
Abstract
Blocking autophagy with hydroxychloroquine (HCQ) augments cell death associated with alkylating chemotherapy in preclinical models. This phase I study evaluated the maximum tolerated dose (MTD), safety, preliminary activity, pharmacokinetics, and pharmacodynamics of HCQ in combination with dose-intense temozolomide (TMZ) in patients with advanced solid malignancies. Forty patients (73% metastatic melanoma) were treated with oral HCQ 200 to 1200 mg daily with dose-intense oral TMZ 150 mg/m2 daily for 7/14 d. This combination was well tolerated with no recurrent dose-limiting toxicities observed. An MTD was not reached for HCQ and the recommended phase II dose was HCQ 600 mg twice daily combined with dose-intense TMZ. Common toxicities included grade 2 fatigue (55%), anorexia (28%), nausea (48%), constipation (20%), and diarrhea (20%). Partial responses and stable disease were observed in 3/22 (14%) and 6/22 (27%) patients with metastatic melanoma. In the final dose cohort 2/6 patients with refractory BRAF wild-type melanoma had a near complete response, and prolonged stable disease, respectively. A significant accumulation in autophagic vacuoles (AV) in peripheral blood mononuclear cells was observed in response to combined therapy. Population pharmacokinetics (PK) modeling, individual PK simulations, and PK-pharmacodynamics (PD) analysis identified a threshold HCQ peak concentration that predicts therapy-associated AV accumulation. This study indicates that the combination of high-dose HCQ and dose-intense TMZ is safe and tolerable, and is associated with autophagy modulation in patients. Prolonged stable disease and responses suggest antitumor activity in melanoma patients, warranting further studies of this combination, or combinations of more potent autophagy inhibitors and chemotherapy in melanoma.
Introduction
Temozolomide (TMZ) is an oral alkylating agent that is used widely to treat glioma,Citation1 metastatic melanoma,Citation2 breast cancer,Citation3 and non-small cell lung cancer.Citation4,Citation5 Although response rates are low in most of these diseases, TMZ is prescribed due to ease of administration, tolerability, and its known capacity to cross the blood-brain barrier, providing modest antitumor activity against brain metastases. Numerous schedules of TMZ administration have been developed and compared, especially for metastatic melanoma.Citation2,Citation6 Unfortunately, dose intensity of TMZ has failed to improve outcomes thus far compared with standard dosing of TMZ in or intravenous dacarbazine in melanoma.Citation7
Preclinical studies have demonstrated that autophagy, a lysosome-dependent degradation process within the cell, may play a significant role in limiting the efficacy of alkylating chemotherapy. Autophagy is a mechanism by which damaged intracellular organelles and macromolecules are sequestered and degraded. The clearance of damaged organelles and recycling of macromolecules to generate energy or building blocks for further growth is the presumed mechanism by which therapy-induced autophagy can promote survival in cancer.Citation8 Alkylating chemotherapy drugs such as TMZ have been shown to induce cytoprotective autophagy in cancer cells.Citation9-Citation11 Autophagy inhibition with chloroquine derivatives in combination with alkylating chemotherapies has been shown to produce additive or synergistic cytotoxicity.Citation11,Citation12 This phase I study was conducted to determine the recommended phase II dose (RPTD) of daily oral HCQ administered in combination with fixed dose, dose-intense TMZ in patients with advanced solid malignancies.
Results
Patient characteristics
A total of 40 patients with advanced solid malignancies were consented and 37 patients received treatment between June 2008 and June 2012 onto 5 dose cohorts (Table S1 and Materials and Methods). All 37 patients were evaluated for safety, with 3 patients never receiving any study drug (screen failure). Twenty-nine patients were evaluable for response. For 8 nonevaluable patients, the reasons for exiting the study prior to receiving 2 wk of combined therapy (evaluable for response) were disease progression (n = 6) and patient intercurrent illness (n = 2: Chronic obstructive pulmonary disease flare and bone fracture from mechanical fall). Demographic and clinical characteristics of all enrolled patients are listed in , and melanoma patients in Table S2. Given that the most common use of dose-intense TMZ is for metastatic melanoma, 73% of patients had stage IV melanoma. Other malignancies represented in this trial included breast cancer, lung cancer, and sarcoma. While 68% of patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0, 43% had brain metastases. Eighteen patients had received prior treatments, whereas 19 were previously untreated. Among the patients who had received prior therapy, 6 patients had 3 or more regimens. Of 25 melanoma patients, the median age was 61, 69% of patients were male, 92% of patients had stage IV M1c disease, and 75% of patients had an ECOG performance status 0 (Table S2). Since TMZ was considered first-line therapy for melanoma, the median number of prior therapies for melanoma patients was 0, with 37% of patients having received prior chemotherapy. Only 2 patients had received prior ipilimumab. Lactate dehydrogenase was elevated in 49% of patients and 48% of patients had brain metastases including 2 patients who did not receive radiation therapy prior to starting treatment on study. Mutational status of the tumor was missing for 44% of patients, but of the 14 with available genotyping for BRAF, 36% of patients were found to have a BRAF mutation.
