
Abstract
Autophagy is a lysosomal degradation process that may act as a mechanism of survival in a variety of cancers. While pharmacologic inhibition of autophagy with hydroxychloroquine (HCQ) is currently being explored in human clinical trials, it has never been evaluated in canine cancers. Non-Hodgkin lymphoma (NHL) is one of the most prevalent tumor types in dogs and has similar pathogenesis and response to treatment as human NHL. Clinical trials in canine patients are conducted in the same way as in human patients, thus, to determine a maximum dose of HCQ that can be combined with a standard chemotherapy, a Phase I, single arm, dose escalation trial was conducted in dogs with spontaneous NHL presenting as patients to an academic, tertiary-care veterinary teaching hospital. HCQ was administered daily by mouth throughout the trial, beginning 72 h prior to doxorubicin (DOX), which was given intravenously on a 21-d cycle. Peripheral blood mononuclear cells and biopsies were collected before and 3 d after HCQ treatment and assessed for autophagy inhibition and HCQ concentration. A total of 30 patients were enrolled in the trial. HCQ alone was well tolerated with only mild lethargy and gastrointestinal-related adverse events. The overall response rate (ORR) for dogs with lymphoma was 93.3%, with median progression-free interval (PFI) of 5 mo. Pharmacokinetic analysis revealed a 100-fold increase in HCQ in tumors compared with plasma. There was a trend that supported therapy-induced increase in LC3-II (the cleaved and lipidated form of microtubule-associated protein 1 light chain 3/LC3, which serves as a maker for autophagosomes) and SQSTM1/p62 (sequestosome 1) after treatment. The superior ORR and comparable PFI to single-agent DOX provide strong support for further evaluation via randomized, placebo-controlled trials in canine and human NHL.
Introduction
(Macro) autophagy is a lysosomal degradation process which allows for the recycling of cytosolic proteins and organelles.Citation1 Autophagy is therefore often upregulated in response to stress, acting as a quality control mechanism as well as replenishing amino acid, lipid, and nucleic acid pools.Citation2-Citation5 These roles for autophagy are thought to enhance survival and therapy resistance in a variety of cancers.Citation6,Citation7 A number of preclinical cancer therapy mouse xenograft models have been used to demonstrate the enhanced efficacy of cancer therapies when combined with the autophagy inhibitors chloroquine (CQ) or hydroxychloroquine (HCQ).Citation8-Citation10 Thus, autophagy appears to be an attractive pathway to target. However, autophagy has been shown to have a protective role in a variety of organs including the gut, kidneys, and liver and combining HCQ with other drugs may exacerbate the toxicities of chemotherapy especially in these tissues.Citation11-Citation13 Induction of autophagy may also enhance the cytotoxicity of certain drugs.Citation14-Citation16 Therefore, establishing the safe and tolerable dose of HCQ in combination with standard of care drugs for canine cancers is an important first step in demonstrating the potential benefit of autophagy inhibition for these diseases.
Currently, the only autophagy inhibitor being used clinically is HCQ, a 4-aminoquinolone drug historically used in treatment of malaria and autoimmune diseases such as systemic lupus erythematosis and rheumatoid arthritis. HCQ pharmacokinetics are characterized by an extremely prolonged terminal half-life (up to 40 d) and a very large volume of distribution, in part due to the partitioning of the drug into red blood cells and strong binding to heme proteins.Citation17,Citation18 In humans, HCQ is rapidly and almost completely absorbed following an oral dose with approximately 50% being bound to plasma proteins. Three HCQ metabolites have been identified, including N-desethylchloroquine, desethylhydroxychloroquine (DHCQ), and bidesethylchloroquine.Citation18,Citation19 However, little is known about HCQ pharmacokinetics in dogs. HCQ has been used in canine discoid and cutaneous lupus erythematosus, similar to humans, at dosages of 5 to 10 mg/kg/d with some evidence of clinical efficacy, suggesting adequate blood and tissue concentrations for that specific indication.Citation20,Citation21 An early laboratory study evaluating chloroquine and HCQ administration in dogs demonstrated that doses up to 32 mg/kg/d for 13 wk were well tolerated, with no significant alterations of liver function or hematology.Citation22 When compared with chloroquine administration, equivalent doses of HCQ were better tolerated and provided higher blood and tissue concentrations. However, the study did not evaluate pharmacokinetic parameters of HCQ in dogs following repeat administration. Additionally, the use of HCQ in treatment of canine cancer has yet to be studied.
The translational utility of the canine cancer model is based in the greater similarity to humans in terms of carcinogenesis and tumor biology. Dogs are relatively outbred, immunocompetent animals that share environments with humans, and experience spontaneously developing tumors with spontaneous metastasis and therapy resistance, representing a spectrum of tumor histotypes similar to humans. The relatively large size of canine tumors closely approximates human solid tumors in regard to biological factors such as clonal variation and hypoxia, and this relatively large size allows for multiple sampling of tumor tissues over time for pharmacodynamic assessments.Citation23,Citation24 In addition, when compared with humans, disease progression in dogs is accelerated which allows for more rapid assessment of therapeutic endpoints than might be possible in similar human trials.Citation23,Citation24 Pet dogs with spontaneous cancer have been utilized for the clinical evaluation of multiple novel cytotoxic, targeted, and immunomodulatory therapies. These studies have then been used to inform the design of further human clinical trials as well as determining treatment protocols in veterinary medicine.Citation25-Citation32
Therefore, canine clinical trials are designed like and have the same objectives (e.g., a Phase I trial is intended to determine doses and assess pharmacokinetics) as human clinical trials but with the possibility of more rapid and complete assessments than is often possible in humans.
