Aldrich, C6628] in acetonitrile), the sample was vigorously mixed with 300 µL of acetonitrile, then centrifuged (10,000 g, 5 min). The supernatant fraction (250 µL) was combined with 200 mM triethylamine (Sigma-Aldrich, T0886), 272 mM formic acid (Sigma-Aldrich, F0507) in water (25 µL) and 100 µL of this solution was injected onto a 150 × 4.6 mm Luna 5 µm CN HPLC column (Phenomenex, Torrance, CA) eluted with a binary mobile phase composed of 30% acetonitrile (Sigma-Aldrich, 271004) and 70% 10 mM triethylamine, 13.6 mM formic acid in water (pH 4.0) delivered at 1.0 mL/min. An Agilent 1100 Series LC/MSD system with an electrospray ionization interface (Agilent Technologies, Palo Alto, CA) was used for detection. Nitrogen was used as the nebulizing gas (60 p.s.i.) and as the drying gas (10 L/min, 350 °C). The single-quadrupole mass spectrometer was operated in the selected ion-monitoring mode to measure positive ions corresponding to [M + H]+ ions of HCQ and the IS at m/z 336.2 and 320.2, respectively. Additional operating parameters were: capillary voltage, 1,500 V; fragmentor voltage, 150 V; peak width, 0.1 min; dwell time, 114 msec. Extracted ion chromatograms were integrated to provide peak areas.
Calibration standards of HCQ in human whole blood had concentrations ranging from 10 to 1,000 ng/mL. Study samples were independently assayed in duplicate, on different ds, together with a set of calibration standards, drug-free blood prepared for analysis with and without addition of the IS, and a set of 3 quality control samples (HCQ 30, 450, and 900 ng/mL in whole blood). The calibration curves were best described by the equation, y = a + bxc, where y is the HCQ/IS peak area ratio and x is the known analyte concentration in each calibration standard. Nonlinear regression was performed using WinNonlin Professional version 5.0 software (Pharsight Corp., Cary, NC), with weighting in proportion to the reciprocal of the HCQ concentration, normalized to the number of calibration standards, to determine the y-intercept (a), coefficient (b) and exponent (c), of the best-fit curve. Values of the parameters describing the best-fit line were used to calculate the analyte concentration in study samples. Specimens with concentrations exceeding the upper range of the standard curve were reassayed upon appropriate dilution with drug-free human plasma or whole blood. The average of the 2 initial determinations of each study sample was calculated. Samples were reassayed in cases where the individual determinations differed from their average by more than 15%. During the analysis of samples from this study, the calibration curves exhibited correlation coefficients ranging from 0.995 to 1.000 and the inter-d accuracy was within ± 8% of the known concentration of the calibration standards and quality-control solutions with a precision ≤ 4.2%.
Population PK modeling
Initial estimates for a base population model were derived from a naïve-pooled data analysis of individual patient blood concentration time data. One and 2-compartment models with first-order absorption and elimination, with and without a lag time, were evaluated as the potential pharmacokinetic structural model. Visual inspection of conditional weighted residuals vs. individual predicted values plots suggested that a multiplicative error model was appropriate, Diagnostic scatter plots and Akaike information criteria were used to select the base model, a 2-compartment model with a lag-time variable.
Visual inspections of scatter and box plots for eta (random effect) values were used to explore potential continuous (age, weight, KPS, minimental exam log weight, log age) and categorical (corticosteroid use, anticonvulsant use, sex) covariates. Covariates were centered on their median values. Stepwise covariate selection processes were conducted to build the full model. Model building criteria were based on covariate models associated with an increase in objection function value greater than 3.84 with 1 degree of freedom (P < 0.05) using the likelihood ratio test. No covariant interactions significantly improved the model. The first order with conditional estimates extended least squares method was used to confirm the final model, which was as follows: Cl/f = tvCl/F * exp(nCl)/F; Q = tvQ * exp(nQ); V/F = tvV/F * exp(nV)/F; V2/F = tvV2/F * exp(nV2)/F; Ka = tvKa * exp(nKa), tlag = tvtlag * exp(ntlag). The predictive ability of the final model was evaluated using a Monte Carlo simulation with 200 simulated data sets based on the parameters and time points. Bootstrapping with 1000 subjects per each bootstrap run was conducted to provide estimates of the precision of fixed effects.
Individual pharmacokinetic parameters (Cl/F, Q, V/F, V2/F, Ka, tlag) for each patient were derived from the final population model and used to simulate time-concentration profiles using a one-compartment model with WinNonlin® 6.2 (Pharsight Corporation, Cary, NC). The simulated blood HCQ concentrations were compared with observed concentrations to determine the predictive performance of the model. HCQ pharmacokinetic parameter estimates (peak blood concentration, Cmax; area under the concentration-vs.-time curve, AUC) from these simulations were used to explore pharmacokinetic-pharmacodynamic (PK-PD) relationships.
