
Abstract
Autophagy is a lysosomal degradative pathway that has diverse physiological functions and plays crucial roles in several viral infections. Here we examine the role of autophagy in the life cycle of JEV, a neurotropic flavivirus. JEV infection leads to induction of autophagy in several cell types. JEV replication was significantly enhanced in neuronal cells where autophagy was rendered dysfunctional by ATG7 depletion, and in Atg5-deficient mouse embryonic fibroblasts (MEFs), resulting in higher viral titers. Autophagy was functional during early stages of infection however it becomes dysfunctional as infection progressed resulting in accumulation of misfolded proteins. Autophagy-deficient cells were highly susceptible to virus-induced cell death. We also observed JEV replication complexes that are marked by nonstructural protein 1 (NS1) and dsRNA colocalized with endogenous LC3 but not with GFP-LC3. Colocalization of NS1 and LC3 was also observed in Atg5 deficient MEFs, which contain only the nonlipidated form of LC3. Viral replication complexes furthermore show association with a marker of the ER-associated degradation (ERAD) pathway, EDEM1 (ER degradation enhancer, mannosidase α-like 1). Our data suggest that virus replication occurs on ERAD-derived EDEM1 and LC3-I-positive structures referred to as EDEMosomes. While silencing of ERAD regulators EDEM1 and SEL1L suppressed JEV replication, LC3 depletion exerted a profound inhibition with significantly reduced RNA levels and virus titers. Our study suggests that while autophagy is primarily antiviral for JEV and might have implications for disease progression and pathogenesis of JEV, nonlipidated LC3 plays an important autophagy independent function in the virus life cycle.
Introduction
Autophagy is an important cellular process that maintains cellular homeostasis. Autophagic cargo such as long-lived cytoplasmic proteins and dysfunctional organelles are sequestered by double-membrane vesicles called autophagosomes and are degraded after autophagosome-lysosome fusion.Citation1 The autophagic mechanism is constitutive and generally occurs at a basal level in all cells, but is upregulated in response to extracellular or intracellular stress and pathogen infection. It is also an important component of the innate and adaptive immune response against a variety of viral and bacterial pathogens.Citation2,Citation3
Several viruses can lead to the induction of an autophagic response in infected cells which is closely linked to their propagation and/or pathogenesis.Citation4-Citation7 Signaling downstream of virus-receptor interactions,Citation8-Citation10 induction of the UPR leading to ER stressCitation11,Citation12 and the activation of immune sensorsCitation13,Citation14 have been shown to be the key autophagic triggers in response to viral infections. Accumulation of autophagosomes in virus-infected cells can also be caused by block of autophagosomal-lysosomal fusion.Citation5,Citation15,Citation16 Functional lysosomes are critical for maturation of autophagosomes and for degradation of their cargo.
Autophagy can function as a mechanism of host defense by clearing the pathogen or its proteins through degradation, enhancement of type I interferons, or processing and presentation of antigens for MHC presentation and T cell priming.Citation4,Citation17,Citation18 Autophagy assumes prime importance in protecting the CNS and neurons from viral infections, since these cells have very limited regenerative capacity. On the other hand microbes can abrogate and/or exploit the autophagic process to enhance their replication or transmission.Citation2,Citation5,Citation15 Recent studies suggest crosstalk between autophagic and apoptotic pathways,Citation16,Citation19 and it is likely that autophagy in response to virus infection can regulate apoptosis and virus propagation from infected cells.
Although microtubule-associated protein 1 light chain 3 (MAP1LC3, and henceforth LC3 in the text), a mammalian ortholog of yeast Atg8, was originally characterized as a microtubule-associated proteinCitation20 it has been extensively studied as a part of the autophagy machinery and its nonautophagic role is not well understood. Some recent studies have provided evidence for an autophagy-independent role of LC3 in cellular processes. In the yeast Pichia pastoris, nonlipidated LC3 plays a crucial role in vacuole fusion.Citation21 Native LC3 also interacts with inclusion bodies in the bacteria Chlamydia trachomatis.Citation22 Studies on the ERAD pathway have demonstrated that nonlipidated LC3 coats ER-derived vesicles referred to as EDEMosomes.Citation23 The primary function of the EDEMosomes is for ERAD tuning which involves selective clearance of short-lived ER chaperones from the ER to the endolysosomal system.Citation23,Citation24 EDEM1, a marker of EDEMosomes,Citation25,Citation26 is selectively cleared from the ER to tune down the ERAD activity.Citation23 Recent studies have shown that these vesicles are co-opted by the mouse hepatitis coronavirus and equine arteritis virus for replication.Citation27,Citation28 Downregulation of LC3 markedly reduces replication of both viruses implicating a crucial role of nonlipidated LC3 in virus replication.Citation27,Citation28 SEL1L, an ERAD tuning receptor component, also localizes to these vesicles and is involved in replication of mouse hepatitis coronavirus.Citation29
JEV, a flavivirus spread through mosquitoes, is responsible for frequent epidemics of viral encephalitis in South East Asia, China, and India.Citation30 JEV is a neurotropic virus and clinical manifestations of the disease range from febrile syndromes to multifocal CNS disorders and death.Citation30 In the present study we have explored the relevance of autophagy and the role of the crucial autophagy protein LC3 in the context of JEV replication. We observe that autophagy primarily functions to restrict viral replication. Autophagy-deficient neuronal cells and mouse embryonic fibroblasts show increased viral replication and titers and are highly susceptible to virus-induced cell death. Further, by immunofluorescence microscopy we observe that viral replication complexes marked by JEV NS1 and the replicative intermediate dsRNA also contain markers of the ERAD tuning LC3-I and EDEM1. JEV replication thus appears to occur on ER-derived EDEMosomes similar to what has been described for the mouse hepatitis coronavirus and equine arteritis virus.Citation27,Citation28 Downregulation of LC3 in Neuro2a cells reduces JEV titers significantly, implying that nonlipidated LC3 acts upstream of the cellular autophagic pathway as a crucial host factor for the virus life cycle.
