
Abstract
The efficacy of proteasome inhibition for myeloma is limited by therapeutic resistance, which may be mediated by activation of the autophagy pathway as an alternative mechanism of protein degradation. Preclinical studies demonstrate that autophagy inhibition with hydroxychloroquine augments the antimyeloma efficacy of the proteasome inhibitor bortezomib. We conducted a phase I trial combining bortezomib and hydroxychloroquine for relapsed or refractory myeloma. We enrolled 25 patients, including 11 (44%) refractory to prior bortezomib. No protocol-defined dose-limiting toxicities occurred, and we identified a recommended phase 2 dose of hydroxychloroquine 600 mg twice daily with standard doses of bortezomib, at which we observed dose-related gastrointestinal toxicity and cytopenias. Of 22 patients evaluable for response, 3 (14%) had very good partial responses, 3 (14%) had minor responses, and 10 (45%) had a period of stable disease. Electron micrographs of bone marrow plasma cells collected at baseline, after a hydroxychloroquine run-in, and after combined therapy showed therapy-associated increases in autophagic vacuoles, consistent with the combined effects of increased trafficking of misfolded proteins to autophagic vacuoles and inhibition of their degradative capacity. Combined targeting of proteasomal and autophagic protein degradation using bortezomib and hydroxychloroquine is therefore feasible and a potentially useful strategy for improving outcomes in myeloma therapy.
Keywords: :
Introduction
Multiple myeloma is an incurable plasma cell malignancy. Proteasome inhibition with bortezomib is now standard therapy for myeloma, based on its ability to induce clinical responses and confer a survival benefit in both relapsed myeloma and as initial therapy.Citation1,Citation2 Despite this efficacy, bortezomib does not lead to any cures, only 6% of patients with relapsed myeloma achieve a complete response with single-agent bortezomib, and only 38% have a partial response. Therefore, improvements in therapy are needed.
The unique biology of plasma cells appears to confer particular susceptibility to proteasome inhibition. Plasma cells are terminally differentiated B-cells, and bortezomib is also active in other B-cell malignancies,Citation3-Citation8 but has limited efficacy in solid tumors.Citation9-Citation12 One possible explanation for this difference in efficacy is the unique burden of misfolded proteins faced by malignant plasma cells, which generate large amounts of a single amino acid sequence as they produce the monoclonal immunoglobulin characteristic of myeloma. Preclinical evidence suggests that the efficacy of proteasome inhibitor therapy depends on the ability of malignant plasma cells to clear misfolded proteins through proteasomal degradation.Citation13
We hypothesized that resistance to bortezomib therapy might be due to activation of autophagy, the other major cellular mechanism for protein degradation. Autophagy is a process by which cells traffic organelles and large proteins to membrane-bound degradative vacuoles known as autolysosomes for degradation by acid-dependent enzymes and recycling to the cell’s catabolic mechanisms.Citation14 Autophagy occurs as part of normal cell physiology but can also contribute to cell survival in the setting of nutrient or growth factor deprivation, oxidative stress, and accumulation of protein aggregates.Citation15 When proteasomal degradation is either overwhelmed or inhibited, misfolded and ubiquitinated proteins that accumulate in the cytosol are transported to perinuclear aggresomes, which then are sequestered within autophagosomes that subsequently fuse with lysosomes for degradation of their contents.Citation16-Citation18 Simultaneous proteasome and autophagy inhibition therefore leads to accumulation of ubiquitinated proteins and synergistic cytotoxicity in preclinical models.Citation19,Citation20
Hydroxychloroquine (HCQ), a drug with a favorable and well-defined toxicity profile that is commonly used to treat autoimmune diseases, is a known inhibitor of autophagy.Citation21 The autophagy-specific mechanism of action of chloroquine derivatives is not fully understood, but they are known to be weak bases that are trapped in acidic cellular compartments, such as lysosomes, and increase the pH of those compartments.Citation22 Deacidification of lysosomes inhibits their function, blocking the last step in autophagy and leading to a cytosolic accumulation of autophagic vacuoles (AVs) with undigested contents. Autophagy inhibition augments the efficacy of many anticancer therapies in cell lines and animal models.Citation23-Citation27 Specifically, preclinical evidence suggests that autophagy inhibition synergistically increases bortezomib cytotoxicity in laboratory models of myeloma,Citation28-Citation30 colon carcinoma,Citation31 and hepatocellular carcinoma.Citation32
Based on myeloma cells’ dependence on protein degradative capacity, the role of autophagy in degrading misfolded proteins, and the strong preclinical rationale, we have employed a strategy of combined targeting of proteasomal and autophagic protein degradation for myeloma therapy. Here we report a phase I trial assessing the safety and preliminary efficacy of hydroxychloroquine and bortezomib in patients with relapsed or refractory multiple myeloma.
Results
Study population
We enrolled 25 patients between January 2008 and February 2011 () to 6 pre-specified dose levels () of a treatment regimen consisting of a 2-wk run-in of single-agent HCQ followed by combined therapy with bortezomib (). Three patients were not evaluable for dose-limiting toxicity (DLT) or response from the combined regimen because of progression (2 patients) or a severe infection (one patient with pneumonia) during the HCQ run-in. One additional patient (at dose level 4) was not evaluable for DLT from the combined regimen because of symptomatic progression after only one dose of bortezomib. We therefore had 21 patients evaluable for toxicity, including 3 at each of the first 5 dose levels and 6 at the top dose level.
Table 1. Subject characteristics
Table 2. Pre-specified dose levels
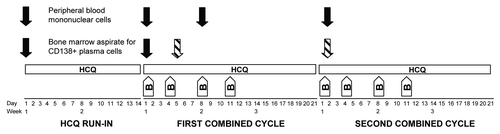
Figure 1. Schema of study treatment and acquisition of correlative samples. Patients started a 2-wk run-in of single-agent hydroxychloroquine (HCQ), which they continued during 3 wk cycles of standard bortezomib (B) on d 1, 4, 8, and 11. Solid arrows indicate time points at which samples were obtained for all patients; this included peripheral blood mononuclear cells obtained at baseline, on d 1 and 8 of cycle 1, and on d 1 of cycle 2, as well as bone marrow samples obtained at baseline and on d 1 of cycle 1. The hatched arrows indicate that the bone marrow samples obtained on combined HCQ and bortezomib were either on d 5 of cycle 1 (the final 3 patients) or d 1 of cycle 2 (all previous patients).