Table 1. Patient characteristics (n = 37)
Dose-escalation
A 2 wk run-in of single agent HCQ was followed by combined therapy with dose intense TMZ. The pretreatment with HCQ served 2 potential purposes: 1) to achieve steady-state concentrations so that autophagy was blocked at the onset of TMZ therapy, and 2) to assess the single agent pharmacodynamics effects of HCQ in surrogate tissues. Three patients were enrolled to dose level 1 at study initiation; no dose-limiting toxicity (DLT) was observed. HCQ dosing was escalated to dose level 2 with no DLT observed. HCQ dosing was then escalated to dose level 3 during which a serious adverse event involving third-degree heart block was observed. This patient was receiving sotalol for chronic atrial fibrillation and within 8 d of starting HCQ 400 mg twice daily he developed third-degree heart block. This significant adverse event was attributed to dehydration and not to the study drug, but this event prompted a cohort expansion of an additional 10 patients at dose level 3 and serial electrocardiograms on all subsequent patients. No further electrocardiogram abnormalities were observed. HCQ dose was escalated to dose level 4 during which one grade 3 rash was observed and mandated enrollment of 3 additional patients. No additional grade 3 or 4 rashes were observed. The HCQ dose was escalated to dose level 5, where one patient experienced grade 3 nausea and vomiting during the first cycle of administration of 1200 mg/d HCQ requiring cohort expansion with an additional 3 patients. No additional dose-limiting nausea and vomiting was observed in the expanded cohort. No maximal tolerated dose for HCQ when combined with dose intense TMZ and dose level 5 (HCQ 600 mg twice daily) was declared the RPTD for the study combination.
Toxicity
Thirty-seven patients received at least one dose of HCQ and were evaluable for toxicities. Besides the DLTs and serious adverse events noted above, the incidence of treatment related toxicities greater than 5% are summarized in . The most common (> 10%) ≥ grade 1 treatment-related adverse events in all patients included fatigue (97%), nausea (65%), constipation (22%), diarrhea (24%), and anorexia (35%). Dose delays of temozolomide were uncommon, but dose reductions of HCQ especially after 3 mo of therapy and at doses above 400 mg twice daily were common.
Table 2. Adverse events by treatment cohort and grade
Efficacy
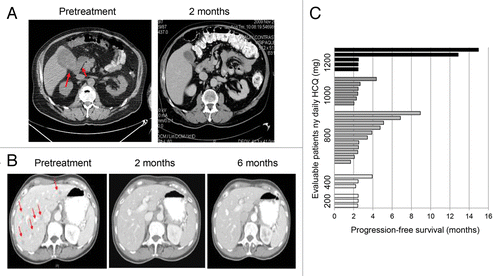
Partial response (PR) was observed in 3/29 (10%) and stable disease was observed in 8/29 (27%) of patients (). The 3 PRs were observed in patients with advanced melanoma. One melanoma patient with brain metastases and large 18-fluorodeoxyglucose-avid gallbladder and mesenteric metastases had complete resolution of extracranial disease when treated with HCQ 400 mg twice daily with dose-intense TMZ. After 4 mo of treatment he progressed in one persistent brain lesion (). A second melanoma patient treated with HCQ 600 mg twice daily with dose-intense TMZ experienced complete resolution of multiple liver metastases that lasted for more than 1 y (). She had persistent leptomeningeal enhancement on brain MRI, which was unchanged for more than a y despite being untreated with radiation therapy. Prolonged stable disease > 4 mo was observed in patients with metastatic melanoma and breast cancer, with 9/29 (31%) evaluable patients surviving progression-free beyond 4 mo (). Three out of 22 evaluable patients with advanced melanoma (14%) had PR and 6/22 (27%) had stable disease (SD) as their best response (Table S3). One patient with stage IV melanoma who had stable disease with TMZ and HCQ 200 twice daily had a PET/CT scan which demonstrated central necrosis of multiple tumors (Fig. S1).
Table 3. RECIST response in evaluable patients
Figure 1. Antitumor activity of dose-intense TMZ and HCQ. (A) Treatment induced clearance of 2 large FDG-avid gallbladder and mesenteric metastases in a melanoma patient with brain metastases. (B) Durable near complete response of all extra CNS lesions in a metastatic melanoma patient with leptomeningeal disease. Red arrows: melanoma metastases. (C) Progression-free survival of patients.
Pharmacokinetics
The population pharmacokinetics HCQ analysis was performed using 127 nonbaseline blood samples from 35 patients collected over a period up to 196 d. The population model PK parameters do not specifically represent steady-state values, as they were determined from multiple repeated single doses taken by individual patients during their period of participation in the study. The model that best described the disposition of HCQ blood concentrations was a 2-compartment model with first-order absorption. Inclusion of a lag time did not improve the model. No covariate interactions were identified that significantly improved the model. A nondiagonal fit was not superior to a diagonal fit based on Akaike information criteria, -2(LL), and Bayesian information criteria. The final model was as follows: first order absorption rate constant (Ka) = typical value (tv)Ka * exp(nKa); apparent volume of distribution in central compartment = tvV * exp(nV)/F; apparent volume of distribution in peripheral compartment = tvV2 * exp(nV2)/F; apparent oral clearance = tvCl * exp(nCl)/F; intercompartmental clearance (Q) = tvQ * exp(nQ). The residual error was supported by an additive error model, as described by: Cobs = C + CEps, where Cobs is the observed concentration, C is the predicted concentration, and Ceps is the zero mean normally distributed random variable. shows the individual predicted concentrations vs. the observed concentrations from the population PK model. HCQ population PK parameters are presented in .
Figure 2. Pharmacokinetic analysis of HCQ in patients receiving dose-intense TMZ and HCQ. (A) Observed vs. individually predicted concentrations of HCQ based on the population PK model. (B) Estimated peak concentrations (Cmax). (C) Estimated average concentrations (Cavg). (D) PK-response relationship. AUC, area under curve.