Results
Autophagosomes accumulate in JEV-infected cells
To investigate the cellular autophagy level changes in response to JEV infection we monitored the processing of LC3-I to its lipidated membrane-bound form LC3-II. The mouse neuroblastoma cell line Neuro2a was transfected with GFP-LC3 and set up for serum-starvation or JEV-infection (). In mock-infected Neuro2a cells, GFP-LC3 shows a diffuse distribution (, left panel). JEV infection leads to accumulation of autophagosomes in cells as detected by GFP-LC3 punctate distribution (). Similarly, on serum-starvation which leads to induction of autophagy, GFP-LC3 can be seen in a punctate pattern which represents autophagosomes (). More than 75% of cells that were JEV-infected or serum-starved showed a punctate distribution of GFP-LC3 compared with ~20% of mock-infected cells (). There were 40 to 50 GFP-LC3 puncta per cell in JEV-infected and serum-starved cells compared with ~10 in mock-infected cells (). Autophagosome accumulation was further validated by western blotting which showed an increase in LC3-II form in serum-starved and JEV-infected cells (). A time course of JEV infection showed that autophagosome accumulation could be detected by 8 to 12 h pi, and it increased as infection progressed (; Fig. S1). Autophagy was not a rapid response to viral infection and its induction paralleled the accumulation of JEV proteins in cells.
Figure 1. JEV infection leads to accumulation of autophagosomes in host cells. (A–C) Neuro2a cells transfected with GFP-LC3 (green, left panels), were mock-infected (A), infected with JEV (MOI 5, 24 h) (B), or serum-starved for 12 h (C). Cells were fixed and stained with JEV-E antibody (blue, middle panels). The merging of the 2 signals is shown in the right panels. Scale bar: 10 µm. (D and E) Quantification of cells showing punctate distribution of GFP-LC3 (D), and the number of GFP-LC3 puncta per cell (E). (F) Mock-infected, serum-starved and JEV-infected Neuro2a cells were lysed at the indicated times postinfection and poststarvation. Lysates were analyzed by western blotting with LC3, JEV, NS5 (infection control), and GAPDH (loading control) antibodies. (G) Quantitative PCR (qPCR) of autophagy genes in Neuro2a cells that were mock-infected, serum-starved (12 h), or infected with JEV (MOI 5, 24 h). The graph shows the relative increased expression of gene transcription normalized to mock-infected samples. Values represent mean ± SD of 3 independent experiments. (H and I) Vero cells (H), WT and atg5−/− MEFs (I) were mock-infected, serum-starved (12 h) or infected with JEV (MOI 5, 24 h). Western blots were done using LC3, JEV, NS5, and GAPDH antibodies. The ratio of LC3-I/GAPDH and LC3-II/GAPDH was calculated as shown below the representative blots. The Student t test was used to calculate P values. *P < 0.05, **P < 0.01.
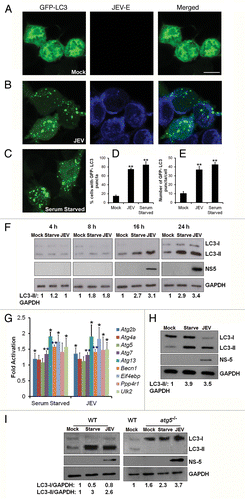
A transcriptome study was performed in Neuro2a cells and analyzed for changes in gene expression profiles of host autophagy genes after JEV infection (Bhattacharya et al., manuscript in preparation). The following genes which have been directly or indirectly shown to be involved at different steps of the autophagy pathway,Citation31,Citation32 were significantly upregulated in JEV-infected cells—Atg2b, Atg4a-ps (autophagy related 4A, pseudogene), Atg13, Eif4ebp1, Pp4r1, and Ulk2. We further validated these observations and analyzed the mRNA levels of a group of 9 genes reported to be involved in several steps of the autophagic pathway. SYBR-green based qPCR assays were performed using gene-specific primers and total RNA from serum-starved, mock-, or JEV-infected Neuro2a cells. Additionally, as a control, qPCR was performed for genes that did not show any deregulation. The cycle threshold (Ct) value for each gene was normalized to that of Gapdh and the fold change in the expression level of each gene was determined relative to mock-infected cells (). We observed a clear transcriptional reprogramming of several autophagy genes in response to serum-starvation and JEV-infection, suggesting that virus leads to induction of a robust autophagic response in host cells.