Among the entire enrollment cohort of 25 patients, the median age was 61 y (range 43 to 69); 56% were male, 76% were Caucasian, 16% African-American, and 8% Asian. The paraprotein type was IgG in 60%, IgA in 12%, and light chain only in 24%, with one patient having non-secretory myeloma. The median number of prior therapies was 3 (range 1 to 7). Of the 16 patients who had received prior bortezomib (64%), 11 were refractory to their most recent bortezomib-containing regimen, one had achieved only a minimal response, 3 had achieved a partial response or better, and one was not assessed for response because of non-secretory disease. All but one patient (96%) had received prior thalidomide or lenalidomide (72% had received prior thalidomide and 72% prior lenalidomide). All but one patient (96%) had received an autologous stem cell transplant, and 2 (8%) had undergone allogeneic transplantation.
Dose escalation and toxicity
After 6 cohorts of dose escalation (), we identified as the recommended phase 2 dose the top dose level: HCQ 600 mg twice daily and bortezomib 1.3 mg/m2. No protocol-defined dose-limiting toxicities occurred at any dose level. The most common adverse events in patients treated with this regimen were bone marrow suppression and fatigue (), though for most patients these were more likely related to their underlying disease than to specific toxicities of therapy. Gastrointestinal toxicities generally appeared during the 2nd cycle of combined therapy and worsened into the 3rd cycle and therefore did not meet our formal definition of a DLT. However, at the top dose level, gastrointestinal toxicities predominated, including 4 out of 6 evaluable patients with grade 3 gastrointestinal toxicity (one with constipation and nausea, one with diarrhea and nausea, one with anorexia, and one with an ileus), all manageable with symptomatic therapy. The patient with grade 3 anorexia also had grade 2 nausea and constipation, as well as grade 3 hypotension and syncopal episodes, which were related to dehydration, autonomic neuropathy, or both; for this patient, we reduced the dose of bortezomib from 1.3 to 1.0 mg/m2 during cycle 3 of combined therapy. We reduced the dose of bortezomib from 1.3 to 1.0 mg/m2 for 4 additional patients because of grade 2 fatigue (one patient in cohort 4 during cycle 4) or grade 2 peripheral neuropathy (one patient in dose cohort 2 during cycle 7, one patient in dose cohort 4 during cycle 3, and one patient in dose cohort 5 during cycle 6).
Table 3. Clinically significant adverse events during combined bortezomib and hydroxychloroquine, number of patients by dose level
Hematologic abnormalities were generally more attributable to disease progression than to treatment toxicity (especially in the lower dose cohorts). However, at the top dose level one patient who developed grade 3 thrombocytopenia and grade 4 neutropenia (nadir platelet count of 26,000/mm3 platelets and absolute neutrophil count of 250/mm3 during cycle 2) had started with normal platelets (156,000/mm3) and neutrophils (3680/mm3) and had stable myeloma serum markers through therapy, suggesting that his profound cytopenias were treatment-related.
Clinically significant infections occurred in a total of 5 patients, distributed among cohort 1 (1 pneumonia), cohort 2 (1 pneumonia and 1 Clostridium difficile colitis after antibiotic therapy for an upper respiratory tract infection), cohort 4 (1 pneumococcal pharyngitis and bacteremia), and cohort 6 (1 patient with pneumococcal pneumonia and sepsis during the HCQ run-in). All patients with infectious complications responded appropriately to antibiotics, and 3 of the 5 were able to continue on study therapy; these infections were thought to be more likely related to the underlying immunosuppression of myeloma than to any specific immunosuppression from study therapy.
Treatment-emergent peripheral neuropathy was generally mild and within the range expected with single-agent bortezomib. The few visual complaints that occurred during study therapy were self-limited and not thought to be related to study treatment. Eleven patients underwent end-of-study ophthalmologic exams, none of which revealed any retinal toxicity.
The median duration of study participation was 14 wk (range 1 to 77). Reasons for study discontinuation were side effects of therapy (3 patients), adverse events unrelated to therapy (3 patients), insufficient response (4), disease progression (14), and noncompliance (one). Of the 3 patients who stopped study therapy because of related side effects, one (at the top dose level) had severe constipation and painful neuropathy during the second cycle of combined therapy, one (at dose level 4) had grade 3 fatigue after 4 cycles of combined therapy, and one (at dose level 1) had grade 1 peripheral neuropathy, grade 2 weakness, grade 3 anemia, and grade 2 anorexia.
Disease response
Of the 25 patients enrolled in the study, 22 were assessable for response to the combination therapy. The other 3 patients had adverse events during the HCQ run-in and came off study without receiving any bortezomib doses. Of the 22 evaluable patients, 3 (14%) had very good partial responses (VGPR, all with the M-spike faintly visible on serum protein electrophoresis or detectable by immunofixation only), 3 (14%) had minor responses (MR), and 10 (45%) had stable disease (SD) for at least one cycle; 6 (27%) had immediate progression without achieving a period of stable disease.
We analyzed response according to dose level () and prior bortezomib exposure (). The 3 VGPRs occurred in patients treated at the higher dose levels who had never before received bortezomib. Two patients who had previously progressed while receiving weekly bortezomib had MRs while on study therapy; one (on dose level 1) achieved this response after 13 cycles of therapy and then maintained it for an additional 7.3 mo, and the other patient (on dose level 2) achieved a MR after 3 cycles of therapy but went off the study for antibiotic-related C. difficile colitis resulting in a prolonged hospitalization, during which her disease progressed. Four subjects previously refractory to bortezomib initially achieved SD during study treatment, with times to progression on study of 9 wk (on dose level 1), 15 wk (on dose level 2) and 9 and 14 wk (on dose level 3). At the top dose level and recommended phase 2 dose, 2 of 6 evaluable subjects had a VGPR (which lasted 18 and 36 wks), and one had SD lasting 8 wk.