Canine non-Hodgkin lymphoma (NHL) is a useful naturally occurring model of NHL in humans owing to significant similarities in pathogenesis, histology/biology, and response to treatment. Correlations between genetic factors and the development or progression of NHL have been identified in both canine and human NHL. In dogs, the main aneuploidies observed in NHL include gains of chromosomes 13 and 31, which are analogous to the partial gains of human chromosomes 4 and 8 and a gain of chromosome 21.Citation33 Subchromosomal regions of CFA 13/HSA8 and CFA31/HSA21 harbor genes important in tumorigenesis such as c-myc, frequently involved in human B-cell lymphomas through aberrant fusion with immunoglobin genes.Citation34 Canine and human peripheral T-cell lymphomas also demonstrate some conservation of copy number aberrations with both having deletions in chromosomal regions leading to loss of CDKN2A/B and CDKN2A/p16-RB1 pathway activity.Citation35,Citation36 In addition to chromosomal aberrations, epigenetic changes such as methylation of CpG islands and promoter hypermethylation of orthologous tumor-suppressor genes have been identified in human and canine NHL.Citation37,Citation38 In dogs, NHL is one of the most prevalent tumor types, making up 7–24% of all cancers and 83% of hematopoietic cancers.Citation39 The incidence of canine NHL (15–30/100,000) is very similar to that seen in human NHL (15.5–29.9/100,000).Citation23 Based on the REAL/WHO or National Cancer Institute Working Formulation schema, canine NHL represents a relatively homogenous population with respect to histologic type with 70% classified as medium to high grade B-cell NHL and the majority being diffuse large B-cell lymphoma.Citation40 At presentation, most dogs with NHL are asymptomatic and have generalized, nonpainful enlargement of peripheral lymph nodes. As in humans, canine NHL is initially highly responsive to multiagent, CHOP-based chemotherapy which is likely to yield complete responses in approximately 90% of dogs.Citation39 However, the duration of first remission is short and 85% of cases will relapse within 6 to 11 mo.Citation39 Owing to toxicity and cost of multiagent chemotherapy, single agent doxorubicin is a frequently used alternative that produces a substantially lower response rate of 63 to 85% and a median progression-free survival of approximately 5 mo.Citation41-Citation43
Although there have been reports of enhancement of antitumor activity with combinations of cytotoxic drugs and autophagy inhibition in vitro and with in vivo mouse models, there are no reports on the clinical utility of autophagy inhibition using HCQ in canine cancer patients. Here we report the results of a Phase I/II clinical trial of oral HCQ given continuously, starting 72 h prior to a standard dose of DOX. This trial was conducted in client-owned (pet) dogs with spontaneous neoplasia presenting as patients to an academic, tertiary-care veterinary teaching hospital. As would be the case in early-phase human clinical trials, primary endpoints included maximum tolerated dose, dose-limiting toxicities, and pharmacokinetic/pharmacodynamic relationships. Preliminary evidence of antitumor activity was also assessed.
Results
Dose-escalation trial
A 3 × 3 dose escalation trial design was used to govern dose escalation toward a maximum tolerated dose (MTD) of HCQ that could be tolerated when administered concurrently with standard dosages of DOX (30 mg/m2 given once every 3 wk) in dogs with any spontaneously occurring tumor. In all, 30 dogs met the inclusion criteria and were enrolled in the study beginning in February 2011 and running through September 2013. All dogs underwent pretreatment evaluation and blood chemistry and CBC, and pretreatment biopsies were obtained from accessible tumors. Patient parameters such as age, sex, weight, breed, and tumor type were recorded for each patient (). In all, 27 of 30 (90%) dogs presented with multicentric lymphoma; 24 (88.9%) and 3 (11.1%) were identified as B- and T-cell lymphoma, respectively. At enrollment, 2 of 30 dogs (6.7%) had documented pulmonary metastasis (1 fibrosarcoma and 1 osteosarcoma) and 2 of 30 had received prior chemotherapy. One dog with lymphoma had been previously enrolled in, and failed, a separate clinical trial.
Table 1. Patient characteristics
Oral HCQ was well tolerated in dogs with no grade 3 or 4 toxicities attributable to the HCQ in any of the dose cohorts in the 3 d prior to DOX administration. Side effects of the HCQ were generally mild and self-limiting and mostly grade 1 or 2 lethargy and/or gastrointestinal upset (). A total of 112 treatment cycles were administered with an average of 3.7 per patient (range, 1–5). In all, 9 of 30 dogs (30%) required dose reductions in DOX following the first treatment cycle because of grade 3 or 4 toxicities attributable to DOX. The first HCQ dose cohort (5 mg/kg) was expanded to 6 dogs because of a grade 4 neutropenia in the first cycle in one dog. Upon subsequent genetic testing this dog was found to be a heterozygous ABCB1-1Δ mutant, a mutation leading to reduced expression and function of the drug transporter ABCB1/P-glycoprotein, which predisposes to increased toxicity of substrate drugs, including DOX.Citation44 No other grade 3 or 4 toxicities were encountered in the cohort. One dog in the 7.5 mg/kg HCQ cohort developed grade 4 neutropenia following the first cycle of DOX resulting in expansion of that cohort to 6 patients; this patient was found to be homozygous for the ABCB1-1Δ mutation leading to a complete lack of ABCB1 expression. Of the first 3 dogs enrolled in the 12.5 mg/kg HCQ cohort, 2 treatment-related deaths occurred following the first cycle of DOX and were related to severe neutropenia, gastrointestinal signs (vomiting and diarrhea), and sepsis; one dog developed grade 5 disseminated intravascular coagulation. As the oral HCQ at 12.5 mg/kg was well tolerated and the toxicities were associated with DOX administration, this cohort was expanded with an initial reduction in DOX dose to 25 mg/m2. Therefore, 12.5 mg/kg HCQ and 25 mg/m2 DOX was determined to be the MTD of the combination. This combination was well tolerated, as 12 dogs were enrolled with only one grade 4 neutropenia and all other toxicities being grade 1 or 2 gastrointestinal effects ().
Table 2. Hydroxychloroquine and doxorubicin adverse events by HCQ dose cohort
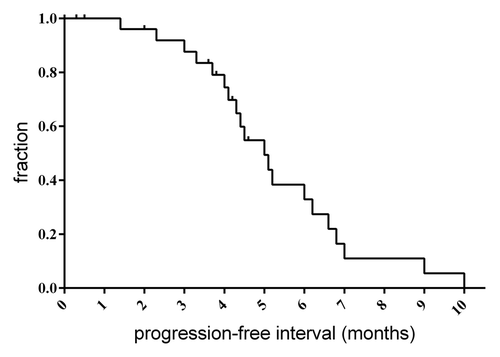
Responses were evaluated by Response Evaluation Criteria in Solid TumorsCitation45 and for the 27 dogs with lymphoma, best responses included 22 of 27 (81.5%) complete response, 3 of 27 (11.1%) partial response and 2 of 27 (7.4%) stable disease (). A total of 17 dogs (56.7%) completed 5 cycles of DOX. Of the 13 dogs that did not complete 5 cycles, 7 (53.8%) were due to progressive disease; 2 of these dogs had an initial complete response prior to being removed because of progressive disease. In 2 cases, dogs were removed from the trial by owner choice due to perceived reduction in quality of life. The overall median progression-free interval was 5.0 mo, which is similar to single agent DOX when given at 30 mg/m2 ().Citation41,Citation42
Table 3. Treatment efficacy of hydroxychloroquine and doxorubicin in dogs with multicentric lymphoma
Figure 1. Progression-free interval. Progression-free interval was determined for the 27 lymphoma patients receiving the HCQ and DOX combination. Median time to progression was 4.9 mo.
Pharmacokinetics
For determination of plasma trough HCQ and N-desethylHCQ levels in the highest dose cohort (12.5 mg/kg), plasma was collected 72 h prior to initiation of therapy and prior to the first dose of DOX. Eleven of the dogs in this cohort had samples available for evaluation. Substantial inter-individual variation was noted in these plasma samples and concentrations (mean ± SD) of HCQ and its metabolite were 105.1 ± 73.1 ng/mL and 16.6 ± 5.4 ng/mL, respectively. In 6 of these dogs, plasma was also collected for determination of DOX exposure via a validated limited-sampling method. Predicted plasma DOX exposure (AUC0–6h) in these dogs (mean ± SD) was 615.8 ± 208.6 ng/mL. Comparison of the DOX exposure in dogs in this study receiving HCQ followed by DOX at 25 mg/m2 with historical controls receiving single agent DOX at 30 mg/m2 demonstrated an approximate 25% reduction in AUC (); however, the dose-normalized exposure (AUC divided by dose) was not significantly different from that observed in dogs receiving single-agent therapy.Citation46,Citation47 This suggests no pharmacokinetic interaction between HCQ and DOX that would result in altered plasma DOX exposure.