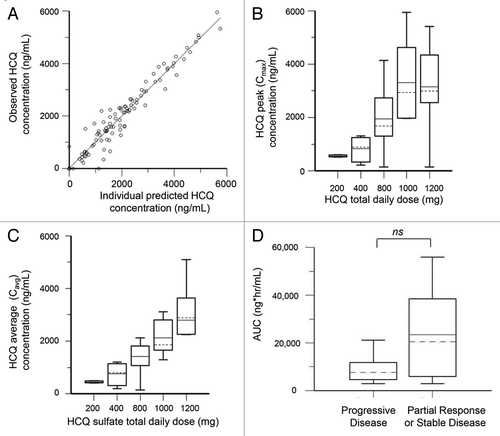
Table 4. Population pharmacokinetic parameters estimates for HCQ
The final PK model developed was used to simulate HCQ blood concentrations for individual patients at steady-state, which was achieved on average after 400 h (16 d). Individual PK parameter estimates derived from the population were most variable for apparent peripheral volume of distribution (Vp/F) and intercompartmental clearance (Q) (). Blood HCQ concentration relationships for area under the concentration-time curve, Cmax, and Cavg were proportional to daily HCQ dose (). An exploratory unplanned analysis of associations between estimated PK individual parameters and clinical response (), the median HCQ exposure (area under the curve [AUC]) in patients that had either a PR or SD (those who derived clinical benefit) was 2.4-fold higher compared with PD patients (median AUC [PR + SD] 205,211 [interquartile range 80,939–356,502] vs. [PD] 85,810 [49,265–114,799]). Although this finding (1-sided P = 0.0635, exact Wilcoxin test) was not statistically significant, this was a notable difference that suggests HCQ exposure may have played a role on antitumor activity of this regimen. No other individual PK parameter significantly correlated with clinical response in ths study (Table S4).
Pharmacodynamics
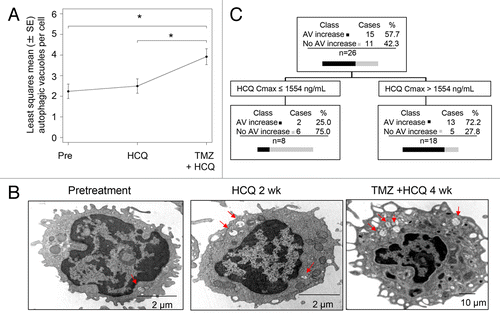
The mean number of AVs per cell was scored in serially collected peripheral blood mononuclear cells (PBMC) from patients at the following time points: 1) pretreatment; 2) after the 2 wk run-in of HCQ; 3) after 4 wk of combined HCQ and TMZ therapy. In the 37 patients that received treatment, 96/111(86%) possible PBMC samples were analyzed. Four patients (400 mg/d [1]; 800 mg/d [2], 1200 mg/d [1]) had complete sets of 3 samples missing due to sample mishandling. Four samples from patients that had 2/3 PBMC analyzed were missing due to sample mishandling (3) or patient refusal (1). In the overall population, there was a significant therapy-induced accumulation of AVs after 4 wk of combined therapy reflecting simultaneous autophagy induction by TMZ and distal blockade by HCQ (). Mean AV counts increased from 2.19 at baseline to 2.45 after HCQ treatment, and to 3.84 after treatment with HCQ and TMZ. The difference between HCQ plus TMZ and baseline was significant (P = 0.0007), as was the difference between HCQ plus TMZ and HCQ only (P = 0.0034). Representative images of serial PBMC demonstrate the stepwise accumulation of AVs that could be seen in individual patients (). After 2 wk of single agent HCQ, CART analysis identified a threshold for HCQ Cmax of 1554 ng/mL that correlated significantly with AV accumulation. Patients with Cmax below 1554 ng/mL (n = 8) were found to have a median AV change of −0.21 (interquartile range [IQR]: −1.35, 0.03), while patients with Cmax above 1554 ng/mL (n = 18) had a median AV change of 0.89 (IQR: -0.45, 1.37) (Kruskal-Wallis P = 0.0227; Kolmogorov-Smirnov P = 0.0319; ). The CART analysis was also conducted using PK and pharmacodynamic (PD) data of 21 patients, with the target variable defined as the change in number of AV from baseline to 4 wk of combined therapy (TMZ + HCQ) and Cmin, Cmax, and AUC as predictors. Patients with Cmax below 856 ng/mL (n = 2) produced a median AV change of −0.43 (IQR: −0.78, −0.09), while patients with Cmax above 856 ng/mL (n = 19) produced a median AV change of 1.30 (IQR: 0.04, 3.21). Statistical inference was not performed due to the small size of one of the groups.
Figure 3. Pharmacodynamic effects of TMZ and HCQ on autophagic vacuole accumulation in PBMC, and PK-PD correlation. (A) Mixed-effects model of mean ± SD autophagic vacuoles (AVs)/cell in PBMC. Dotted line: regression line. (B) Representative electron micrographs of serial PBMC; red arrows: AV (C) Classification tree from CART analysis. (D) Histogram of AV change at 2 wk in patients with estimated HCQ Cmax above or below 1554 ng/mL.