| Abbreviations: | ||
| ABTC | = | Adult Brain Tumor Consortium |
| AE | = | adverse effect |
| AUC | = | area under the curve |
| AV | = | autophagic vacuole |
| Cl/F | = | apparent oral clearance |
| CQ | = | chloroquine |
| CART | = | classification and regression trees analysis |
| DLT | = | dose-limiting toxicity |
| EM | = | electron microscopy |
| EORTC | = | European Organization for Research and Treatment of Cancer |
| GB | = | glioblastoma multiforme |
| HCQ | = | hydroxychloroquine |
| IS | = | internal standard |
| Ka | = | first-order absorption rate constant |
| KPS | = | Karnofsky performance status |
| MTD | = | maximal tolerated dose |
| PBMC | = | peripheral blood mononuclear cells |
| PD | = | pharmacodynamics |
| PK | = | pharmacokinetics |
| Q | = | intercompartmental clearance |
| RT | = | radiation therapy |
| Tlag | = | lag time |
| TMZ | = | temozolomide, V/F, apparent volume of distribution in central compartment |
| V2/F | = | apparent volume of distribution in peripheral compartment |
Additional material
Download Zip (434.3 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NCI 1R21CA133898 (RKA), NCI 3R21CA133898 (RKA), 1K23CA120862 (RKA), CTEP grant. We thank the ABTC Central Office for data collection and integration. We thank Ray Meade and biomedical imaging core for EM processing and technical advice.
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987 - 96; http://dx.doi.org/10.1056/NEJMoa043330; PMID: 15758009
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448 - 57; http://dx.doi.org/10.1038/sj.cdd.4401359; PMID: 14713959
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548 - 58; http://dx.doi.org/10.1038/sj.cdd.4402030; PMID: 16946731
- Zhao H, Cai Y, Santi S, Lafrenie R, Lee H. Chloroquine-mediated radiosensitization is due to the destabilization of the lysosomal membrane and subsequent induction of cell death by necrosis. Radiat Res 2005; 164:250 - 7; http://dx.doi.org/10.1667/RR3436.1; PMID: 16137197
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene 2010; 29:3241 - 51; http://dx.doi.org/10.1038/onc.2010.74; PMID: 20348950
- O’Neill PM, Bray PG, Hawley SR, Ward SA, Park BK. 4-Aminoquinolines--past, present, and future: a chemical perspective. Pharmacol Ther 1998; 77:29 - 58; http://dx.doi.org/10.1016/S0163-7258(97)00084-3; PMID: 9500158
- Romanelli F, Smith KM, Hoven AD. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr Pharm Des 2004; 10:2643 - 8; http://dx.doi.org/10.2174/1381612043383791; PMID: 15320751
- Katz SJ, Russell AS. Re-evaluation of antimalarials in treating rheumatic diseases: re-appreciation and insights into new mechanisms of action. Curr Opin Rheumatol 2011; 23:278 - 81; http://dx.doi.org/10.1097/BOR.0b013e32834456bf; PMID: 21448012
- Marmor MF, Kellner U, Lai TY, Lyons JS, Mieler WF, American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology 2011; 118:415 - 22; http://dx.doi.org/10.1016/j.ophtha.2010.11.017; PMID: 21292109
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709 - 30; http://dx.doi.org/10.1038/nrd3802; PMID: 22935804
- Leone RD, Amaravadi RK. Autophagy: a targetable linchpin of cancer cell metabolism. Trends Endocrinol Metab 2013; 24:209 - 17; http://dx.doi.org/10.1016/j.tem.2013.01.008; PMID: 23474062
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120:237 - 48; http://dx.doi.org/10.1016/j.cell.2004.11.046; PMID: 15680329
- Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2006; 144:337 - 43; http://dx.doi.org/10.7326/0003-4819-144-5-200603070-00008; PMID: 16520474
- Smith ER, Klein-Schwartz W. Are 1-2 dangerous? Chloroquine and hydroxychloroquine exposure in toddlers. J Emerg Med 2005; 28:437 - 43; http://dx.doi.org/10.1016/j.jemermed.2004.12.011; PMID: 15837026
- Gunja N, Roberts D, McCoubrie D, Lamberth P, Jan A, Simes DC, Hackett P, Buckley NA. Survival after massive hydroxychloroquine overdose. Anaesth Intensive Care 2009; 37:130 - 3; PMID: 19157361
- Gerber DE, Grossman SA, Zeltzman M, Parisi MA, Kleinberg L. The impact of thrombocytopenia from temozolomide and radiation in newly diagnosed adults with high-grade gliomas. Neuro Oncol 2007; 9:47 - 52; http://dx.doi.org/10.1215/15228517-2006-024; PMID: 17108062
- Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit 2003; 25:671 - 81; http://dx.doi.org/10.1097/00007691-200312000-00005; PMID: 14639053
- Lim HS, Im JS, Cho JY, Bae KS, Klein TA, Yeom JS, Kim TS, Choi JS, Jang IJ, Park JW. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob Agents Chemother 2009; 53:1468 - 75; http://dx.doi.org/10.1128/AAC.00339-08; PMID: 19188392
- Khoury H, Trinkaus K, Zhang MJ, Adkins D, Brown R, Vij R, Goodnough LT, Ma MK, McLeod HL, Shenoy S, et al. Hydroxychloroquine for the prevention of acute graft-versus-host disease after unrelated donor transplantation. Biol Blood Marrow Transplant 2003; 9:714 - 21; http://dx.doi.org/10.1016/j.bbmt.2003.08.006; PMID: 14652855
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369 - 79; PMID: 24991839
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380 - 90; http://dx.doi.org/10.4161/auto.29264; PMID: 24991834
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391 - 402; PMID: 24991838
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403 - 14; http://dx.doi.org/10.4161/auto.29231; PMID: 24991835