Increased LC3-II accumulation was also observed in JEV-infected Vero cells (), suggesting that autophagy is a generic response to JEV infection in different cell types. Autophagic induction in response to JEV infection has also been reported in NT-2 (pluripotent human testicular embryonal carcinoma), N18 and Neuro2a (mouse neuroblastoma) and A549 (human lung carcinoma) cell lines in 2 earlier studies.Citation33,Citation34
To examine the relevance of the cellular autophagy pathway in JEV infection we also used wild-type (WT) and atg5−/− MEFs.Citation35 ATG5 is an essential protein for autophagosome formation, and processing of LC3-I to LC3-II is greatly reduced or absent in atg5−/− MEFs.Citation35 As expected, WT MEFs showed accumulation of LC3-II in response to serum-starvation and JEV infection (, left panel), whereas, atg5−/− MEFs did not show LC3-II (, right panel). Interestingly, atg5−/− MEFs showed higher basal levels of LC3-I compared with WT MEFs consistent with the fact that LC3-I cannot be processed to LC3-II in these cells.
Autophagy restricts JEV replication and influences viral yields
ATG7 is crucial for elongation and closure of the autophagosome and for the conversion of LC3-I to its lipidated LC3-II form.Citation36,Citation37 To elucidate the significance of autophagy in JEV life cycle, we specifically depleted key autophagy protein ATG7 in Neuro2a cells by RNA interference (). In ATG7-depleted Neuro2a cells higher levels of LC3-I was observed, similar to what was seen for atg5−/− MEFs. While the JEV-infection efficiency in both control and Atg7 siRNA-treated cells was similar (Fig. S2), JEV RNA levels were enhanced more than 4-fold in the ATG7-depleted background and virus titers were significantly higher by 2.5-fold (). This amplification of JEV RNA levels and titers in ATG7-deficient cells was observed consistently in cells infected across different multiplicities of infection (MOIs).
Figure 2. Autophagy restricts JEV replication and influences viral yields. (A) Western blot showing levels of ATG7 and LC3 in control nontargeting (NT) and Atg7 siRNA-transfected Neuro2a cells at 48 h post-transfection. The ratio of ATG7/GAPDH and LC3-I/GAPDH is shown below the blot. (B) Control (NT) /Atg7 siRNA-transfected Neuro2a cells were infected with JEV (MOI 5) and 24 h pi, viral RNA levels were determined by qRT-PCR. (C) JEV titers in culture supernatants of infected control and ATG7-deficient cells. (D) Western blot showing levels of ATG5 in WT and atg5−/− MEFs. (E and F) WT and atg5−/− MEFs were infected with JEV (MOI 1). (E) Viral RNA levels were determined by qRT-PCR at indicated times postinfection. (F) JEV titers in culture supernatants at 24 h pi were determined. The Student t test was used to calculate P values. **P < 0.01.
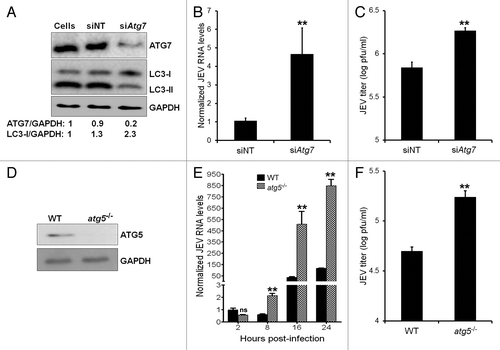
To further validate our observations we studied JEV replication in WT and atg5−/− MEFs (). A time-course analysis of JEV RNA accumulation showed that viral RNA levels were essentially comparable at 2 h pi, indicating similar virus uptake in both cell lines (). Whereas JEV RNA levels increased in WT MEFs by approximately 100-fold in 24 h, a close to 600-to-800 fold increase was seen in atg5−/− MEFs (). This enhancement also manifested in a significant increase (~3.5-fold) in JEV titers in atg5−/− MEFs (). Collectively our data from ATG7-depleted Neuro2a and atg5−/− MEFs suggests that autophagy significantly restricts JEV replication and reduces extracellular virus yields.
We further tested whether pharmacological induction of autophagy also gives a similar effect. For this we employed Torin1, a highly potent and selective MTOR inhibitor.Citation38,Citation39 Treatment with Torin1, led to rapid accumulation of LC3-II in cells (Fig. S3A). Torin1, however, significantly enhanced viral protein translation (Fig. S3A) and JEV RNA levels in Neuro2a cells (Fig. S3B). This enhancement in JEV RNA levels was also observed both in WT and atg5−/− MEFs (Fig. S3C). These observations imply that increase in virus replication by Torin 1 is independent of autophagic induction and could be potentially mediated by other effects of MTOR inhibition on cellular physiology like inhibition of cell growth and/or cell cycle arrest.