Table 4. Best responses to combined bortezomib/hydroxychloroquine, by dose level, including all subjects evaluable for response
Table 5. Best responses to combined bortezomib/hydroxychloroquine, by prior response to bortezomib, including all subjects evaluable for response
Autophagy assessments
To explore the effects of this regimen on the autophagy pathway, we assessed mean AVs per cell in serial samples of bone marrow plasma cells and peripheral blood mononuclear cells (PBMCs) obtained during study therapy. We expected the number of AVs per cell to increase with HCQ alone (which inhibits the degradation of vesicular contents and clearance of AVs), with a further increase after the addition of bortezomib (which induces the formation of AVs by blocking proteasomal degradation of misfolded proteins). Ten subjects had complete sets of 3 bone marrow aspirates, and all 21 subjects who completed at least one full cycle of study therapy had complete sets of 4 PBMC samples.
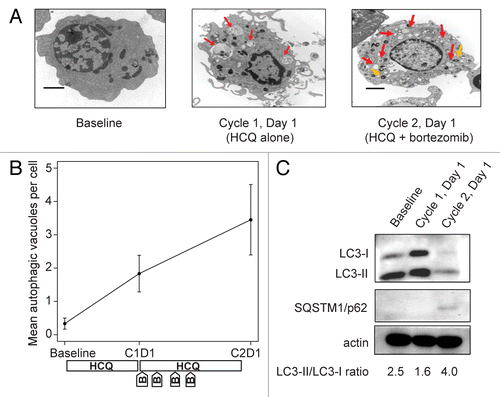
Therapy-associated accumulation of AVs was apparent in bone marrow plasma cells. In one patient with a VGPR to study therapy (patient number 13), striking step-wise accumulation of vacuoles with undigested contents was observed after 2 wk of HCQ therapy and with combination therapy (). For this patient, we performed confirmatory immunoblotting for MAP1LC3A/LC3 and SQSTM1/p62 (). MAP1LC3A is a cytoplasmic protein that is conjugated to the surface of AVs during their formation; the cytoplasmic form migrates as LC3-I on gel electrophoresis whereas the AV-conjugated form migrates as LC3-II, so the LC3-II/LC3-I ratio correlates directly with the number and size of accumulated AVs. SQSTM1 binds to aggregated proteins and is degraded by autophagy, so inhibition of autophagic degradation leads to intracellular accumulation of SQSTM1. For patient 13, increases in both SQSTM1 and the LC3-II/LC3-I ratio are evident from baseline to d 1 of cycle 2 (after 3 wk of combined bortezomib and HCQ), consistent with the accumulation of AVs seen on electron micrographs.
Figure 2. Therapy-associated autophagy modulation in myeloma cells from a patient treated with hydroxychloroquine 400 mg daily and standard bortezomib. (A) Representative electron micrographs of CD138-selected bone marrow plasma cells. Red arrows indicate AVs; orange arrows indicate mitochondria. Scale bar: 2 µm. Samples were obtained from a single patient (patient 13, on dose cohort 4) prior to treatment (baseline), after the 2-wk HCQ run-in on d 1 of cycle 1, and prior to therapy on d 1 of cycle 2. (B) Quantification of AVs in bone marrow plasma cells from patient 13 obtained at baseline, d 1 of cycle 1 (C1D1), and d 1 of cycle 2 (C2D1). Vacuole counts from 2 assessors blinded to patient and time point were averaged, with error bars reflecting the standard error of measurement across 25 cells per sample. The treatment schema shows the timing of administration of daily oral hydroxychloroquine (HCQ) and intravenous bortezomib (B). (C) Immunoblotting of bone marrow plasma cells from patient 13 at the same time points.
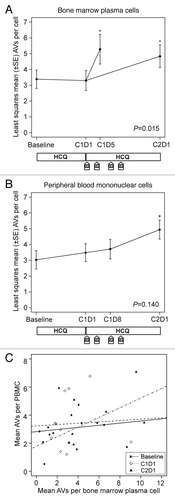
Across all patients with available samples, we observed a significant therapy-associated accumulation of AVs in bone marrow plasma cells (). Results from mixed-effects models indicated a significant time effect in bone marrow plasma cells (P = 0.015), with mean AVs per cell elevated on cycle 1 d 5 and cycle 2 d 1 compared with baseline and cycle 1 d 1. In contrast, we did not observe accumulation of AVs in PBMCs (), and the corresponding mixed effects models showed no significant time effect in mean AVs per cell measured in PBMCs (P = 0.140). Mean vacuole counts in PBMCs did not correlate well with counts in bone marrow plasma cells obtained at the same time, as shown in .
Figure 3. Therapy-associated autophagy modulation in myeloma and peripheral blood mononuclear cells from patients treated with hydroxychloroquine (HCQ) and bortezomib. Shown are mean autophagic vacuole counts in (A) bone marrow plasma cells and (B) PBMCs sampled during therapy. The bone marrow results were taken at baseline, on d 1 of cycle 1 (C1D1, after a 2-wk HCQ run-in), and either on d 5 of cycle 1 (C1D5, ~24 h after the d 4 bortezomib dose) or on d 1 of cycle 2 (C2D1, ~10 d after the d 11 bortezomib dose). Peripheral blood samples were obtained at baseline, on C1D1, on d 8 of cycle 1 (C1D8), and on C2D1. P values are shown for comparisons across all time points, and asterisks indicate time points that are significantly (P < 0.05) different from baseline. The treatment schema below each panel shows the timing of administration of daily oral hydroxychloroquine (HCQ) and intravenous bortezomib (B). (C) Correlation between autophagic vacuole counts in peripheral blood mononuclear cells and bone marrow plasma cells for all time points at which patients had both samples obtained at the same time. Correlation coefficients were not statistically significant at baseline (0.34, P = 0.21), C1D1 (0.10, P = 0.77), or C2D1 (0.65, P = 0.18).