Table 4. Comparison of predicted and dose-normalized doxorubicin exposure between dogs administered hydroxychloroquine at 12.5 mg/kg daily and historical controls receiving single agent doxorubicin
Tumor tissue concentrations of HCQ and its metabolite were also determined in the 11 dogs in which plasma concentrations were available. Results indicate a significant accumulation of HCQ and the metabolite in tumor tissues, with an approximate 100-fold increase in tumor compared with plasma (). Although tumor tissue concentrations were consistently higher, there was no significant correlation between the plasma and tumor concentration for individual dogs (Pearson correlation = 0.143, P = 0.695, ).
Figure 2. Assessment of pharmacodyanmic response. Concentrations of hydroxychloroquine (HCQ) and the metabolite N-desethylhydroxychloroquin (DHCQ) in dogs administered 12.5 mg/kg HCQ daily were significantly higher in tumor tissues compared with plasma [HCQ (P < 0.0001) and DHCQ (P = 0.0003)] (A). There was no significant correlation between plasma and tumor HCQ or DHCQ concentrations (Pearson correlation = 0.143 and 0.238, P = 0.695 and 0.537) (B). PBMCs were isolated from whole blood before and 3 d post HCQ administration. The representative plot shows gating of cells and histograms demonstrate the increase of LC3 positive cells after HCQ administration (C). Overall there was a significant increase in the mean fluorescence intensity post HCQ. Representative EM images of PBMCs taken pre and post HCQ administration. There is an increase in autophagic vesicles after HCQ treatment, indicated by arrows (D). Tumor biopsies were taken before and 3 d post HCQ administration. Western blot analysis was performed on biopsies to determine expression of LC3-II. Though not significant, both increased overall after treatment (E).
![Figure 2. Assessment of pharmacodyanmic response. Concentrations of hydroxychloroquine (HCQ) and the metabolite N-desethylhydroxychloroquin (DHCQ) in dogs administered 12.5 mg/kg HCQ daily were significantly higher in tumor tissues compared with plasma [HCQ (P < 0.0001) and DHCQ (P = 0.0003)] (A). There was no significant correlation between plasma and tumor HCQ or DHCQ concentrations (Pearson correlation = 0.143 and 0.238, P = 0.695 and 0.537) (B). PBMCs were isolated from whole blood before and 3 d post HCQ administration. The representative plot shows gating of cells and histograms demonstrate the increase of LC3 positive cells after HCQ administration (C). Overall there was a significant increase in the mean fluorescence intensity post HCQ. Representative EM images of PBMCs taken pre and post HCQ administration. There is an increase in autophagic vesicles after HCQ treatment, indicated by arrows (D). Tumor biopsies were taken before and 3 d post HCQ administration. Western blot analysis was performed on biopsies to determine expression of LC3-II. Though not significant, both increased overall after treatment (E).](/cms/asset/c0b1635c-b3dc-479f-bf3c-121cad792cc1/kaup_a_10929165_f0002.gif)
Pharmacodynamic response in peripheral blood and tumor tissue
The pharmacodynamic response to HCQ was evaluated in peripheral blood mononuclear cells (PBMCs) through flow cytometric evaluation of changes in LC3 for 6 dogs treated at 12.5 mg/kg po qd HCQ/25 mg/m2 DOX. All 6 dogs had a complete response and minimal toxicity (). Comparison of pre- and post-treatment mean fluorescence intensity (MFI) for LC3 by flow cytometry in PBMC revealed a significant increase of nearly 2-fold (P = 0.033) (). Additionally, we employed the gold standard, electron microscopy (EM), to visualize the formation and accumulation of autophagosomes, which is indicative of a blockade in the autophagic pathway by a drug such as HCQ, which alters the lysosomal pH and thus prevents fusion with autophagosomes leading to increased accumulation of autophagosomes ().Citation48 In tumor tissues, the pharmacodynamic response was evaluated by western analysis for changes in LC3 and SQSTM1. All biopsies were from involved lymph nodes in patients with NHL in the 12.5 mg/kg dose cohort. Though not significant, there was a trend toward increases in LC3-II and SQSTM1 expression in biopsy samples as well as tissue aspirates following HCQ administration (). Taken together, these data indicate that a variety of pharmacodynamic assays, that are feasible in the clinical setting, show strong evidence of autophagy inhibition in PBMCs and also in tumor tissue. It should however be noted that in the tumor tissue these responses were less robust than in the blood.
Discussion
Autophagy inhibition is thought to be a mechanism of survival and drug resistance for many types of cancers. Thus combining the autophagy inhibitor HCQ with cytotoxic chemotherapy may enhance efficacy. Here we report the results of a phase I/II clinical trial evaluating the use of combined HCQ and DOX in dogs with spontaneously occurring cancer, aimed at defining a safe and potentially biologically effective dose of HCQ. The rationale for the dosing scheme of 72 h pretreatment prior to the first dose of DOX followed by continuous daily dosing with HCQ was based on the reported long half-life and time to reach steady-state in humans, and the lack of any corresponding pharmacokinetic data in dogs.Citation19-Citation21 Four dose cohorts, ranging from 5 mg/kg/d up to 12.5 mg/kg/d, were evaluated. This study identified maximum tolerated doses of HCQ in combination with DOX of 12.5 mg/kg/d, and 25 mg/m2, respectively. Consistent with previous reports of the clinical use of HCQ in dogs, toxicities attributable to HCQ alone were generally mild with the most commonly reported adverse events being grade 1 lethargy and gastrointestinal disturbance.Citation20,Citation21 Dose-limiting toxicities following DOX administration were observed in 1 dog in each of the 5 mg/kg and 7.5 mg/kg dose cohorts; however, the finding that both of these dogs were carriers of the 1Δ mutation of the ABCB1/MDR1 gene (termed Abcb1a in mice and rats), would support the argument that these toxicities were not due to the combination of HCQ and DOX but an intrinsic sensitivity to DOX because of reduced or absent function of the ABCB1 efflux pump. Our finding of 12.5 mg/kg/d as the MTD of HCQ is based on the necessity to reduce the DOX dose by approximately 20% (30 mg/m2 to 25 mg/m2) in order to avoid the grade 5 adverse events that occurred at the standard DOX dose. Twelve dogs with lymphoma were subsequently enrolled in the 12.5 mg/kg/d HCQ and 25 mg/m2 DOX cohort with 2 requiring an additional 20% dose reduction in DOX due to grade 4 neutropenia. Importantly, 9 of these dogs achieved a complete remission and 8 of these dogs completed the trial, which would indicate that the reduced DOX dose did not result in a reduction in initial response. We did observe a substantial overall response rate in dogs with lymphoma (93.3%). These results are encouraging given that reported response rates with DOX alone for treatment-naive lymphoma in dogs range from 60% to 85%.Citation41,Citation42 The median progression-free interval in dogs with lymphoma in this study was 5.0 mo which is comparable to single agent DOX at 30 mg/m2.Citation41,Citation42 A larger study group with longer follow-up would be required to determine if the combined HCQ and lower DOX (25 mg/m2) dose is superior, or at least as effective, as single agent DOX administered at the standard 30 mg/m2 dose.