Discussion
While the role of autophagy in cancer and cancer therapy is multifaceted, genetic ablation of key autophagy genes in multiple mouse models of oncogene-driven cancer impairs the progression of tumorigenesis.Citation13-Citation15 We have previously shown that autophagy inhibition with antimalarial CQ derivatives could augment cancer cell death when combined with an apoptotic stress such as alkylating chemotherapy in vivo.Citation12 There is now clear evidence that many DNA-damaging chemotherapies induce cytoprotective autophagy including nitrogen mustard,Citation12 doxorubicin,Citation16 melphalan,Citation16 cisplatin,Citation17 and etoposide.Citation10 The rationale for combining a DNA-damaging chemotherapy and a lysosomal autophagy inhibitor is that the former induces massive autophagic flux and the latter prevents autophagic contents from being degraded, leading to an accumulation of ineffective and toxic AVs that leads to cell death. Preclinical studies indicate that lysosomal autophagy inhibition is associated with a burst of reactive oxygen species, presumably from the uncleared and damaged mitochondria within ineffectively cleared AV. This ROS in turn produces DNA damage and activation of apoptosis.Citation18
Among DNA-damaging chemotherapeutics, TMZ has attractive qualities that have led to its widespread use in multiple malignancies, including its oral bioavailability, that it is generally well tolerated, crosses the blood brain barrier and it can be dosed by multiple schedules.Citation19 For these reasons, and because multiple preclinical and correlative studies have demonstrated that TMZ induces cytoprotective autophagy, and its activity can be augmented by combination with CQ derivatives,Citation9-Citation11,Citation20,Citation21 we chose to determine the safety of combining TMZ and HCQ (a safer derivative than CQ to dose escalateCitation8) in patients with advanced solid tumors. Although previous randomized phase III studies have failed to show improved survivals with dose-intense TMZ regimens, we postulated that autophagy induction may have contributed to the resistance even in the face of higher dose exposure of the chemotherapeutic. We chose a dose-intense TMZ regimen to serve as the backbone for this combination, assuming the higher dose exposure of the chemotherapy agent would induce higher levels of autophagy than standard TMZ dosing regimens, thereby sensitizing cancer cells further to the effects of HCQ. This could also lead to enhanced toxicity, with myelosuppression a key concern.
Surprisingly, in this phase I study of dose-intense TMZ and HCQ there were no significant recurring dose-limiting toxicities, and as was observed with other HCQ combinations with bortezomib (Vogl et al., this issueCitation22) or temsirolimus (Rangwala et al., this issueCitation23), the highest planned doses of HCQ 1200 mg/d in combination with dose intense TMZ was well tolerated. One elderly metastatic melanoma patient was treated at these doses for over 1 y with a near-complete response, and another patient with metastatic melanoma patient who had failed 2 prior therapies has had stable disease for over 8 mo. In both cases reduction of the HCQ dose was necessary after 3 to 4 mo due to late appearing grade 3 fatigue, nausea, and vomiting, suggesting that further dose escalation beyond 1200 mg/d HCQ (600 mg twice daily) may not be feasible in combination with dose-intense TMZ. This is the highest dose reported in use for patients with rheumatoid arthritis and produced dose-limiting nausea in the population after 6 wk as a single agent.Citation24 The lack of excess myelosuppression was also unexpected since significant dose-limiting myelosuppression was observed in the phase I trial of concomitant and adjuvant TMZ with radiation therapy for newly resected glioma (Rosenfeld et al., this issueCitation25). In that study, the maximal tolerated dose of HCQ was 600 mg daily when it was combined with TMZ 75 mg/m2 daily for 6 wk with concomitant brain radiation. Patients treated at HCQ 800 mg in that combination regimen all experienced grade 4 myelosuppression. This indicates that either the involvement of whole brain radiation somehow magnifies the toxicity of TMZ and HCQ or that the 6-wk continuous daily dosing of TMZ as opposed to the intermittent dosing of the dose-intense regimen studied in this trial, was the reason why myelosuppression was observed with the former and not the latter approach to TMZ dosing. This finding has significant implications for how future studies involving autophagy inhibitor and chemotherapy combinations should be approached, and suggests that intermittently dosed chemotherapy backbones are the best approach for these combinations. The population PK estimates for HCQ are consistent with those reported with single dose and prolonged HCQ administration to healthy subjects and patients with Plasmodium vivax malaria, although HCQ concentrations were measured in plasma in those studies rather than whole blood, as was done in this study.Citation26 The prolonged HCQ elimination half-life necessitates at least 2 wk of continuous drug administration to achieve steady-state blood concentrations. Despite pharmacokinetic variability of HCQ in this population that was also taking dose-intensive TMZ, dose-exposure relationships were proportional for area under the concentration-time curve, peak, trough, and average blood HCQ concentrations at steady-state.
The pharmacokinetic variability of HCQ in this population may be the key reason that although a greater than 2-fold difference in the median HCQ AUC was found in patients that derived clinical benefit compared with those that did not from this regimen, this finding was not statistically significant. Further study of this regimen in a larger phase II trial may uncover a significant association between HCQ exposure and clinical benefit. Our PBMC data demonstrated significant accumulation of AV in PBMC. The magnitude of significant AV accumulation in the 1200 mg cohort was a 2-fold accumulation of AV only observable after 6 wk of HCQ and 4 wk of combination therapy of TMZ and HCQ. Given that the largest change was observed when TMZ was added to HCQ, we cannot be certain that the TMZ-associated induction of autophagy and not HCQ-associated blockade of AV clearance was the main contributing factor to the increase in AV at 6 wk compared with baseline. However, similar to the results of the Adult Brain Tumor Consortium TMZ, HCQ, radiation study in glioma (Rosenfeld et al., this issueCitation25), PK-PD analysis was able to identify a threshold of estimated peak HCQ concentration that predicted therapy-induced AV accumulation. This finding provides evidence that autophagy modulation with this regimen is possible. Moreover, as demonstrated in other clinical trials in this series (Vogl et al., this issueCitation22; Mahalingam et al., this issueCitation27; Barnard et al., this issueCitation28), HCQ may accumulate within tumor tissue and the magnitude of AV accumulation may be higher within the tumor microenvironment. This study did not have any tumor tissue analysis. Future studies in melanoma patients should focus on tumor biopsy material to study the direct effects of autophagy modulators rather than relying on surrogate tissues such as PBMC. It is likely that even higher doses of HCQ or a more potent aminoquinoline autophagy inhibitor would produce a greater degree of cell death when combined with dose-intense TMZ. This dose escalation trial was not designed to look at efficacy, but there were signs of clinical activity with this combination. Randomized studies would be necessary to determine if this signal of activity with TMZ + HCQ is actually attributable to the addition of HCQ. This study did not have genotype information on the majority of patients and there was no correlation with BRAF mutation and activity of TMZ and HCQ. It may be that a subset of melanoma patients is more or less susceptible to autophagy inhibition. The tolerability of this combination provides an opportunity to consider a 3-drug regimen combining a second apoptotic stressor, which may further sensitize cells to HCQ without overlapping toxicities. For instance TMZ + HCQ could be combined with an anti-angiogenesis inhibitor, targeted therapy, or immunotherapy in future studies.