Autophagy is functional in early stages of JEV infection
Since our results indicate that autophagy restricts viral replication, we analyzed the turnover of autophagosomes in JEV-infected cells. Autophagosomes fuse with late endosomes and lysosomes to form autolysosomes.Citation1 To distinguish between autophagosome accumulation caused by autophagy induction vs. a block in autophagosome degradation, we used the vacuolar ATPase inhibitor bafilomycin A1 which blocks vesicle acidification, and thus prevents autophagosome maturation into lysosomes and subsequent degradation of cargo and pepstatin A which is an inhibitor of lysosomal-mediated proteolysis.Citation40,Citation41 Mock- or JEV-infected cells were treated with bafilomycin A1 or pepstatin A and cell lysates were analyzed by western blotting. In mock-infected cells, bafilomycin A1 and pepstatin A treatment led to rapid accumulation of LC3-II, indicating that autophagosomes are not being turned over by lysosomal proteolysis (). A comparison of LC3-II signals in JEV-infected cells (24 h pi), showed an enhancement of LC3-II levels in bafilomycin A1 and pepstatin A-treated sample (, quantification). This suggests that at 24 h post-infection (pi) lysosomal proteolysis was not completely blocked, indicative of a functional autophagic pathway. However by 48 h pi, no further accumulation of LC3-II could be observed in response to bafilomycin A1 and pepstatin A treatment, demonstrating an autophagosomal-lysosomal block in JEV-infected cells ().
Figure 3. Autophagy-deficient cells are highly susceptible to virus-induced cell death. (A) Mock and JEV-infected (MOI 5, 24 and 48 h), Neuro2a cells were treated with DMSO (Ctl), 100 nM bafilomycin A1 (Baf) for 2 h before fixation or 50 µg/ml pepstatin A (Pep) for 12 h before fixation, and protein extracts were analyzed by western blotting with LC3, SQSTM1, NS1, and GAPDH (loading control) antibodies. (B) Percentage of death in JEV-infected (MOI 1) control/ ATG7-depleted Neuro2a cells at 48 h pi. (C and D) WT (black bar) and atg5−/− (hashed bar) MEFs were infected with JEV at MOI 0.1 (C) and MOI 1 (D), and percentage of cell death was analyzed at 24 and 48 h pi. Presented are mean ± standard error of values obtained from 3 independent experiments. The Student t test was used to calculate P values. *P < 0.05, **P < 0.01.
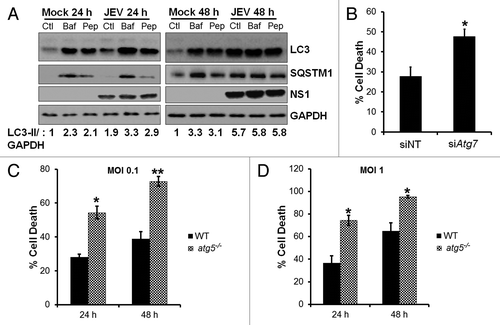
Autophagy is also responsible for degradation of autophagosome cargo SQSTM1/p62. Inhibition of autophagy in cells leads to an increase in the size and number of the autophagosome cargo and protein levels of SQSTM1.Citation42 In mock-infected cells that were treated with bafilomycin A1 or pepstatin A, we observed accumulation of autophagosome cargo SQSTM1 (). JEV-infected cells at 24 h pi showed levels of SQSTM1 similar to that in mock-infected cells (), and it increased on bafilomycin A1 and pepstatin A treatment in a pattern similar to LC3-II accumulation, indicative of a functional autophagic pathway at 24 h pi. However by 48 h pi increased levels of SQSTM1 are seen in JEV-infected cells indicative of accumulation of misfolded proteins and a block in autophagosome maturation ().