HCQ pharmacokinetics and pharmacodynamics
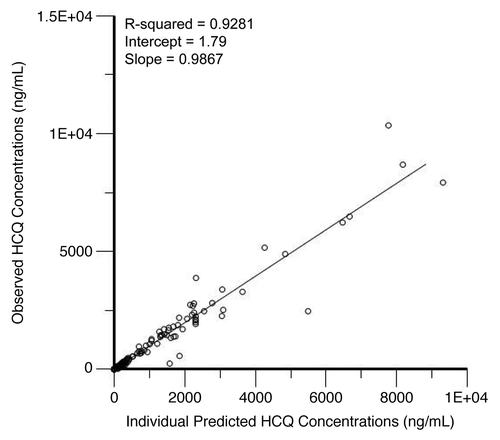
We performed population pharmacokinetic (PK) analysis using 238 non-baseline blood HCQ levels from 21 patients collected over a period up to 169 d. shows the individual observed concentrations vs. the predicted concentrations from the population PK model. The population model PK parameters do not specifically represent steady-state values, as they were determined from multiple repeated single doses taken by individual patients during their period of participation in the study. The model that best described the disposition of HCQ blood concentrations was a 2-compartment model with first-order absorption and a lag time. The inclusion of weight as a factor on clearance significantly improved the model. A nondiagonal fit was not superior to a diagonal fit based on Akaike information criterion, -2(LL), and Bayesian information criterion. The final model, specified as the population or typical value (tv) and random effect (n) for each parameter was as follows: first order absorption rate constant (Ka) = tvKa * exp(nKa); apparent volume of distribution in central compartment (Vc/F) = tvV * exp(nV)/F; apparent volume of distribution in peripheral compartment (Vp/F) = tvV2 * exp(nV2)/F; apparent oral clearance = tvCl * weightdCldweight *exp(nCl)/F; intercompartmental clearance (Q) = tvQ*exp(nQ); lag time (tLag) = tvtLag = exp(ntLag). The residual error was supported by a multiplicative error model, as described by: Cobs = C *(1 + Ceps), where Cobs is the observed concentration, C is the predicted concentration, and Ceps is the zero mean normally distributed random variable.
Figure 4. Correlation of predicted and observed hydroxychloroquine (HCQ) levels. Shown are observed HCQ whole blood concentrations vs. individual predicted concentrations from a population pharmacokinetic model in patients treated with HCQ and bortezomib.
We used the final PK model to simulate HCQ blood concentrations for individual patients at steady-state, which was achieved on average after 21 d. Blood HCQ concentration relationships for area under the concentration-time curve (AUC), maximum concentration (Cmax), trough concentration (Cmin), and average concentration (Cavg) were proportional to daily HCQ dose. shows results for Cavg, which is representative of the other parameters. Individual PK parameter estimates derived from the population were most variable for central volume of distribution (Vc/F) and intercompartmental clearance ().
Figure 5. Hydroxychloroquine (HCQ) levels in patients receiving HCQ and bortezomib for myeloma. Shown are steady-state HCQ whole blood concentrations, by average daily HCQ dose. Steady-state HCQ concentrations are estimated from a population pharmacokinetic model. The graph shows the median (solid horizontal line), mean (dashed horizontal line), 25th–75th percentiles (box), and 5th–95th percentiles (whiskers).
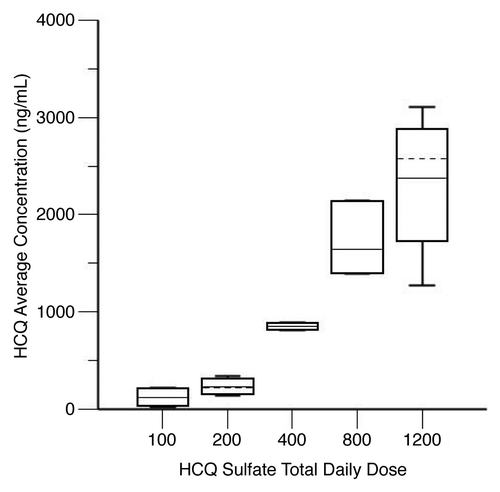
Table 6. Final HCQ population pharmacokinetic parameter estimates
We could not identify a statistically significant relationship between HCQ levels and pharmacodynamic markers. A classification and regression tree analysis of 11 patients did not identify a threshold at which there was a significant association between HCQ concentrations (Cmax, Cmin, or AUC) and change in AVs in bone marrow plasma cells from baseline to after the 2-wk HCQ run-in. An equivalent analysis of AV accumulation in PBMCs yielded similar results. Mixed-model analysis also revealed no statistically significant association of PK parameters with AV accumulation in bone marrow plasma cells or PBMCs.
Discussion
Preclinical studies demonstrating increased antitumor efficacy of combined proteasome and autophagy inhibition provided a strong rationale for conducting this phase I trial of bortezomib and hydroxychloroquine in multiple myeloma.Citation16,Citation19,Citation24-Citation29 Our results demonstrate the feasibility of this approach, as well as intracellular accumulation of AVs during combined stimulation of the autophagy pathway and inhibition of its final step.
Our clinical results demonstrate that the combination of bortezomib and HCQ is feasible and well tolerated. Unlike other clinical trials combining HCQ either with vorinostat (Mahalingam et al., this issueCitation33) or with temozolomide and cranial radiation (Rosenfeld et al., this issueCitation34), we observed no adverse events that met our definition of a dose-limiting toxicity, with only 3 subjects stopping therapy because of side effects of the combined regimen. The likely DLTs for this combination are gastrointestinal and hematologic, and we did not observe any evidence that HCQ potentiates the sensory neuropathy that is the primary dose-limiting toxicity for bortezomib. We observed several responses, including 2 minor responses in patients previously refractory to bortezomib and 3 near-complete responses in bortezomib-naïve patients, as well as stable disease lasting 9 to 17 wk in 4 patients previously refractory to bortezomib. These responses and periods of stable disease suggest improved efficacy with the combination over bortezomib alone. However, some patients received a higher bortezomib dose intensity during this treatment than when they previously progressed on bortezomib therapy, so it remains unclear whether the HCQ truly increased responses beyond what would be expected with single-agent bortezomib.
There are several possible reasons that we did not see more robust clinical responses. While the combination of bortezomib and HCQ clearly modified autophagy dynamics in tumor cells, a higher degree of autophagy modulation may be necessary to elicit more prominent cell death. This could be achieved with higher doses of HCQ, the use of a more potent autophagy inhibitor, or the use of a more potent proteasome inhibitor. The toxicities that we observed at our top dose level (which is the highest tolerated dose of HCQ in rheumatologic illnesses) suggest that higher doses of HCQ would not be well tolerated in this combination. HCQ’s long-terminal half-life results in a time to steady-state concentrations of several weeks,Citation35 so some of our patients may not have achieved sufficient HCQ concentrations during their time on study therapy, and an autophagy inhibitor with more favorable pharmacokinetic characteristics may be necessary to successfully implement this strategy. Other mechanisms of resistance to bortezomib, such as drug efflux pumps or proteasome mutations, may be present in individual patients, especially those previously treated with bortezomib. If one of these resistance mechanisms is active, bortezomib would not actually inhibit the proteasome, and autophagy inhibition, which is unlikely to be effective alone, would not be expected to augment bortezomib efficacy. Therefore, it is possible that a more potent and pharmacologically active proteasome inhibitor could produce more antitumor activity in combination with an autophagy inhibitor. In addition, other cell survival pathways such as the ER stress and unfolded protein responses may allow malignant plasma cells to avoid apoptosis, even in the presence of combined proteasome and autophagy inhibition.