Materials and Methods
Patient eligibility
Patients ≥ 18 y, with histologically confirmed metastatic or unresectable solid tumor for which standard-of-care options were exhausted or there were no agreed upon standard-of-care options, measurable disease, an ECOG performance status of < 2, adequate hematological (White Blood Count ≥ 3,000/mm3, absolute neutrophil count ≥ 1,500/mm3, platelets ≥ 100,000/mm3), renal (serum creatinine ≤ 2.0 × upper limit of normal [ULN]), hepatic (bilirubin ≤ 1.5 × ULN transaminases ≤ 2.5 × ULN or ≤ 5.0 ULN in the presence of liver metastases), and coagulopathic (international normalized ratio ≤ 1.5 and partial thromboplastin time < ULN;) function were eligible. There was no limit on the number of prior therapies, except that prior TMZ was not allowed. Patients must have discontinued active immunotherapy or chemotherapy ≥ 4 wk prior to entering the study and oral targeted therapies ≥ 2 wk prior to starting the study treatment, and must have recovered from adverse events due to those agents. Patients with brain metastases were also eligible; however, the patients must have completed radiation therapy, if radiation was clinically indicated at the time of diagnosis, and discontinued steroids prior to enrollment. Patients with active, clinically significant and/or uncontrolled medical conditions were excluded, including human immunodeficiency virus, psoriasis, and porphyria, the latter 2 due to the risk of disease exacerbation. The study protocol was reviewed and approved by the Institutional Review Board at the University of Pennsylvania; written informed consent was mandatory and obtained from all enrolled patients.
Treatment
This was an open-label, single-institution, phase I dose-escalation trial of oral HCQ (patients obtained HCQ by submitting prescriptions to retail pharmacies; HCQ is manufactured by a number of generic pharmaceutical companies) with oral, dose-intense TMZ in patients with advanced solid tumors. TMZ was supplied by Schering/Plough/Merck. Treatment consisted of HCQ monotherapy for the first 14 d (HCQ run-in), followed by the addition of dose-intense TMZ, 150 mg/m2 daily for 7 out of every 14 d. The starting dose for HCQ was 200 mg daily. The planned dose escalation schema (Table S1); for doses greater than 200 mg, HCQ was administered in divided, twice-daily doses, taken approximately 12 h apart. On the days when both drugs were administered, the HCQ and TMZ could not be taken at the same time. Antiemetics including ondansetron, were scheduled 30 min prior to administration of TMZ. One cycle was considered 28 d. Treatment was administered until disease progression (as defined by a greater than 20% increase in measurable disease, or the appearance of a new lesion), treatment delay due to toxicity for > 28 d from both TMZ and HCQ, withdrawal of consent, or intercurrent illnesses. Hematological toxicities were attributed to TMZ. Complete blood counts were monitored weekly for the first 6 wk and biweekly thereafter. Redosing of TMZ required an absolute neutrophil count > 1000/mm3, a platelet count > 100,000/mm3, and resolution of all nonhematological toxicities to grade ≤ 2. For dose delays lasting more than 7 d due to hematological toxicities, TMZ was dose reduced from 150 mg/m2 to 125 mg/m2. If a dose delay of > 7 d due to hematological toxicity was observed after the first dose reduction of TMZ, then the HCQ dose was reduced by one dose level and TMZ redosed at 125 mg/m2. If a dose delay of > 7 d due to hematological toxicity was observed after the first dose reduction of TMZ and HCQ, then TMZ was dose reduced to 100 mg/m2. If there was a dose delay of > 7 d after this second dose reduction, the patient remained on single-agent HCQ until disease progression. If after the second dose reduction of TMZ the patient experienced grade 4 neutropenia > 7 d, grade 4 neutropenia with fever (100.5 °F or higher), grade 4 thrombocytopenia, lack of recovery of ANC or platelet count to retreatment levels by the end of the 28 d, or any grade 3 or 4 nonhematological toxicity attributed to TMZ, TMZ was discontinued. All dose reductions in TMZ were permanent. If any adverse events of ≥ grade 3 were observed and attributed to HCQ, HCQ was held until the adverse events resolved to ≤ grade 1 or baseline. If the adverse event resolved, reinstitution of treatment occurred at a dose reduced by 200 mg per d from the starting dose. Two dose reductions of HCQ were allowed before removal from the study. Any adverse events that led to a dose delay of > 28 consecutive d of HCQ led to withdrawal from the study.