Autophagy-deficient cells are highly susceptible to virus-induced cell death
Since virus RNA replication and titers were significantly enhanced in autophagy-deficient cells, we also examined virus-induced cell death in these cells. ATG7 depletion was not toxic to cells (Fig. S4A) and no significant difference in cell viability was seen between mock-infected control and ATG7-depleted cells (Fig. S4B). However, in response to JEV infection Neuro2a cells depleted of ATG7 showed greater cell death (~1.9-fold more) compared with control (). This enhancement in cell death was consistently seen in cells infected across different MOI and at different time pi (data not shown). A similar increase in the number of apoptotic and secondary necrotic cells (~2-fold) was also seen in atg5−/− MEFs compared with WT MEFs, infected with JEV at MOI 0.1 and l and at both 24 and 48 h pi (). Our data suggest that enhanced viral replication in autophagy-deficient cells potentially leads to a stronger, robust, and earlier cell death response in these cells. It is also possible that autophagy operates as a survival pathway in JEV-infected cells and its deficiency leads to increased susceptibility to cell death. This is similar to what is observed in case of influenza and chikungunya viruses, where compared with WT MEFs, atg5−/− MEFs have been shown to display increased cell death in response to virus infection.Citation5,Citation19 JEV induced enhancement of cell death in autophagy-deficient cells has also been reported in an earlier study.Citation33
JEV replication complexes localize with endogenous LC3 but not with GFP-LC3
Flavivirus replication occurs on replication complexes that are associated with the dsRNA template located in induced membranes in the cytoplasm. Several of the viral NS proteins colocalize to these replication complexes.Citation43-Citation49 Recent electron tomography studies on the flaviviruses West Nile, dengue, and tick-borne encephalitis have demonstrated that viral replication complexes are a highly organized network of rearranged membranes that are derived from the ER. These replication complexes contain dsRNA and appear as invaginations of the ER membrane with a pore-like connection to the cytoplasm.Citation47,Citation50,Citation51 The subcellular sites of RNA replication can be probed by immunolabeling with anti-dsRNA antibodies which recognize the double-stranded replicative form RNA.Citation52 We analyzed the distribution of dsRNA and JEV NS1, NS3, and NS5 proteins in virus-infected Neuro2a cells by immunofluorescence (Fig. S5). The dsRNA stained as discrete puncta in JEV-infected cells that potentially represent sites of viral replication. All 3 NS proteins were also strongly labeled at 24 h pi, in an irregular-shaped punctate network that extended throughout the cell. The staining pattern of the JEV NS proteins was more widespread and variable than that for dsRNA; however, all 3 NS proteins showed significant overlap with dsRNA (Pearson coefficient > 0.5) (Fig. S5). Immunofluorescence analysis of components of replication complexes—NS1, NS3, and dsRNA in JEV-infected WT and atg5−/− MEFs showed that these markers are similarly distributed to numerous punctate structures and membranes in both cell lines (Fig. S6 and S7). atg5−/− MEFs showed very well-defined, numerous vesicular structures that represent viral replication complexes. The Pearson coefficient of colocalization of NS1 and dsRNA ranged from 0.43 to 0.5, and that of NS1 and NS3 from 0.62 to 0.7 in WT and atg5−/− MEFs infected with JEV.
A closer examination of localization of JEV replication complexes with autophagy markers showed a remarkable overlap between endogenous LC3 and NS1 in infected cells (). Colocalization analysis of LC3 and NS1 showed a Pearson coefficient close to 0.6 (). Overlap between endogenous LC3 and NS1 was detected very early during infection (~10 to 12 h pi) and was maintained throughout the infectious cycle (48 h pi). To further analyze if these structures are autophagosomes, we checked for colocalization of NS1 with the markers of late endosomes-LAMP1 (marks membrane of late endosomes) and LysoTracker Red (marks acidic compartments) (). While NS1 was significantly concentrated in LC3-positive compartments, we did not observe overlap of NS1 in LAMP1 and LysoTracker Red compartments () suggesting that LC3-NS1 structures are neither acidic nor late endosomal structures. We further checked the colocalization of NS1 with GFP-LC3, and observed significantly reduced overlap between the 2 proteins (). Several studies have shown that GFP-LC3 localization in cells is restricted exclusively to autophagosomes.Citation41,Citation53 Thus, while endogenous LC3 extensively colocalizes with JEV replication complexes, ectopically expressed GFP-LC3 does not.
Figure 4. JEV NS1 colocalizes with endogenous LC3 but not with GFP-LC3. (A and B) JEV-infected Neuro2a cells (MOI 5, 24 h pi) were stained with mouse NS1 (red) and rabbit LC3 (green) (A), or with mouse NS1 (red) and rabbit LAMP1 (green) antibodies (B). Nuclei were visualized by DAPI staining (blue). (C) JEV-infected Neuro2a cells were given a pulse of LysoTracker Red, fixed and stained with NS1 antibody (green). (D) GFP-LC3 (green)-transfected Neuro2a cells were infected with JEV and stained with NS1 antibody (red). Color merged images are shown in the third panels. Extreme right panels show a magnified view of the region corresponding to the asterisk (*). Scale bar: 10 µm and 3 µm (extreme right panels). (E) Bar graph showing the extent of colocalization of JEV NS1 with LC3, LAMP1, LysoTracker Red and GFP-LC3-positive compartments. Colocalization index between NS1 and endogenous LC3 is significantly higher than with LAMP1, LysoTracker Red, and GFP-LC3. The Student t test was used to calculate P values. *P < 0.05, **P < 0.01.
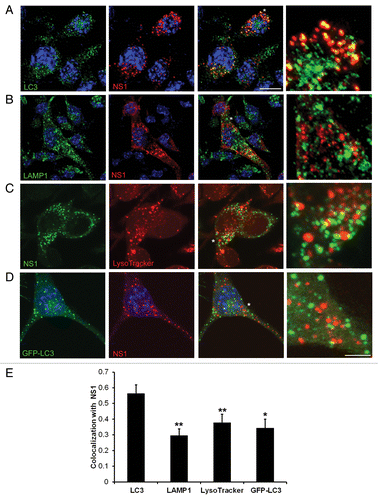
Overexpression of HA-fused NS1 in Neuro2a cells did not lead to its accumulation in LC3-positive structures (Pearson coefficient of 0.38), suggesting that observed colocalization occurs in replication complexes, and, is a result of infection-induced events and not NS1 expression alone (Fig. S8). It is also possible that HA-NS1 localizes differently than NS1 produced in response to viral infection.