We were able to evaluate plasma exposure (area under the concentration-time curve) of DOX in 6 dogs within the highest HCQ dose cohort. Samples from this cohort were chosen as it was assumed that if changes in DOX PK were to be seen it would most likely occur with the highest HCQ dose. This was evaluated with the use of a validated limited-sampling model to predict overall exposure to DOX. We were able to demonstrate that overall plasma DOX exposure (dose-normalized area under the plasma concentration-time curve) was not significantly different in our group from dogs receiving DOX alone.Citation46,Citation47
In our study we were able to detect pharmacodynamic activity in PBMC and in tumor tissue following 72 h of oral HCQ administration, although the extent of autophagy inhibition, at least as determined by analysis of LC3, was less robust and at a lower level of statistical significance in tumor tissue compared with blood. Importantly, we found significant accumulation of HCQ in tumor tissue relative to plasma. A substantial fraction of HCQ partitions to red blood cells, and it is possible that whole blood levels would correlate with concentrations in homogenized tumor especially since the tumor samples also include some blood cells in them and do not reflect only tumor cells; however, this seems unlikely as 4-aminoquinolone binding to red blood cells has been reported to be linear with concentration and red blood cell density.Citation49 This infers a constant relationship between total concentration and the fraction bound to red blood cells, and while whole blood levels are certain to be higher than plasma, the same relationship to tumor concentrations would hold true. It is important to note that this study also evaluated plasma HCQ concentrations after 72 h of therapy and, based on the reported long half-life in humans (see Rosenfeld et al., this issueCitation50; Vogl et al., this issueCitation51; Rangwala et al., this issueCitation52,Citation53), it is possible samples were taken before dogs achieved steady-state concentrations. Variability and delays in achieving steady-state concentrations have been reported to contribute to the variability in tissue PD response in humans.Citation17 However, it does appear that plasma HCQ concentration is not an adequate surrogate for concentrations within the tumor and, similarly, evidence of autophagy inhibition in PBMCs is not necessarily sufficient to infer that autophagy was effectively inhibited in the tumor. These data suggest that although HCQ is capable of inhibiting autophagy in tumors of cancer patients, autophagy inhibitors with better pharmacokinetics and tumor bioavailability would be useful. Additionally our results suggest that one should be cautious in inferring from surrogate markers in the blood that sufficient tumor drug levels for effective autophagy inhibition have been achieved and emphasize the value in future clinical trials of attempting to make such measurements in tumor tissue.
In conclusion, this is the first study to evaluate the safety and potential clinical utility of autophagy inhibition using HCQ combined with cytotoxic chemotherapy in dogs with spontaneous cancer. We used continuous oral administration of HCQ combined with DOX on a 21-d cycle and showed that HCQ at doses up to 12.5 mg/kg/d are well tolerated but necessitate a reduction in the standard dose of DOX used in order to avoid unacceptable toxicity. Importantly, this reduction in DOX still provided a superior ORR and comparable PFI to dogs receiving single-agent, standard dose DOX. We were able to show target modulation in both PBMC and tumor tissue. The superior overall response rate and comparable progression-free interval in our study provide strong support for further evaluation of this combination in randomized, placebo-controlled studies in canine lymphoma, and the strength of the canine model of NHL supports initiation of similar clinical trials in human lymphoma patients.
Materials and Methods
Patient recruitment
All dogs in this study were client-owned, pet dogs presenting to the Colorado State University Flint Animal Cancer Center. Study participation was offered in cases where standard therapy had been declined by the dog’s owner or such therapy had previously failed, or in cases of advanced disease where no meaningful standard therapy exists. Protocol approval was obtained from the Institutional Animal Care and Use Committee and the Colorado State University Veterinary Teaching Hospital Clinical Review Board. All dogs were treated in accordance with the NIH Guidelines for Care and Use of Laboratory Animals. Signed informed consent and consent to necropsy were obtained from all owners prior to enrollment in the study.
This study was initially open to dogs with histologically or cytologically confirmed neoplasia of any histotype for which single-agent DOX would be an acceptable therapy. Dogs with regional or distant metastasis or locally advanced disease were included if a survival time of > 6 wk was anticipated. Dogs were required to be free of other severe complicating concurrent disease conditions, and were required to have adequate clinical indices to safely undergo chemotherapy and, in some cases, sedation for tumor biopsy acquisition (specifically, total bilirubin not exceeding 1.5× normal; creatinine no exceeding 2× normal; at least 2,500 neutrophils/µL, 75,000 platelets/µL, and a hematocrit of at least 28%). A Veterinary Comparative Oncology Group performance status of 0 or 1 was required for study inclusion (0, normal activity; 1, restricted activity [decreased from predisease status]; 2, compromised [ambulatory only for vital activities, consistently defecates and urinates in acceptable areas]; 3, disabled [requires force feeding, is unable to confine urination and defecation to acceptable areas]; 4, dead). Prior chemotherapy and radiation therapy were allowed with a 3- and 6-wk washout period, respectively. If prednisone was utilized as an anti-neoplastic agent, a 72 h washout was required and no concurrent antineoplastic therapy was allowed. For dogs previously administered DOX, prior cumulative exposure could not exceed 90 mg/m2.
Pretreatment procedures and evaluations
A complete blood count (CBC), serum biochemistry profile, and urinalysis were done prior to enrollment. Staging and immunophenotyping were performed as appropriate for the specific tumor type. Heparinized whole blood (10 to 12 mL) was collected for separation of peripheral blood mononuclear cells (PMBC), and a 14-gauge needle core biopsy was obtained from accessible tumors using local anesthesia or brief sedation, as necessary.
Treatments
All dogs were given oral HCQ sulfate tablets (Ranbaxy Pharmaceuticals Inc., NDC 63304-0296-01) once daily, beginning 72 h prior to DOX (Pfizer Inc., NDC 0069-3034-20) administration and continuing through the remainder of the study. An initial dose of 5 mg/kg was chosen as the starting point and doses were escalated according to a standard 3 × 3 dose-escalation protocol whereby 3 dogs were enrolled in each dose cohort and the cohort expanded to 6 if dose limiting toxicity (grade 3 or higher) was encountered in 1 of the first 3 dogs. Dogs were scheduled to receive a standard dose of DOX (30 mg/m2 intravenously, or 1 mg/kg if < 15 kg) as initial treatment on d 4 and continued on a 21-d cycle for a maximum of 5 treatments or disease progression. As with single agent DOX, dose reductions of 20% were instituted for subsequent treatments if grade 3 or 4 toxicities were observed after the first dose.