Dose-limiting toxicities and dose-escalation plan
DLT was defined as any treatment-related adverse event ≥ grade 3 that occurred during the first 6 wk of this study that was at least possibly related to the study drugs. Nausea and vomiting were considered DLTs if their severity was ≥ grade 3 in the setting of multimodal antiemetic prophylaxis. Any grade 3 nonhematological toxicity was considered a dose-limiting toxicity. Hematological effects were considered a DLT if any of the following occurred in the first cycle of combined therapy: grade 4 neutropenia > 7 ds, febrile neutropenia, or platelet count < 10,000/mm3. Any DLT that led to a dose delay of > 28 consecutive d of HCQ resulted in the patient being taken off treatment. Patients were evaluable for toxicity if they had taken at least one dose of HCQ. Patients were evaluable for dose escalation decision-making and response evaluation if they completed at least 2 wk of concurrent HCQ and TMZ. Nonevaluable patients were replaced. The maximum tolerated dose (MTD) was defined as the highest dose level at which ≤ 1 of 6 patients experienced DLT during the first 6 wk of the study.
The trial used a standard 3 + 3 dose escalation design as previously described,Citation29 with only the HCQ dose subject to dose-escalation rules. No intra-patient dose escalation of HCQ was allowed. If a serious adverse event was observed at any dose level that was not related to study drugs, but was serious or life threatening in any way, 4 to 10 additional patients were enrolled at that dose level to explore safety. Dose escalation beyond 1200 mg HCQ daily was not pursued because this is the highest administered dose typically used in other disorders such as rheumatoid arthritis.Citation24 If no MTD was established, 1200 mg dose level would be the RPTD.
Assessment of response and toxicity
Response assessments consisted of history and physical exam every 4 wk and CT scans of the chest, abdomen, and pelvis every 8 wk (one cycle). Responses were investigator-assessed using the Response Evaluation Criteria in Solid Tumors (RECIST 1.0) guidelines,Citation30 MRI scans of the brain were obtained in follow-up only if clinically indicated. Toxicities were graded using the National Cancer Institute Common Toxicity Criteria Version 3.0. Genotyping of BRAF mutation status in melanoma patients was performed as previously described.Citation31
Pharmacokinetic and pharmacodynamic sampling and assays
All patients enrolled had whole blood drawn for PK analysis at the following time points: pre-dose, 2 wk, 4 wk, 8 wk, 12 wk, and 6 mo. Blood was collected in tubes containing sodium heparin, and stored at −70 °C until analysis.
Whole blood concentrations of HCQ were measured using high-performance liquid chromatography with tandem mass spectrometry detection (HPLC-MS/MS). Sample aliquots of 100 µL containing 500 ng of internal standard (IS) (d4-HCQ) were vortexed with acetonitrile/methanol, then centrifuged. An aliquot of the supernatant fraction was withdrawn, dried under nitrogen gas, then reconstituted with mobile phase and 10 µL injected onto a Kinetex 50 × 3 mm 2.6 um 100A HPLC column (Phenomenex, Torrance, CA). Samples were eluted with a gradient mobile phase of 0.1% formic acid in acetonitrile and water using a 1200 series Agilent HPLC system with an API 4000™ (AB SCIEX, Foster City, CA) mass spectrometer and electrospray interface operated in positive mode with multiple reaction monitoring detection. The capillary voltage was 4000 V with a source temperature of 500 °C. Mass spectrometer parameters were adjusted to maximize the intensity of the [M + H]+ ions in quadrupole 1 and the m/z transition ions of HCQ (337.275 → 248.152) and internal standard (341.150 → 252.035) in quadropole 3.
The HPLC system and mass spectrometer were controlled by AB SCIEX Analyst® software (Version 1.6.1), and data collection and analyses were conducted with the same software. Standard curves were constructed by plotting the analyte to internal standard ratio vs. the known concentration of HCQ (x) in each sample. Standard curves were fit by linear regression with weighting by 1/x. Samples were assayed in duplicate; samples for which the percent difference exceeded 15% were reanalyzed and samples for which concentrations exceeded the range values for the calibration curve were diluted appropriately and reanalyzed.
The calibration curve was linear from 1 to 5000 ng/mL with correlation coefficients ranging from 0.9990 to 0.9999. The lower limit of quantification was 1.0 ng/mL. The correlation coefficients for both inter- and intra-day variability were < 5.6% for each concentration (15 ng/mL, 150 ng/mL, and 1500 ng/mL) studied. The mean accuracy for inter- and intra-day evaluations was between 97.2 and 102%.
Measurement of AV accumulation as a surrogate for autophagic flux was assessed in PBMC and serial tumor biopsies. Venous blood samples were collected in 2 BD Vacutainer® CPT tubes at the following time points: 1) pre-dose, 2) 2 wk (after single-agent HCQ run-in), and 3) 4 wk of combination therapy. Manufacturer’s instructions were followed to collect PBMC in 2 cell pellet fractions. PBMCs were immediately fixed with 2% glutaraldehyde and stored at 4 °C until embedding. Embedding and image capture were performed as previously described.Citation12 For quantification of AV in PBMC using electron microscopy, high-powered micrographs (10,000–12,000×) of 20 to 25 mononuclear cells from multiple distinct low-powered fields in each sample were obtained. Autophagic vacuoles were scored by 2 independent investigators who were masked to treatment time points. Morphological criteria for AV included 1) circularity, 2) contrast with structures that were white or lighter than the cytoplasm, 3) vacuoles with contents, 4) vacuoles > 200 nm in size and, 5) vacuoles > 200 nm interior to the plasma membrane. Vesicular structures with cristae characteristic of mitochondria in cross section were excluded. The average of 2 investigators counts are presented as mean ± standard error of the mean.