Our correlative results suggest that we successfully targeted protein degradation pathways in patients’ tumor cells. We observed therapy-associated changes in AVs in myeloma cells in individual patients, with an overall increase over time in AVs during study therapy. Perhaps due to our small sample size, we were unable to correlate changes in AVs with clinical response or HCQ exposure, though a clear correlation between HCQ exposure and AV accumulation was observed in larger HCQ trials in other malignancies (Rosenfeld et al., this issueCitation34; Rangwala et al., this issueCitation36). We also may have missed peak changes in AVs due to the timing of sample acquisition.
A unique feature of this study was the analysis of autophagy dynamics simultaneously in tumor cells and in PBMCs, which demonstrated that tumor cell autophagy was more significantly perturbed with treatment than PBMC autophagy. This was also observed in a phase I trial of vorinostat and HCQ in advanced solid tumors (Mahalingam et al., this issueCitation33), and a trial of HCQ in combination with doxorubicin for canine lymphoma showed a much higher concentration of HCQ in tumor tissue than in plasma (Barnard et al. this issueCitation37). These findings suggest that direct measures of tumor cell autophagy should continue to be incorporated into future clinical trials of autophagy inhibitors, rather than using analyses of surrogate tissue.
Our trial results suggest several avenues for further investigation. A more thorough characterization of treatment-associated changes in AVs in patients treated with single-agent proteasome inhibition would determine how common and significant autophagy induction is in these patients. Based on this study, future trials of novel proteasome inhibitors with either HCQ or a novel autophagy inhibitor are also warranted. Preclinical studies are identifying more potent inhibitors of autophagy,Citation38 which may be useful in such combinations. In addition, although lysosomal targeting with HCQ is a rational approach to blocking the final degradative step in the autophagy pathway, another critical step to target in this protein-degradation pathway may be the trafficking of ubiquitinated proteins to the aggresome, a process that is mediated by histone deacetylase (HDAC) 6.Citation16 HDAC inhibitors have synergistic preclinical activity in combination with bortezomib.Citation20,Citation39 The pan-HDAC inhibitors vorinostat and panobinostat have clear activity in relapsed/refractory myeloma, though their clinical utility may be limited by fatigue and thrombocytopenia.Citation40,Citation41 ACY-1215, a specific inhibitor of HDAC6, is currently in early clinical trials (clinicaltrials.gov identifier: NCT01323751); the combination of this drug with bortezomib should result in an accumulation of intracellular ubiquitinated proteins but decreased formation of AVs.
Beyond protein degradation, autophagy also has a role in allowing malignant cells to survive other apoptotic stresses and therefore has a wider potential to improve treatment efficacy across tumor types.Citation42 Trials evaluating hydroxychloroquine as an autophagy inhibitor in combination with cytotoxic chemotherapy (Rangwala et al., this issueCitation36; Rosenfeld et al., this issueCitation34), cell signaling inhibitors (Rangwala et al., this issueCitation43), or epigenetic modifiers (Mahalingam et al., this issueCitation33) have shown encouraging early results. Therefore, autophagy inhibition remains a promising strategy for improving the efficacy of cancer therapy.
Materials and Methods
Patients
We enrolled patients with relapsed or refractory myeloma after at least one prior treatment regimen. Prior bortezomib therapy was permitted if it did not result in dose-limiting toxicity; prior autologous or allogeneic hematopoietic stem cell transplants were also permitted. Myeloma was considered refractory if it progressed while the patient was receiving prior therapy or within 60 d of stopping it. Participants met the following inclusion criteria: no baseline peripheral neuropathy of grade 2 or higher, no known macular degeneration or retinopathy, porphyria, or uncontrolled psoriasis, Eastern Cooperative Oncology Group performance status ≤ 2, adequate bone marrow function (absolute neutrophil count ≥ 500/μl, hemoglobin ≥ 7 g/dL, platelets ≥ 25,000/μl), adequate organ function (serum creatinine and total bilirubin ≤ 2 times upper limit of normal, and aspartate aminotransferase and alanine aminotransferase ≤ 2.5 times upper limit of normal), no known central nervous system involvement, and no treatment with other antimyeloma agents within 14 d or corticosteroids within 7 d. All patients provided written informed consent prior to participation, and the study protocol was approved by the University of Pennsylvania Institutional Review Board.
Study design
Patients enrolled on a standard 3 + 3 dose escalation design using the pre-specified dose levels shown in . Patients received a 2-wk run-in of single-agent oral HCQ (Watson, 00591-0698-01), followed by the addition of intravenous bortezomib (Millennium Pharmaceuticals) on d 1, 4, 8, and 11 of each 21-d cycle (see for treatment schema).
Dose escalation was permitted if 0 of 3 or < 2 of 6 patients experienced a dose-limiting toxicity (DLT). DLT was defined as a grade ≥ 3 toxicity during the first cycle of combined therapy that was probably or definitely related to study therapy. Because of the known toxicity profile of bortezomib and the commonly accepted toxicities of therapy for relapsed and refractory myeloma, we did not consider the following to be DLTs, regardless of grade: anemia, lymphopenia, neutropenia responsive to growth factor, platelets > 10,000/mm3 not associated with bleeding, or gastrointestinal complaints relieved by symptomatic therapy. Toxicity was graded using version 3.0 of the National Cancer Institute Common Terminology Criteria for Adverse Events.
Patients who experienced a DLT received no further therapy until the toxicity resolved to grade 1 or better, with any toxicity-related treatment delay of ≥ 14 d resulting in termination of study treatment. Dose reductions of bortezomib were from 1.3 to 1.0 mg/m2, then to 0.7 mg/m2, and then discontinuation; for HCQ, reductions were by 200 mg/d. For development of neuropathy on study, the protocol required reduction of the bortezomib dose for grade 2 neuropathy, discontinuation of both agents for grade 3 neuropathy (with the possibility of restarting bortezomib at 0.7 mg/m2), and discontinuation of study therapy for grade 4 neuropathy. Gastrointestinal symptoms were managed with symptomatic therapy, holding treatment for grade 3 or 4 toxicity until it returned to grade ≤ 2, then restarting with a dose reduction of hydroxychloroquine (for grade 3) or both agents (for grade 4). For all other DLTs, the protocol required dose reduction of both agents.