Monitoring procedures and evaluations
Adverse events were recorded on d 4, 11, and at each subsequent visit. All treatment-related adverse events were graded based upon the guidelines set forth in the Veterinary Comparative Oncology Group-Common Terminology Criteria for adverse events v1.0.Citation54 A CBC and blood chemistry were obtained 72 h after initiation of HCQ therapy. Serum, plasma, and heparinized whole blood were collected prior to DOX administration on d 4 for determination of trough HCQ and DHCQ levels and evaluation of HCQ pharmacodynamics in blood. For 6 dogs in the highest HCQ dose cohort (12.5 mg/m2), plasma was obtained at 5, 45, and 60 min for evaluation of DOX exposure utilizing a previously published limited-sampling model.Citation55 Tumor biopsies were obtained following the initial 72 h therapy with HCQ for determination of HCQ and DHCQ levels and pharmacodynamics. A CBC and blood chemistry were obtained 7 and 21 d following DOX administration. Owners were asked to fill out quality of life/pain questionnaires prior to the study, after the initial 72 h of HCQ therapy, and again at each subsequent visit. The HCQ-DOX combination was continued on an every-3-wk basis until disease progression, maximal cumulative DOX dosage (> 150 mg/m2), or owner request. Tumor responses were evaluated using Response Evaluation Criteria in Solid Tumors criteriaCitation45 on measured lymph nodes.
PBMC Isolation
Twenty mL of whole blood were collected from dogs and divided. Peripheral blood mononuclear cells (PBMC) were isolated using lymphocyte separation media (Cellgro, 25-072-CV). Briefly, one volume of phosphate-buffered saline containing 5 mmol/L EDTA was added to heparinized blood samples, which were then underlaid with 2 mL of lymphocyte separation media. The samples were centrifuged at 400 × g for 20 min and the lymphocyte layer was aspirated and washed 3 times in one volume of PBS/EDTA. Isolated PBMCs were stored at −80 °C until processing for flow cytometry, or EM analysis.
Flow cytometry
After PBMCs were isolated, cells were immediately resuspended in 200 μL of FACs buffer (2% FBS and 0.05% sodium azide in phosphate buffered saline). Cells were centrifuged at 700 × g for 2 min and then fixed and permeabilized for 18 h at 4 °C in diluted Fix/Perm buffer (eBiosciences, 00-5123-43 and 00-5223-56). Cells were washed once in diluted Perm Buffer (eBiosciences, 00-8333-56) and then stained with anti-LC3 (Novus Biologicals, NB100-2220) at 1:40 for 30 min at room temperature. Two wash steps were done in Perm Buffer and cells were stained with anti-rabbit secondary conjugated to FITC (Bethyl Laboratories, A120-101F) at 1:40 for 30 min at room temperature then washed twice with FACs buffer. Cells were then analyzed by flow cytometry using a Cyan cytometer (DakoCytomation, Carpenteria, CA) with Summit version 4.3.02 and FlowJo version 7 analysis software. The MFI between pre- and post-treatment with HCQ was used to compare samples for evidence of autophagy inhibition.
Electron microscopy
PBMC samples were fixed in Karnovsky fixative (3% glutaraldehyde, 2% formaldehyde, 0.1 M sodium phosphate buffer, pH 7.4; Electron Microscopy Sciences, 15720) and stored at 4 °C until processing. Following fixation, samples were washed 3 × 10 min with buffer, and postfixed for 1 h with 1% osmium tetroxide in 0.1 M sodium phosphatebuffer. After osmication, samples were again washed 3 times with buffer, dehydrated through a graded ethanol series (10 min each in 50%, 70%, 80%, and 90% ethanol, 2 × 10 min in 100% ethanol), transferred to propylene oxide (10 min in 1:1 ethanol:propylene oxide, 2 × 10 min in 100% propylene oxide), and infiltrated with Eponate 12 resin (medium hardness 45% glycerol polyglycidyl, 30.5% dodecenyl succinic anhydride, 23% NMA methylnadic anhydride, and 1.5% DMP-30; Ted Pella, Inc., 18010). Resin-embedded samples were polymerized for 24 h at 65 °C. Ultrathin sections 60 to 90 nm in thickness were cut from the embedded samples using a Diatome diamond knife and a Reichert Ultracut E ultramicrotome, mounted on formvar-coated slot grids (Electron Microscopy Sciences, 2010-Cu), and poststained with uranyl acetate and lead citrate. Sections were examined and photographed at 12,000× using a JEOL JEM-2000EX II electron microscope (JEOL USA, Inc., Peabody MA) operated at 100 kV. Negatives were scanned at 1200 ppi using an Epson Perfection flatbed scanner (Epson America, Inc., Long Beach, CA).
Western blot analysis
For protein extraction from tumors, snap-frozen biopsies were placed in 500 μL of lysis buffer (0.01% Triton X-100, 150 mM NaCl, 10 mM Tris pH 7.5, 0.2 mM Na-orthovanadate [Alexis Biochemicals, 400-032-G025], 34.8 μg/mL PMSF [Fluka Biochemica, 78830], and 1× protease inhibitor cocktail [Roche, 11836153001]). Samples were then homogenized for 20 s on ice and sonicated on ice for 3, 3 s pulses. Samples were centrifuged at 21,000 × g for 5 min at 4 °C and supernatant fractions collected.
Protein concentration was determined using a bicinchoninic acid protein assay (Thermo Scientific, 23225). Thirty micrograms of protein was used in SDS-PAGE and transferred onto PVDF membranes (Millipore, IPVH0010). Blots were blocked in 2.5% nonfat dry milk in Tris-buffered saline/Tween 80 (10 mM Tris pH 7.5, 100 mM NaCl, and 0.2% Tween 80; Fisher Chemicals, BP338-500) for 1 h at room temperature. Blots were probed with anti-LC3 (Novus Biologicals, NB100-2220), at 1:1000, anti-SQSTM1 (Abnova, H0008878-M01), at 1:1000, or anti-ACTB/β-actin (Sigma, A5441), at 1:5000, antibodies were incubated overnight at 4 °C. After 3 washes in TBST, membranes were incubated for 1 h at room temperature with either anti-rabbit (Pierce, 31460) or anti-mouse (Pierce, 31430) secondary antibodies conjugated to HRP. Immunoreactive proteins were detected using West Dura (Thermo Scientific, 37071) and imaged in a ChemiDoc XRS+ (Bio Rad, Hercules, CA) using Image Labs version 3.0 software. Densitometry analysis was performed using ImageJ software available online from the NIH (http://rsb.info.nih.gov/ij/index.html). LC3-I (cytosolic form of LC3) or LC3-II (autophagosome-associated) and SQSTM1 were first normalized to actin loading controls and then relative density determined for post HCQ administration samples by comparison to preadministration samples. A relative density of 1.5 in post-treatment samples was chosen as the cut-off for evidence of autophagy inhibition.