Statistical analysis
The primary objective of this study was to determine the MTD of HCQ when given in combination with dose intense TMZ in patients with refractory solid tumors. Secondary endpoints included toxicity rates, and characterization of a dose-response relationship between HCQ exposure and AV accumulation in PBMC. The target DLT rate was 33%. The MTD was defined as a) the dose producing DLT in ≤ one out of 6 patients, or b) the dose level below the dose which produced DLT in ≥ one out of 3 patients, or in ≥ 2 out of 6 patients. Patients were evaluable for toxicity if they received any HCQ. Patients were evaluable for DLT and response if they received at least 2 wk of combined therapy. Descriptive statistics were used. Significance testing was conducted using the Student t test.
Pharmacokinetics
Whole blood HCQ concentration data were analyzed by nonlinear mixed-effect modeling using Phoenix™ NLME 1.2 (Pharsight, Cary, NC). Initial estimates for a base population pharmacokinetic model were derived from a naïve-pooled data analysis of individual patient blood concentration time data. One and 2-compartment models with first-order absorption and elimination, with and without a lag time, were evaluated as the potential pharmacokinetic base structural model. Inter-individual variability of population pharmacokinetic parameters was considered to be log-normally distributed with mean of 0 and variance of ω2. Visual inspection of standard goodness of fit/diagnostic plots and numerical diagnostics were used to determine optimal model fits. The first-order conditional maximum likelihood estimation, The Lindstrom-Bates method was used for the modeling process. Diagnostic scatter plots (individual and population predicted values vs. observed concentrations, conditional weighted residuals vs. time and vs. observed concentrations), Akaike information criteria, and the likelihood ratio test, were used to select the base model. Conditional weighted residuals vs. time and predicted concentration time plots helped confirm that the chosen residual error model was appropriate.
Visual inspections of scatter and box plots for eta (random effect) values were used to explore potential continuous (age, weight) and categorical (sex, dose cohort) covariates. Covariates were centered on their median values. A stepwise covariate selection process was performed to build the full model. Model building criteria were based on covariate models associated with an increase in objection function value greater than 3.84 with one degree of freedom (P < 0.05) using the likelihood ratio test. A visual predictive check with 200 replicates was performed to assess the model performance. A total of 1000 bootstrap runs were performed to provide estimates of the precision of parameter estimates and the 95% confidence intervals for the pharmacokinetic parameters.
Individual pharmacokinetic parameters for each patient were derived from the final population model and used to simulate time-concentration profiles using WinNonlin® 6.2 (Pharsight Corporation, Cary, NC). The simulated blood HCQ concentrations were compared with observed concentrations to determine the predictive performance of the model. HCQ pharmacokinetic parameter estimates (peak blood concentration, Cmax; trough blood concentration, Cmin, average blood concentration, Cavg, area under the concentration-time curve, AUC) from these simulations were used to explore PK-PD relationships.
Pharmacodynamics
We analyzed the AV accumulation data on mean counts of AVs per cell using mixed models including subject id as a random effect and cohort (dose-finding vs. melanoma extension), dose, and time (baseline vs. after HCQ vs. after HCQ + TMZ) as fixed factors. We fit all models in SAS Proc Mixed (SAS Version 9.2; SAS Institute, Inc., Cary, NC).
The PK-PD relationship between HCQ and AV accumulation at 2 wk of single agent HCQ and after 4 wk of combined TMZ and HCQ was first investigated by using an exploratory classification and regression trees analysis (CART; Salford Predictive Modeler Builder v6.6), which identified a threshold effect of the HCQ peak blood concentration (). The CART analysis was conducted using PK and PD data of 26 patients (those with complete PK and PD data), with the target variable defined as the change in number of AV from baseline to 2 wk and Cmin, Cmax, and AUC as predictors. The effect of the threshold value for Cmax on the change in AV from baseline to 2 wk was then investigated using the Kruskal-Wallis test for comparing median values and the Kolmogorov-Smirnov 2-sample test to identify any significant shift in the distribution.
| Abbreviations: | ||
| AUC | = | area under the curve |
| AV | = | autophagic vacuole |
| CQ | = | chloroquine |
| CART | = | classification and regression trees analysis |
| DLT | = | dose-limiting toxicity |
| ECOG | = | Eastern Cooperative Oncology Group |
| HCQ | = | hydroxychloroquine |
| Ka | = | first order absorption rate constant |
| MTD | = | maximal tolerated dose |
| PBMC | = | peripheral blood mononuclear cells |
| PD | = | pharmacodynamics |
| PK | = | pharmacokinetics |
| Q | = | intercompartmental clearance |
| RPTD | = | recommended phase II dose |
| TMZ | = | temozolomide |
| ULN | = | upper limit of normal |
Additional material
Download Zip (530.3 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Investigational temozolomide was provided free of charge to patients by Schering-Plough/Merck. No monetary funding was provided by Schering-Plough or Merck. The conduct of this trial was supported by the Abramson Cancer Center. This research was also supported by NCI SPORE Career Development Award 5 P50 CA 93372-04, NCI-K23-CA-120862, RO1 CA169134 (RKA). The authors would like to thank Ray Meade in the UPENN electron microscopy facility. We would like to thank Kurt D’Andrea, Richard Letrero, and Christopher Watt for providing genotyping analysis for patients in the study.