Response was assessed on d 1 of each cycle according to International Working Group criteria,Citation44 which define a partial response (PR) as a 50% decrease in serum paraprotein and a 90% decrease in urine paraprotein, a very good partial response (VGPR) as a 90% reduction in serum paraprotein or serum and urine paraprotein detectable only by immunofixation, and a complete response as the absence of paraprotein on serum and urine immunofixation and ≤ 5% plasma cells in the bone marrow, with the addition of a category of minor response (MR),Citation45 defined as a 25% reduction in serum paraprotein and 50% decrease in urine paraprotein; disease progression is defined as an increase of 25% from baseline in serum or urine paraprotein or development of new bone lesions, hypercalcemia, or soft tissue plasmacytomas. We therefore measured serum protein electrophoresis (with immunofixation as indicated), quantitative serum immunoglobulins, and serum free light chain levels on d 1 of every cycle, as well as a urine protein electrophoresis (with immunofixation as indicated) every cycle for subjects with a baseline measurable urine paraprotein (or every 4th cycle otherwise). For patients with non-secretory myeloma, a bone marrow biopsy and radiologic evaluation was required every 4 cycles. Safety assessments included a complete blood count and comprehensive metabolic panel on d 1 of each cycle, as well as ophthalmology examinations at baseline and end of study (for any subject completing at least 2 cycles of combined therapy).
Autophagy assessments
We collected peripheral blood mononuclear cells (PBMCs) from all patients at 4 time points: at baseline, prior to bortezomib on d 1 and 8 of cycle 1, and prior to bortezomib on d 1 of cycle 2. For consenting patients, we performed bone marrow aspirates and core biopsies at 3 time points: baseline, prior to bortezomib on d 1 of cycle 1, and prior to bortezomib on d 1 of cycle 2; for the final 3 patients in the study, the third and final bone marrow aspirate was performed on d 5 of cycle 1, approximately 24 h after the d 4 dose of bortezomib. PBMC samples were drawn directly into cell preparation tubes, and the mononuclear cell layer was extracted. Bone marrow aspirate samples were processed using Ficoll (GE Healthcare Bio-Sciences, 17-1440-03) extraction and isolation of plasma cells using CD138 beads (Miltenyi, 130-051-301).
The most reliable measurement of autophagy is quantification of AVs by electron microscopy. After sample processing (using previously described proceduresCitation21), digital electron micrographs were captured for 25 individual cells at 6000× (bone marrow plasma cells) and 12,000 × (PBMCs). AVs were scored by investigators who were blinded to treatment time points. Morphological criteria for AVs included 1) circularity, 2) contrast: white or lighter than the cytoplasm, 3) contents, 4) size > 200 nm, and 5) location: > 200 nm interior to the plasma membrane. Vesicular structures with cristae characteristic of mitochondria in cross section were excluded. We fit mixed-effects models to the mean AV counts in both PBMCs and bone marrow plasma cells. Models included a fixed effect of measurement time (cycle/day) and a random subject effect. We estimated the models using the Proc Mixed procedure in SAS Version 9.3 (SAS Institute).
Western blots were performed as previously described,Citation21 using antibodies to MAP1LC3A/LC3 (generated using QCB biologicalsCitation21), SQSTM1/p62 (Santa Cruz Biotechnology, SC-D3), and ACTB/actin, β (Santa Cruz Biotechnology, SC-I19).
Hydroxychloroquine pharmacokinetics
Whole blood samples were collected on d 1 and 8 of the first cycle and on d 1 of each subsequent treatment cycle for HCQ concentration analyses. Sample aliquots of 100 μL were mixed with 10 μL of internal standard (IS) (d4-HCQ, Toronto research, H916902), then vortexed vigorously with 400 μL of 90:10 acetonitrile (Fluka, 34967-2.5L)/methanol (Fluka, 34966-2.5L), then centrifuged. A 350-μL aliquot of the supernatant fraction was withdrawn and dried under nitrogen gas. The samples were reconstituted with 100 μL of mobile phase, using 90:10:0.1% acetonitrile/water (Fluka, 39253-4L-R)/formic acid (Fluka, 56302-50ML-F). Then, 10 μL was injected onto a Kinetex 50 × 3 mm 2.6 um 100A HPLC column (Phenomenex, 00B 4462-40) eluted with a gradient mobile phase of 0.1% formic acid in acetonitrile and water at 500 µL/min. A 1200 Series Agilent HPLC system was used with an API 4000™ (AB SCIEX, Foster City, CA) mass spectrometer and electrospray interface operated in positive mode with multiple reaction monitoring detection. The capillary voltage was 4000 V with a source temperature of 500 °C. Mass spectrometer parameters were adjusted to maximize the intensity of the [M + H]+ ions in quadrupole 1 and the m/z transition ions of HCQ (337.275 → 248.152) and IS (341.150 → 252.035) in quadrupole 3. The HPLC system and mass spectrometer were controlled by AB SCIEX Analyst® software (version 1.6.1) and data collection and analyses were conducted with the same software. Standard curves were constructed by plotting the analyte-to-IS ratio vs. the known concentration of HCQ (x) in each sample. Standard curves were fit by linear regression with weighting by 1/x. Samples were assayed in duplicate; samples for which the percent difference exceeded 15% were reanalyzed and samples for which concentrations exceeded the range values for the calibration curve were diluted appropriately and reanalyzed. The calibration curve was linear from 1 to 5000 ng/mL with correlation coefficients ranging from 0.9990 to 0.9999. The lower limit of quantification was 1.0 ng/mL. The correlation coefficients for both inter- and intra-day variability were < 5.6% for each concentration (15 ng/mL, 750 ng/mL, and 1500 ng/mL) studied. The mean accuracy for inter- and intra-day evaluations was between 97.2 and 102%.