Hydroxychloroquine analysis in plasma and tumor tissue by liquid chromatography-tandem mass spectrometry
Hydroxychloroquine and the main metabolite DHCQ were measured in dog plasma and tissue biopsies using a liquid chromatography/tandem mass spectrometry assay using chloroquine as an internal standard. Positive ion electrospray ionization mass spectra were obtained with a MDS Sciex 3200 Q-TRAP triple quadrupole mass spectrometer (Applied Biosystems, Inc., Foster City, CA) with a turbo ionspray source interfaced to Shimadzu LC-20AD Series Binary Pump HPLC system (Columbia, MD). Samples were chromatographed with a Sunfire 2.5 µm, C8, 4.6 × 50 mm column. A liquid chromatography gradient was employed with mobile phase A consisting of 20 mM ammonium acetate containing 0.5% acetic acid and mobile phase B consisting of acetonitrile containing 0.5% acetic acid at 800 µL/min. Chromatographic separation was achieved by holding mobile phase B steady at 2% from 0 to 0.25 min, increasing linearly to 95% at 3 min, holding mobile phase B steady at 95% from 3.0 to 3.5 min, decreasing linearly to 2% at 3.75 min, followed by re-equilibration at 2% B until 4.5 min. The sample injection volume was 10 µL and the analysis run time was 4.5 min. The mass spectrometer settings were optimized as follows: turbo ionspray temperature, 550 °C; ion spray voltage, 1500 V; source gas 1, 60 units; source gas 2, 50 units; curtain gas, 45; collision gas, medium. Compound parameters for HCQ were optimized as follows: declustering potential, 47.9 V; entrance potential, 4.30 V; collision cell entrance potential, 14.0 V; collision energy, 29.7 V; collision cell exit potential 3.9 V. Sample concentrations of HCQ and metabolite were quantified by internal standard reference method in the multiple reaction monitoring mode with ion transitions m/z 336.2 → 247.1 atomic mass units (amu) for HCQ, m/z 308.2 → 179.0 and 308.2 → 130.1 amu (summed) for N-DHCQ, and m/z 320.3 → 247.1 amu for the internal standard, CQ. Scan times were 200 ms, and Q1 and Q3 were both operated in unit resolution mode.
Analytical standards (1 ng to 1,000 ng/mL), quality control (5, 50, and 500 ng/mL), and unknown plasma samples were prepared via a liquid-liquid extraction method whereby 180 µL of unknown or fortified plasma samples were added to 1.5 mL polypropylene tubes containing 50 ng of internal standard (CQ) followed by 500 µL of ethyl acetate. Samples were then vortex mixed for 8 min and centrifuged at 21,000 × g for 8 min. The organic phase was transferred to fresh microcentrifuge tubes and evaporated to dryness. Samples and standards were then reconstituted in 200 µL of a 1:1 methanol/20 mM ammonium acetate mixture at pH 4.0 and transferred to autosampler vials containing glass inserts. For analysis in tissue, biopsy samples were homogenized in Milli-Q H20 at a concentration of 100 mg/mL and 180 µL of homogenate was prepared as described above. The lower limit of quantification for HCQ was 5 ng/mL, and for DHCQ it was 10 ng/mL. The standard curves were linear across the range of concentrations utilized. Accuracy of the standard curve and the quality control samples was within 15% at all concentrations and precision was within 15% of the coefficient of variation.
Doxorubicin, doxorubicinol, and aglycone analysis in plasma by liquid chromatography-tandem mass spectrometry
Doxorubicin and its metabolites, doxorubicinol and doxorubicin aglycone, were measured in canine plasma using a liquid chromatography/tandem mass spectrometry assay. Negative ion electrospray ionization mass spectra were obtained with the instrumentation described above. Samples were chromatographed on a Waters Sunfire 5 µm, C8 column (4.6 × 50 mm) (Waters Corporation, Milford, MA) with a Phenomenex C18 filter frit guard cartridge (Torrance, CA). A liquid chromatography gradient was employed with mobile phase A consisting of 10 mM ammonium acetate containing 0.1% formic acid and mobile phase B consisting of methanol at 1200 µL/min. Chromatographic separation was achieved by holding mobile phase B steady at 25% from 0 to 1.0 min, increasing linearly to 98% at 2 min, holding mobile phase B steady at 98% from 2.0 to 3.0 min, decreasing linearly to 25% at 4.0 min, followed by re-equilibration at 25% B until 4.5 min. The sample injection volume was 60 µL and the analysis run time was 4.5 min. The mass spectrometer settings were optimized as follows: turbo ionspray temperature, 575 °C; ion spray voltage, −4500 V; source gas 1, 60 units; source gas 2, 60 units; curtain gas, 10; collision gas, low. Compound parameters for DOX were optimized as follows: declustering potential, −41.7 V; entrance potential, −6.3 V; collision cell entrance potential, −15.9 V; collision energy, −22.5 V; collision cell exit potential −3.9 V. Sample concentrations of DOX and metabolite were quantified by internal standard reference method in the multiple reaction monitoring mode with ion transitions m/z 542.3 → 395.4 amu for DOX, m/z 544.3 → 397.4 and 544.3 → 309.5 amu (summed) for doxorubicinol, and m/z 526.2 → 379.3 amu for the internal standard, daunorubicin (Sigma, 30450). Scan times were 200 ms, and Q1 and Q3 were both operated in unit resolution mode. Analytical standards (1 to 1,000 ng/mL), quality control (5, 100, and 500 ng/mL), and unknown plasma samples were prepared via a liquid-liquid extraction method whereby 100 µL of unknown or fortified plasma samples were added to 1.5 mL polypropylene tubes containing 100 ng/mL of internal standard (daunorubicin) followed by 1,000 µL of ethyl acetate. Samples were then vortex mixed for 10 min and centrifuged at 17,000 × g for 10 min. The organic phase (950 µL) was transferred to fresh microcentrifuge tubes and evaporated to dryness. Samples and standards were then reconstituted in 100 µL of 1:1 methanol/10 mM ammonium acetate with 0.1% formic acid and transferred to autosampler vials containing polypropylene inserts.