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987 - 96; http://dx.doi.org/10.1056/NEJMoa043330; PMID: 15758009
- Yang AS, Chapman PB. The history and future of chemotherapy for melanoma. Hematol Oncol Clin North Am 2009; 23:583 - 97, x; http://dx.doi.org/10.1016/j.hoc.2009.03.006; PMID: 19464604
- Trudeau ME, Crump M, Charpentier D, Yelle L, Bordeleau L, Matthews S, Eisenhauer E. Temozolomide in metastatic breast cancer (MBC): a phase II trial of the National Cancer Institute of Canada - Clinical Trials Group (NCIC-CTG). Ann Oncol 2006; 17:952 - 6; http://dx.doi.org/10.1093/annonc/mdl056; PMID: 16565212
- Adonizio CS, Babb JS, Maiale C, Huang C, Donahue J, Millenson MM, Hosford M, Somer R, Treat J, Sherman E, et al. Temozolomide in non-small-cell lung cancer: preliminary results of a phase II trial in previously treated patients. Clin Lung Cancer 2002; 3:254 - 8; http://dx.doi.org/10.3816/CLC.2002.n.009; PMID: 14662033
- Siena S, Crinò L, Danova M, Del Prete S, Cascinu S, Salvagni S, Schiavetto I, Vitali M, Bajetta E. Dose-dense temozolomide regimen for the treatment of brain metastases from melanoma, breast cancer, or lung cancer not amenable to surgery or radiosurgery: a multicenter phase II study. Ann Oncol 2010; 21:655 - 61; http://dx.doi.org/10.1093/annonc/mdp343; PMID: 19767314
- Neyns B, Tosoni A, Hwu WJ, Reardon DA. Dose-dense temozolomide regimens: antitumor activity, toxicity, and immunomodulatory effects. Cancer 2010; 116:2868 - 77; http://dx.doi.org/10.1002/cncr.25035; PMID: 20564393
- Patel PM, Suciu S, Mortier L, Kruit WH, Robert C, Schadendorf D, Trefzer U, Punt CJ, Dummer R, Davidson N, et al, EORTC Melanoma Group. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer 2011; 47:1476 - 83; http://dx.doi.org/10.1016/j.ejca.2011.04.030; PMID: 21600759
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448 - 57; http://dx.doi.org/10.1038/sj.cdd.4401359; PMID: 14713959
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548 - 58; http://dx.doi.org/10.1038/sj.cdd.4402030; PMID: 16946731
- Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478 - 89; http://dx.doi.org/10.1158/1078-0432.CCR-10-2372; PMID: 21325076
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447 - 61; http://dx.doi.org/10.1101/gad.219642.113; PMID: 23824538
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013; 3:1272 - 85; http://dx.doi.org/10.1158/2159-8290.CD-13-0397; PMID: 23965987
- Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, Dong B, Chen X. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res 2011; 17:3248 - 58; http://dx.doi.org/10.1158/1078-0432.CCR-10-0890; PMID: 21288924
- Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F, Xia Q. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun 2012; 423:826 - 31; http://dx.doi.org/10.1016/j.bbrc.2012.06.048; PMID: 22713463
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Tatar Z, Thivat E, Planchat E, Gimbergues P, Gadea E, Abrial C, Durando X. Temozolomide and unusual indications: review of literature. Cancer Treat Rev 2013; 39:125 - 35; http://dx.doi.org/10.1016/j.ctrv.2012.06.002; PMID: 22818211
- Carmo A, Carvalheiro H, Crespo I, Nunes I, Lopes MC. Effect of temozolomide on the U-118 glioma cell line. Oncol Lett 2011; 2:1165 - 70; PMID: 22848283
- Natsumeda M, Aoki H, Miyahara H, Yajima N, Uzuka T, Toyoshima Y, Kakita A, Takahashi H, Fujii Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011; 31:486 - 93; http://dx.doi.org/10.1111/j.1440-1789.2010.01197.x; PMID: 21269334
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380 - 90; http://dx.doi.org/10.4161/auto.29264; PMID: 24991834
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391 - 402; PMID: 24991838
- Munster T, Gibbs JP, Shen D, Baethge BA, Botstein GR, Caldwell J, Dietz F, Ettlinger R, Golden HE, Lindsley H, et al. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum 2002; 46:1460 - 9; http://dx.doi.org/10.1002/art.10307; PMID: 12115175
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359 - 68; http://dx.doi.org/10.4161/auto.28984; PMID: 24991840
- Lim HS, Im JS, Cho JY, Bae KS, Klein TA, Yeom JS, Kim TS, Choi JS, Jang IJ, Park JW. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob Agents Chemother 2009; 53:1468 - 75; http://dx.doi.org/10.1128/AAC.00339-08; PMID: 19188392
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403 - 14; http://dx.doi.org/10.4161/auto.29231; PMID: 24991835
- Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014; 10:1415 - 25; http://dx.doi.org/10.4161/auto.29165; PMID: 24991836
- Lin Y, Shih WJ. Statistical properties of the traditional algorithm-based designs for phase I cancer clinical trials. Biostatistics 2001; 2:203 - 15; http://dx.doi.org/10.1093/biostatistics/2.2.203; PMID: 12933550
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92:205 - 16; http://dx.doi.org/10.1093/jnci/92.3.205; PMID: 10655437
- Amaravadi RK, Schuchter LM, McDermott DF, Kramer A, Giles L, Gramlich K, Carberry M, Troxel AB, Letrero R, Nathanson KL, et al. Phase II Trial of Temozolomide and Sorafenib in Advanced Melanoma Patients with or without Brain Metastases. Clin Cancer Res 2009; 15:7711 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-09-2074; PMID: 19996224