Whole blood HCQ concentration data were analyzed by nonlinear mixed-effect modeling using Phoenix™ NLME 1.2 (Pharsight, Cary, NC). Initial estimates for a base population pharmacokinetic model were derived from a naïve-pooled data analysis of individual patient blood concentration time data. One and 2-compartment models with first-order absorption and elimination, with and without a lag time, were evaluated as the potential pharmacokinetic base structural model. Inter-individual variability of population pharmacokinetic parameters was considered to be log-normally distributed with mean of 0 and variance of ω2. Visual inspection of standard goodness of fit/diagnostic plots and numerical diagnostics were used to determine optimal model fits. The first-order conditional maximum likelihood estimation, the Lindstrom-Bates method, was used for the modeling process. Diagnostic scatter plots (individual and population predicted values vs. observed concentrations, conditional weighted residuals vs. time and conditional weighted residuals vs. observed concentrations), Akaike information criterion, and the likelihood ratio test, were used to select the base model. Conditional weighted residuals vs. time and predicted concentration time plots helped confirm that the chosen residual error model was appropriate.
Visual inspections of scatter and box plots for eta (random effect) values were used to explore potential continuous (age, weight) and categorical (sex, dose cohort) covariates. Covariates were centered on their median values. A stepwise covariate selection process was performed to build the full model. Model building criteria were based on covariate models associated with an increase in objection function value greater than 3.84 with 1 degree of freedom (P < 0.05) using the likelihood ratio test. A visual predictive check with 200 replicates was performed to assess the model performance. A total of 1000 bootstrap runs were performed to provide estimates of the precision of parameter estimates and the 95% confidence intervals for the pharmacokinetic parameters.
Individual pharmacokinetic parameters for each patient were derived from the final population model and used to simulate time-concentration profiles using WinNonlin® 6.2 (Pharsight Corporation, Cary, NC). The simulated blood HCQ concentrations were compared with observed concentrations to determine the predictive performance of the model. HCQ pharmacokinetic parameter estimates (peak blood concentration, Cmax; trough blood concentration, Cmin; average blood concentration, Cavg; area under the concentration-time curve, AUC) from these simulations were used to explore pharmacokinetic-pharmacodynamic relationships.
The pharmacokinetic-pharmacodynamic relationship between HCQ and AV accumulation at 2 wk was investigated by using an exploratory classification tree (Salford Predictive Modeler Builder v6.6) to identify a threshold effect of Cmax on AV accumulation in each of peripheral blood mononuclear cells and bone marrow plasma cells. The effect of the threshold value for Cmax on the change in AV from baseline to 2 wk was then investigated using the Kruskal-Wallis test for comparing median values and the Kolmogorov-Smirnov 2-sample test to identify any significant shift in the distribution.
| Abbreviations: | ||
| AUC | = | area under the concentration-time curve |
| AV | = | autophagic vacuole |
| Cavg | = | average concentration |
| Cmax | = | maximum concentration |
| Cmin | = | trough concentration |
| DLT | = | dose-limiting toxicity |
| HDAC | = | histone deacetylase |
| HCQ | = | hydroxychloroquine |
| IS | = | internal standard |
| Ka | = | absorption rate constant |
| MAP1LC3A | = | microtubule-associated protein 1 light chain 3 alpha |
| MR | = | minor response |
| n | = | random effect |
| PBMC | = | peripheral blood mononuclear cells |
| PK | = | pharmacokinetic |
| SD | = | stable disease |
| SQSTM1 | = | sequestosome 1 |
| tLag | = | lag time |
| tv | = | population or typical value |
| Vc/F | = | volume of distribution in the central compartment |
| VGPR | = | very good partial response |
Disclosure of Potential Conflicts of Interest
The research described in this manuscript was supported in part by a research grant from Millennium Pharmaceuticals, which manufactures and distributes bortezomib (Velcade). DTV currently holds a separate research grant from Millennium Pharmaceuticals. DTV has in the past worked as a consultant for Millennium Pharmaceuticals. DTV currently holds a research grant from Acetylon Pharmaceuticals, which manufactures the HDAC6 inhibitor referred to in the Discussion section. DTV is currently participating as an investigator in the clinical trial referred to in the Discussion.
Acknowledgments
The authors thank Cezary R Swider, Joy Cannon, and Martin Carroll at the University of Pennsylvania Stem Cell and Xenograft Core, for processing samples. One of the authors (RR) is now an employee of Merck. This research was supported by a Special Fellow in Clinical Research award from the Leukemia & Lymphoma Society (DTV), a research grant from Millennium Pharmaceuticals, a pilot project grant from the University of Pennsylvania Abramson Cancer Center, National Cancer Institute grants K23CA120862 (RKA), K23CA130074 (DTV), and P30CA016520 (DFH, KST), and National Center for Research Resources grant UL1-RR-024134. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, the National Center for Research Resources, or the National Institutes of Health.