Staining of LC3 in WT and atg5−/− MEFs revealed LC3 in numerous well-defined punctate and vesicular structures. Since atg5−/− MEFs do not process LC3-I to its lipidated form, the LC3 puncta in these cells represent LC3-I and not LC3-II. We further checked the colocalization between NS1 and LC3 in WT and atg5−/− MEFs. Similar to what was observed in Neuro2a cells there was significant overlap (Pearson coefficient of 0.6) between endogenous LC3 and NS1 in both MEF cell lines (). NS1-LC3 complexes showed a punctate distribution in Neuro2a cells (), while their staining pattern was more defined and vesicular in MEFs (). These compartments did not show any overlap with GFP-LC3 in WT MEFs indicating that these are not autophagosomes (Fig. S9).
Figure 5. JEV NS1 colocalizes with endogenous LC3 in WT and atg5−/− MEFs. (A) Wt and (B) atg5−/− MEFs were mock-or JEV-infected (MOI 5, 24 h) and stained with antibodies against LC3 (green) and NS1 (red). Merging of the 2 signals and DAPI (blue) for nuclear staining is shown in the extreme right panels. Scale bar: 10 µm.
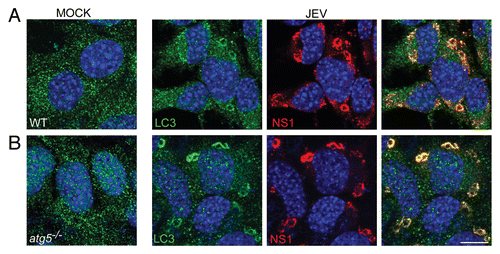
The presence of LC3-positive puncta and vesicular membranes in atg5−/− MEFs demonstrates that LC3-I form is associated with NS1 containing replication complexes. Lipidation is thus not a requirement for LC3 association with JEV replication complexes. Collectively these data suggest that LC3-I associates with NS1 marking viral replication complexes, and ATG5 and a functional autophagy pathway were not required for the association of LC3 with NS1.
Viral replication complexes are marked by EDEM1
Recent studies have demonstrated that mouse hepatitis virus and equine arteritis virus utilize ER-derived components of the ERAD pathway, EDEMosomes that are marked by the proteins EDEM1 and LC3-I, for their replication.Citation27,Citation28 Importantly, the replication complexes for these viruses stain with antibodies against endogenous LC3 but not with GFP-LC3. The LC3-I coat is a key feature of these replication complexes and EDEMosomes,Citation23 whereas autophagosomes, are associated with LC3-II and can be marked with GFP-LC3.Citation53 SEL1L, a critical component of the ERAD tuning receptor, also associates with mouse hepatitis virus replication platforms and is essential for the virus life cycle.Citation29
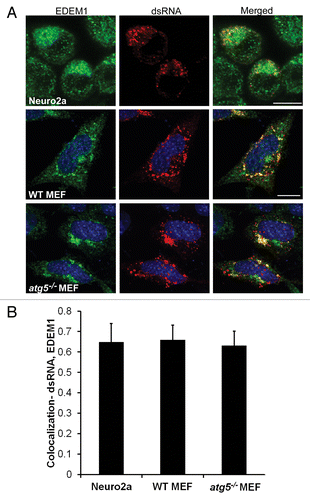
We thus tested whether JEV replication complexes also localize with EDEM1 containing membranes in Neuro2a cells and MEFs. There was significant colocalization between NS1 and EDEM1 in all cell types () strongly suggesting that JEV replication complexes are associated with markers of ERAD tuning. Density gradient fractionation of cell lysates from JEV treated Neuro2a cells revealed significant enrichment of virus nonstructural proteins in denser membrane fractions that were also enriched in EDEM1 and LC3-I. These fractions were clearly separated from the lighter autophagosomal/late endosomal fraction containing LC3-II and LAMP1 that floated to the top of the gradient (fraction 1) (). Significant overlap was also observed between EDEM1 and dsRNA in Neuro2a cells, WT and atg5−/− MEFs (). The presence of numerous EDEM1-positive vesicles in atg5−/− MEFs indicates that EDEM1 turnover is independent of the autophagy pathway as shown for the mouse hepatitis coronavirus.Citation54 The strong parallels seen between our observations and those described for the mouse hepatitis coronavirus and equine arteritis virus suggest that JEV also utilizes the ERAD tuning machinery to derive membranes for replication.