Prediction of doxorubicin exposure by limited sampling
Six dogs in the 12.5 mg/kg dose cohort had plasma samples collected at 5, 45, and 60 min following the first dose of DOX. Plasma DOX concentrations at these time points were used to predict the overall DOX exposure (AUC) using a limited sampling method that was previously validated and published by the authors.Citation40 In this model, overall DOX exposure was predicted using the equation:
AUC = 46.9 + 0.63 ∙ C5min + 1.96 ∙ C45min + 6.63 ∙ C60min
where C5min, 45min, 60min refer to plasma DOX concentrations at those time points. These AUC values were dose-normalized and compared with historical control data generated in our laboratory for dogs receiving DOX alone to evaluate for potential interactions between HCQ and doxorubicin that might result in alterations in overall exposure.Citation47
Statistical analysis
All statistical analysis was performed in Prism version 5.0 (GraphPad Software). A paired, 2-tailed Student t test was used to compare average MFI pre- and post-HCQ administration in PBMCs and lymph node aspirates. An unpaired, 2-tailed Student t test was used to compare DOX exposure between study subjects and historical controls. Pearson correlation was performed to determine correlations between pharmacokinetic and pharmacodynamic endpoints. A P value of less than 0.05 was considered significant.
| Abbreviations: | ||
| amu | = | atomic mass units |
| AUC | = | areas under the curve |
| CBC | = | complete blood cell count |
| CQ | = | chloroquine |
| CHOP | = | cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin (vincristine), prednisone |
| DHCQ | = | desethylhydroxychloroquine |
| DOX | = | doxorubicin |
| EM | = | electron microscopy |
| HCQ | = | hydroxychloroquine |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTD | = | maximum tolerated dose |
| MFI | = | mean fluorescence intensity |
| NHL | = | non-Hodgkin lymphoma |
| ORR | = | overall response rate |
| PBMCs | = | peripheral blood mononuclear cells |
| PFI | = | progression-free interval |
| SQSTM1 | = | sequestosome 1 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported by National Cancer Institute grant 1R01CA150925 Role of Autophagy in Tumor Cell Death, Pharmacology Shared Resource, supported by the University of Colorado Cancer Center Grant (P30 CA046934), and the Microscope Imaging Network University Core facility funded in part through a core infrastructure grant from the Office of the Vice President for Research.
References
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Ichimura Y, Komatsu M. Pathophysiological role of autophagy: lesson from autophagy-deficient mouse models. Exp Anim 2011; 60:329 - 45; http://dx.doi.org/10.1538/expanim.60.329; PMID: 21791873
- Suraweera A, Münch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 2012; 48:242 - 53; http://dx.doi.org/10.1016/j.molcel.2012.08.003; PMID: 22959274
- Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, Singh R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 2011; 14:173 - 83; http://dx.doi.org/10.1016/j.cmet.2011.06.008; PMID: 21803288
- Fujiwara Y, Kikuchi H, Aizawa S, Furuta A, Hatanaka Y, Konya C, Uchida K, Wada K, Kabuta T. Direct uptake and degradation of DNA by lysosomes. Autophagy 2013; 9:1167 - 71; http://dx.doi.org/10.4161/auto.24880; PMID: 23839276
- Loos B, Engelbrecht AM, Lockshin RA, Klionsky DJ, Zakeri Z. The variability of autophagy and cell death susceptibility: Unanswered questions. Autophagy 2013; 9:1270 - 85; http://dx.doi.org/10.4161/auto.25560; PMID: 23846383
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3:28 - 31; http://dx.doi.org/10.4161/auto.3269; PMID: 16969128
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res 2013; 19:2995 - 3007; http://dx.doi.org/10.1158/1078-0432.CCR-12-1542; PMID: 23461901
- Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, Gu CY, Yang H, Shi GM, Ke AW, Wang XY, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res 2011; 17:6229 - 38; http://dx.doi.org/10.1158/1078-0432.CCR-11-0816; PMID: 21825039
- Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73:3 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-12-2464; PMID: 23288916
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012; 55:222 - 32; http://dx.doi.org/10.1002/hep.24690; PMID: 21932416
- Cabrera S, Fernández AF, Mariño G, Aguirre A, Suárez MF, Español Y, Vega JA, Laurà R, Fueyo A, Fernández-García MS, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013; 9:1188 - 200; http://dx.doi.org/10.4161/auto.24797; PMID: 23782979
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest 2005; 115:2679 - 88; http://dx.doi.org/10.1172/JCI26390; PMID: 16200202
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448 - 57; http://dx.doi.org/10.1038/sj.cdd.4401359; PMID: 14713959
- Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, Gewirtz DA. Autophagy inhibition for chemosensitization and radiosensitization in cancer: do the preclinical data support this therapeutic strategy?. J Pharmacol Exp Ther 2013; 344:544 - 52; http://dx.doi.org/10.1124/jpet.112.199802; PMID: 23291713
- Lim HS, Im JS, Cho JY, Bae KS, Klein TA, Yeom JS, Kim TS, Choi JS, Jang IJ, Park JW. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob Agents Chemother 2009; 53:1468 - 75; http://dx.doi.org/10.1128/AAC.00339-08; PMID: 19188392
- Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit 2003; 25:671 - 81; http://dx.doi.org/10.1097/00007691-200312000-00005; PMID: 14639053
- Tett SE. Clinical pharmacokinetics of slow-acting antirheumatic drugs. Clin Pharmacokinet 1993; 25:392 - 407; http://dx.doi.org/10.2165/00003088-199325050-00005; PMID: 7904547
- Oberkirchner U, Linder KE, Olivry T. Successful treatment of a novel generalized variant of canine discoid lupus erythematosus with oral hydroxychloroquine. Vet Dermatol 2012; 23:65 - 70, e15-6; http://dx.doi.org/10.1111/j.1365-3164.2011.00994.x; PMID: 21718370
- Mauldin EA, Morris DO, Brown DC, Casal ML. Exfoliative cutaneous lupus erythematosus in German shorthaired pointer dogs: disease development, progression and evaluation of three immunomodulatory drugs (ciclosporin, hydroxychloroquine, and adalimumab) in a controlled environment. Vet Dermatol 2010; 21:373 - 82; PMID: 20374572
- McChesney EWMA. Laboratory Studies of the 4-aminoquinoline antimalarials: I. Some biochemical characteristics of chloroquine, hydroxychloroquine, and SN-7718. Antibiot Chemother 1961; 11:800 - 10
- Hansen K, Khanna C. Spontaneous and genetically engineered animal models; use in preclinical cancer drug development. Eur J Cancer 2004; 40:858 - 80; http://dx.doi.org/10.1016/j.ejca.2003.11.031; PMID: 15120042
- Kimmelman J, Nalbantoglu J. Faithful companions: a proposal for neurooncology trials in pet dogs. Cancer Res 2007; 67:4541 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-06-3792; PMID: 17510377