References
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al, Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352:2487 - 98; http://dx.doi.org/10.1056/NEJMoa043445; PMID: 15958804
- San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, et al, VISTA Trial Investigators. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008; 359:906 - 17; http://dx.doi.org/10.1056/NEJMoa0801479; PMID: 18753647
- de Vos S, Goy A, Dakhil SR, Saleh MN, McLaughlin P, Belt R, Flowers CR, Knapp M, Hart L, Patel-Donnelly D, et al. Multicenter randomized phase II study of weekly or twice-weekly bortezomib plus rituximab in patients with relapsed or refractory follicular or marginal-zone B-cell lymphoma. J Clin Oncol 2009; 27:5023 - 30; http://dx.doi.org/10.1200/JCO.2008.17.7980; PMID: 19770386
- Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009; 113:6069 - 76; http://dx.doi.org/10.1182/blood-2009-01-199679; PMID: 19380866
- Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006; 24:4867 - 74; http://dx.doi.org/10.1200/JCO.2006.07.9665; PMID: 17001068
- Goy A, Younes A, McLaughlin P, Pro B, Romaguera JE, Hagemeister F, Fayad L, Dang NH, Samaniego F, Wang M, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005; 23:667 - 75; http://dx.doi.org/10.1200/JCO.2005.03.108; PMID: 15613697
- Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res 2007; 13:5291 - 4; http://dx.doi.org/10.1158/1078-0432.CCR-07-0871; PMID: 17875757
- O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol 2005; 23:676 - 84; http://dx.doi.org/10.1200/JCO.2005.02.050; PMID: 15613699
- Shah MH, Young D, Kindler HL, Webb I, Kleiber B, Wright J, Grever M. Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clin Cancer Res 2004; 10:6111 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-04-0422; PMID: 15447997
- Kozuch PS, Rocha-Lima CM, Dragovich T, Hochster H, O’Neil BH, Atiq OT, Pipas JM, Ryan DP, Lenz HJ. Bortezomib with or without irinotecan in relapsed or refractory colorectal cancer: results from a randomized phase II study. J Clin Oncol 2008; 26:2320 - 6; http://dx.doi.org/10.1200/JCO.2007.14.0152; PMID: 18467723
- Fanucchi MP, Fossella FV, Belt R, Natale R, Fidias P, Carbone DP, Govindan R, Raez LE, Robert F, Ribeiro M, et al. Randomized phase II study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer. J Clin Oncol 2006; 24:5025 - 33; http://dx.doi.org/10.1200/JCO.2006.06.1853; PMID: 17075122
- Alberts SR, Foster NR, Morton RF, Kugler J, Schaefer P, Wiesenfeld M, Fitch TR, Steen P, Kim GP, Gill S. PS-341 and gemcitabine in patients with metastatic pancreatic adenocarcinoma: a North Central Cancer Treatment Group (NCCTG) randomized phase II study. Ann Oncol 2005; 16:1654 - 61; http://dx.doi.org/10.1093/annonc/mdi324; PMID: 16085692
- Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, Orsi A, Pasqualetto E, Mezghrani A, Calbi V, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009; 113:3040 - 9; http://dx.doi.org/10.1182/blood-2008-08-172734; PMID: 19164601
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest 2005; 115:2679 - 88; http://dx.doi.org/10.1172/JCI26390; PMID: 16200202
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859 - 63; http://dx.doi.org/10.1038/nature05853; PMID: 17568747
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 2005; 280:40282 - 92; http://dx.doi.org/10.1074/jbc.M508786200; PMID: 16192271
- Wu WK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, Wong CC, Li HT, Zhang L, Ren SX, et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy 2010; 6:228 - 38; http://dx.doi.org/10.4161/auto.6.2.11042; PMID: 20087064
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513 - 24; http://dx.doi.org/10.2353/ajpath.2007.070188; PMID: 17620365
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 2005; 102:8567 - 72; http://dx.doi.org/10.1073/pnas.0503221102; PMID: 15937109
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 1981; 90:665 - 9; http://dx.doi.org/10.1083/jcb.90.3.665; PMID: 6169733
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Hui B, Shi YH, Ding ZB, Zhou J, Gu CY, Peng YF, Yang H, Liu WR, Shi GM, Fan J. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer 2012; 118:5560 - 71; http://dx.doi.org/10.1002/cncr.27586; PMID: 22517429
- Jia L, Gopinathan G, Sukumar JT, Gribben JG. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS One 2012; 7:e32584; http://dx.doi.org/10.1371/journal.pone.0032584; PMID: 22393418
- Yao F, Wang G, Wei W, Tu Y, Tong H, Sun S. An autophagy inhibitor enhances the inhibition of cell proliferation induced by a proteasome inhibitor in MCF-7 cells. Mol Med Rep 2012; 5:84 - 8; PMID: 21931937
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-08-2839; PMID: 19417138
- Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, Itoh M, Tomoda A. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol 2011; 38:643 - 54; PMID: 21174067
- Shen, John Paul Y. C., Divakaran S, Ponduru S, Vogl DT, Amaravadi RK, Bradner JE. The rationale for combined proteasome and autophagy inhibition in multiple myeloma established using novel translational platforms. American Society of Hematology 2008; 112:2755
- Escalante AM, McGrath RT, Karolak MR, Dorr RT, Lynch RM, Landowski TH. Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo. Cancer Chemother Pharmacol 2013; 71:1567 - 76; http://dx.doi.org/10.1007/s00280-013-2156-3; PMID: 23572175
- Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, Liu J, Yin XM. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther 2009; 8:2036 - 45; http://dx.doi.org/10.1158/1535-7163.MCT-08-1169; PMID: 19584239
- Hui B, Shi YH, Ding ZB, Zhou J, Gu CY, Peng YF, Yang H, Liu WR, Shi GM, Fan J. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer 2012; 118:5560 - 71; http://dx.doi.org/10.1002/cncr.27586; PMID: 22517429
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403 - 14; http://dx.doi.org/10.4161/auto.29231
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359 - 68; http://dx.doi.org/10.4161/auto.28984
- Tett SE, Cutler DJ, Day RO, Brown KF. A dose-ranging study of the pharmacokinetics of hydroxy-chloroquine following intravenous administration to healthy volunteers. Br J Clin Pharmacol 1988; 26:303 - 13; http://dx.doi.org/10.1111/j.1365-2125.1988.tb05281.x; PMID: 3179169
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369 - 79; http://dx.doi.org/10.4161/auto.29118
- Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014; 10:1415 - 25; http://dx.doi.org/10.4161/auto.29231
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012; 109:8253 - 8; http://dx.doi.org/10.1073/pnas.1118193109; PMID: 22566612
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr., Pal A, Bornmann WG, Chiao PJ, Huang P, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res 2006; 66:3773 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-05-2961; PMID: 16585204
- Dimopoulos M, Siegel DS, Lonial S, Qi J, Hajek R, Facon T, Rosinol L, Williams C, Blacklock H, Goldschmidt H, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol 2013; 14:1129 - 40; http://dx.doi.org/10.1016/S1470-2045(13)70398-X; PMID: 24055414
- Richardson PG, Schlossman RL, Alsina M, Weber DM, Coutre SE, Gasparetto C, Mukhopadhyay S, Ondovik MS, Khan M, Paley CS, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013; 122:2331 - 7; http://dx.doi.org/10.1182/blood-2013-01-481325; PMID: 23950178
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448 - 57; http://dx.doi.org/10.1038/sj.cdd.4401359; PMID: 14713959
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391 - 1402
- Durie BG, Harousseau JL, Miguel JS, Bladé J, Barlogie B, Anderson K, Gertz M, Dimopoulos M, Westin J, Sonneveld P, et al, International Myeloma Working Group. International uniform response criteria for multiple myeloma. Leukemia 2006; 20:1467 - 73; http://dx.doi.org/10.1038/sj.leu.2404284; PMID: 16855634
- Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23:3 - 9; http://dx.doi.org/10.1038/leu.2008.291; PMID: 18971951