Figure 6. JEV NS1 localizes to EDEM1-positive vesicles. (A) JEV-infected Neuro2a cells, and WT and atg5−/− MEFs (MOI 5, 24 h) were stained with EDEM1 (green) and NS1 (red) antibodies. Color-merged images are shown in the third panel. Panels on the extreme right show magnified view of the region marked with *. Scale bar: 10 µm and 3 µm (extreme right panels). (B) Colocalization of NS1 with EDEM1 in Neuro2a, WT and atg5−/− MEFs (C) Postnuclear supernatants of JEV-infected Neuro2a cells were fractionated on a discontinuous Optiprep gradient. Five fractions were collected from top to bottom of the gradient and probed with antibodies against LC3, EDEM1, LAMP1, NS1, NS3, and NS5.
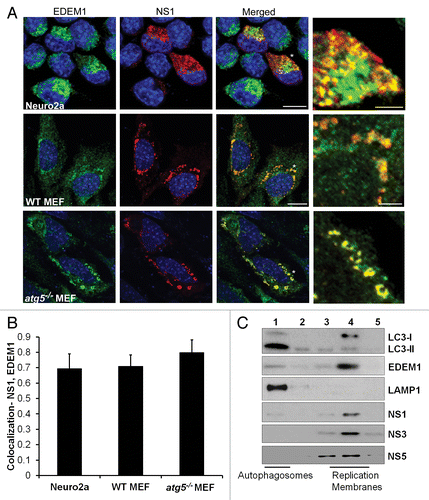
Figure 7. Replication intermediate dsRNA colocalizes with EDEM1 vesicles. (A) JEV-infected (MOI 5, 24 h) Neuro2a cells, WT, and atg5−/− MEFs were stained with EDEM1 (green) and dsRNA (red) antibodies. Merging of the 2 signals is shown in the right panels. (B) Pearson coefficient of colocalization between dsRNA and EDEM1. Scale bar: 10 µm.
EDEM1 localization in mock-infected cells was both ER as seen by colocalization with ectopically expressed Myc-GRP78 (Pearson coefficient ~0.65), and as several discrete independent vesicles in the cytosol (Fig. S10A). In JEV-infected cells EDEM1 showed enrichment in viral replication complexes (; ) and its overlap with ER was reduced as seen indirectly by its colocalization with JEV-envelope protein (Pearson coefficient ~0.46) (Fig. S10B). JEV-envelope protein predominantly localizes to the ER in infected cellsCitation55,Citation56 (Fig. S10C). This suggests enrichment or mobilization of EDEM1 in replication complexes compared with mock-infected cells. However total levels of EDEM1 did not appear to vary significantly between mock and JEV-infected Neuro2a cells (Fig. S10D).
Role of ERAD effectors EDEM1, SEL1L, and LC3-I in JEV replication
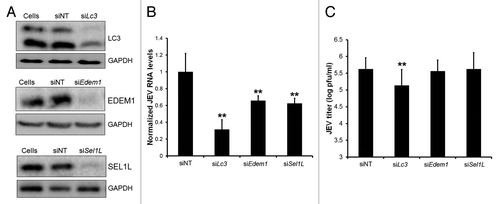
The observation that JEV replication complexes are decorated with anti-LC3 antibodies in cells lacking Atg5 and LC3-II is indicative of an association between LC3-I and JEV replication complexes. To study the relevance of LC3 in JEV replication we specifically depleted LC3A and LC3B from Neuro2a cells by RNA interference. LC3 downregulation was confirmed by western blotting with LC3B antibody (; Fig. S10E) and by qRT-PCR with LC3A- and LC3B-specific primers (data not shown). Reduction of LC3 levels resulted in nearly 70% decrease in JEV RNA accumulation demonstrating an important role of LC3 in viral life cycle (). This decrease also manifested as reduced levels of JEV proteins (Fig. S10E) and titers (). Nonlipidated LC3 thus plays an important role in viral replication upstream of its function in autophagy. Depletion of EDEM1 and SEL1IL () decreased JEV-RNA levels by around 35% (). This decrease, however, was insufficient to manifest as reduction in final JEV titers (). Collectively our data suggests that JEV subverts the ERAD tuning components to establish its replication platforms.
Figure 8. LC3 is an essential host factor for JEV replication. (A) Neuro2a cells transfected with control nontargeting (NT) or Lc3 (Lc3a and Lc3b), Sel1l and Edem1 siRNA were lysed 48 h post-transfection and probed with corresponding antibodies and loading control, GAPDH. (B) Viral RNA levels as determined by qRT-PCR in siRNA treated Neuro2a cells infected with JEV (MOI 5, 24 h). (C) JEV titers in culture supernatants of cells treated as described in (B). The Student t test was used to calculate P values. *P < 0.05, **P < 0.01.