- Vail DM, Chun R, Thamm DH, Garrett LD, Cooley AJ, Obradovich JE. Efficacy of pyridoxine to ameliorate the cutaneous toxicity associated with doxorubicin containing pegylated (Stealth) liposomes: a randomized, double-blind clinical trial using a canine model. Clin Cancer Res 1998; 4:1567 - 71; PMID: 9626479
- Thamm DH, Kurzman ID, King I, Li Z, Sznol M, Dubielzig RR, Vail DM, MacEwen EG. Systemic administration of an attenuated, tumor-targeting Salmonella typhimurium to dogs with spontaneous neoplasia: phase I evaluation. Clin Cancer Res 2005; 11:4827 - 34; http://dx.doi.org/10.1158/1078-0432.CCR-04-2510; PMID: 16000580
- Paoloni MCTA, Tandle A, Mazcko C, Hanna E, Kachala S, Leblanc A, Newman S, Vail D, Henry C, Thamm D, et al. Launching a novel preclinical infrastructure: comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS One 2009; 4:e4972; http://dx.doi.org/10.1371/journal.pone.0004972; PMID: 19330034
- Vail DM, Thamm DH, Reiser H, Ray AS, Wolfgang GH, Watkins WJ, Babusis D, Henne IN, Hawkins MJ, Kurzman ID, et al. Assessment of GS-9219 in a pet dog model of non-Hodgkin’s lymphoma. Clin Cancer Res 2009; 15:3503 - 10; http://dx.doi.org/10.1158/1078-0432.CCR-08-3113; PMID: 19417014
- Thamm DH, Kurzman ID, Clark MA, Ehrhart EJ 3rd, Kraft SL, Gustafson DL, Vail DM. Preclinical investigation of PEGylated tumor necrosis factor alpha in dogs with spontaneous tumors: phase I evaluation. Clin Cancer Res 2010; 16:1498 - 508; http://dx.doi.org/10.1158/1078-0432.CCR-09-2804; PMID: 20160058
- Wittenburg LA, Gustafson DL, Thamm DH. Phase I pharmacokinetic and pharmacodynamic evaluation of combined valproic acid/doxorubicin treatment in dogs with spontaneous cancer. Clin Cancer Res 2010; 16:4832 - 42; http://dx.doi.org/10.1158/1078-0432.CCR-10-1238; PMID: 20705615
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 2010; 107:13075 - 80; http://dx.doi.org/10.1073/pnas.1004594107; PMID: 20615965
- London CA, Hannah AL, Zadovoskaya R, Chien MB, Kollias-Baker C, Rosenberg M, Downing S, Post G, Boucher J, Shenoy N, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res 2003; 9:2755 - 68; PMID: 12855656
- Thomas R, Seiser EL, Motsinger-Reif A, Borst L, Valli VE, Kelley K, Suter SE, Argyle D, Burgess K, Bell J, et al. Refining tumor-associated aneuploidy through ‘genomic recoding’ of recurrent DNA copy number aberrations in 150 canine non-Hodgkin lymphomas. Leuk Lymphoma 2011; 52:1321 - 35; http://dx.doi.org/10.3109/10428194.2011.559802; PMID: 21375435
- Hartmann EM, Ott G, Rosenwald A. Molecular biology and genetics of lymphomas. Hematol Oncol Clin North Am 2008; 22:807 - 23, vii; http://dx.doi.org/10.1016/j.hoc.2008.07.004; PMID: 18954738
- Fosmire SP, Thomas R, Jubala CM, Wojcieszyn JW, Valli VE, Getzy DM, Smith TL, Gardner LA, Ritt MG, Bell JS, et al. Inactivation of the p16 cyclin-dependent kinase inhibitor in high-grade canine non-Hodgkin’s T-cell lymphoma. Vet Pathol 2007; 44:467 - 78; http://dx.doi.org/10.1354/vp.44-4-467; PMID: 17606508
- Zettl A, Rüdiger T, Konrad MA, Chott A, Simonitsch-Klupp I, Sonnen R, Müller-Hermelink HK, Ott G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am J Pathol 2004; 164:1837 - 48; http://dx.doi.org/10.1016/S0002-9440(10)63742-X; PMID: 15111330
- Shaknovich R, Melnick A. Epigenetics and B-cell lymphoma. Curr Opin Hematol 2011; 18:293 - 9; http://dx.doi.org/10.1097/MOH.0b013e32834788cf; PMID: 21577103
- Bryan JN, Jabbes M, Berent LM, Arthur GL, Taylor KH, Rissetto KC, Henry CJ, Rahmatpanah F, Rankin WV, Villamil JA, et al. Hypermethylation of the DLC1 CpG island does not alter gene expression in canine lymphoma. BMC Genet 2009; 10:73; http://dx.doi.org/10.1186/1471-2156-10-73; PMID: 19912643
- Vail DM, Young KM. Canine lymphoma and lymphoid leukemia. In: Withrow SJ, and Vail D M, ed. Withrow and MacEwen's Small Animal Clinical Oncology. St. Louis: WB Saunders, 2007:699-712
- Jacobs RM. MJB, Valli V E. Tumors of the hemolymphatic system. In: D M, ed. Tumors in Domestic Animals. Ames, IA: Iowa State Press, 2002:119-98
- Lori JC, Stein TJ, Thamm DH. Doxorubicin and cyclophosphamide for the treatment of canine lymphoma: a randomized, placebo-controlled study. Vet Comp Oncol 2010; 8:188 - 95; PMID: 20691026
- Postorino NCSJ, Withrow SJ. Single agent therapy with Adriamycin for canine lymphosarcoma. J Am Anim Hosp Assoc 1989; 24:221 - 5
- Valerius KD, Ogilvie GK, Mallinckrodt CH, Getzy DM. Doxorubicin alone or in combination with asparaginase, followed by cyclophosphamide, vincristine, and prednisone for treatment of multicentric lymphoma in dogs: 121 cases (1987-1995). J Am Vet Med Assoc 1997; 210:512 - 6; PMID: 9040837
- Mealey KL. Therapeutic implications of the MDR-1 gene. J Vet Pharmacol Ther 2004; 27:257 - 64; http://dx.doi.org/10.1111/j.1365-2885.2004.00607.x; PMID: 15500562
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45:228 - 47; http://dx.doi.org/10.1016/j.ejca.2008.10.026; PMID: 19097774
- Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci 2002; 91:1488 - 501; http://dx.doi.org/10.1002/jps.10161; PMID: 12115848
- Selting KA, Ogilvie GK, Gustafson DL, Long ME, Lana SE, Walton JA, Hansen RA, Turner AS, Laible I, Fettman MJ. Evaluation of the effects of dietary n-3 fatty acid supplementation on the pharmacokinetics of doxorubicin in dogs with lymphoma. Am J Vet Res 2006; 67:145 - 51; http://dx.doi.org/10.2460/ajvr.67.1.145; PMID: 16426224
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Omodeo-Salè F, Cortelezzi L, Basilico N, Casagrande M, Sparatore A, Taramelli D. Novel antimalarial aminoquinolines: heme binding and effects on normal or Plasmodium falciparum-parasitized human erythrocytes. Antimicrob Agents Chemother 2009; 53:4339 - 44; http://dx.doi.org/10.1128/AAC.00536-09; PMID: 19651905
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359 - 68; http://dx.doi.org/10.4161/auto.28984; PMID: 24991840
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380 - 90; http://dx.doi.org/10.4161/auto.29264; PMID: 24991834
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369 - 79; http://dx.doi.org/10.4161/auto.29118; PMID: 24991839
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391 - 402; PMID: 24991838
- Veterinary Co-operative Oncology Group (VCOG). Veterinary Co-operative Oncology Group - Common Terminology Criteria for Adverse Events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.0. Vet Comp Oncol 2004; 2:195 - 213; http://dx.doi.org/10.1111/j.1476-5810.2004.0053b.x; PMID: 19379294
- Wittenburg LA, Thamm DH, Gustafson DL. Development of a limited-sampling model for prediction of doxorubicin exposure in dogs. Vet Comp Oncol 2014; 12:114 - 9; http://dx.doi.org/10.1111/j.1476-5829.2012.00340.x; PMID: 22747489