Discussion
In the present work we have studied the relevance of the cellular autophagy pathway and the role of key autophagy protein LC3 in the context of JEV infection. We demonstrate that autophagy antagonizes JEV infection and functions to limit viral replication and reduce viral yields. Down modulation of autophagy-related proteins leads to enhanced viral RNA accumulation, elevated virus titers, and increased cell death. We further demonstrate that viral replication complexes colocalize with markers of ERAD tuning EDEM1 and LC3-I, and downregulation of LC3 decreased virus titers. JEV replication occurs on membranes of ERAD tuning referred to as EDEMosomes, as demonstrated for mouse hepatitis coronavirus and equine arteritis virus, both positive strand RNA viruses.Citation27,Citation28,Citation54
The cellular autophagic process has been examined in the context of several viruses. Viruses can induce the accumulation of autophagosomes in infected cells either by de novo formation or by inhibition of their maturation. Induction of autophagic vesicle formation in infected cells can be driven by recognition of viral RNA by innate immune sensors, virus binding to receptors, expression of viral proteins that usually leads to the induction of UPR due to ER stress and/or production of ROS.Citation4 JEV infection also triggers ER stress and UPR,Citation57 and since the autophagic response in infected cells parallels the production of viral proteins, it is likely that ER stress partly contributes to JEV induced autophagy.
Two earlier studies have shown that JEV induces autophagyCitation33,Citation34 but have primarily implicated autophagy as a positive regulator of virus replication. One group has shown reduced JEV replication in NT-2 (pluripotent human testicular embryonal carcinoma) cells with downregulated ATG5 or BECN1/Beclin 1 expression.Citation34 Another study has shown that JEV infection in autophagy knockdown cells reduces JEV replication and leads to upregulation of IFN signaling pathway and greater cell death.Citation33 However, our study with ATG7-depleted Neuro2a cells and Atg5-deficient MEFs clearly indicates that autophagy negatively regulates JEV replication and reduces viral yields. Autophagy-deficient cells have numerous well-defined replication complexes marked by viral NS proteins and dsRNA.
Viral yields are largely dependent on the efficiency of replication within the host cell and the activity of antiviral response mediated by type I IFNs. In their study Li et al., demonstrate that autophagy-deficient A549 cells show enhanced mRNA levels of IFNB, IL6, and IP10 in response to JEV infection.Citation34 Since A549 is a cell line known to produce robust type I interferon response after virus infection, it is possible that interferon levels in these cells are much higher and thus effective at controlling JEV replication. On the other hand neuronal cells including Neuro2a (unpublished data from our lab) are highly restricted in their interferon response.Citation58,Citation59 Recent studies have shown that some cells like neurons have reduced type I IFN-dependent innate responses and are heavily reliant on an alternative prosurvival pathway like autophagy for antiviral defense.Citation58 Further studies need to be done to determine the relative contribution of interferon vs. autophagy in neuronal cells and fibroblasts in response to JEV infection.
A second possibility is that the kinetics and efficiency of virus replication between cell lines varies significantly. Since JEV replication complexes are derived from components of ERAD tuning, the extent of UPR and ERAD tuning activity is likely to determine the availability of host factors like LC3-I that are crucial for virus replication.
Analysis of autophagic flux in cells by use of bafilomycin A1 and pepstatin A demonstrates that autophagy is functional early during infection, but as infection progresses autophagosome maturation is inhibited and level of SQSTM1 increases significantly by 48 h pi. Dysfunctional autophagy leads to accumulation of misfolded proteins and is a prime cause of neurodegeneration.Citation60-Citation62 Since, JEV is primarily a neurotropic virus it is likely that dysfunctional autophagy and accumulation of disordered proteins contribute to the neurological symptoms of JEV infection.
The antiviral potential of autophagy is well documented and has been extensively reviewed.Citation2-Citation4,Citation7,Citation60,Citation63,Citation64 Autophagy can operate as a cell-intrinsic mechanism that inhibits viral replication and/or eliminate viruses from infected cells. Autophagy plays a crucial role in restricting viral infections of the CNS as has been shown for the Sindbis virus and α-herpesvirus, HSV-1.Citation60,Citation65,Citation66 Recent studies have shown that antiviral autophagy is conserved in flies and mammals during infection with Rift Valley fever virus and viral replication was enhanced in the absence of autophagy genes.Citation58 Dorsal root ganglionic neurons also require autophagy to restrict HSV-1 replication both in vivo and in vitro, and produce little type I IFNs in response to virus infection.Citation59 Our data showing that autophagy inhibits viral replication and reduces virus titers in neuronal cells strongly suggests that autophagy could be an important means of controlling JEV replication in the CNS.
Autophagy can operate as a cellular survival mechanism in infection. Studies in influenza and chikungunya have shown that the ability of virus to induce apoptosis is increased in autophagy-protein deficient cells, suggesting that autophagy favors the survival of infected cells.Citation5,Citation19 A similar observation of enhanced cell death has also been reported for JEV-infected, autophagy-deficient cells.Citation33 Indeed we also observe that blocking autophagy by depletion of ATG7 increases JEV-induced cell death, and enhanced viral yields. It is also possible that increased cell death is observed because a greater viral load would result in a stronger apoptotic stimulus in autophagy deficient cells. Further studies will reveal the exact mechanism by which autophagy restricts JEV infection.
Our study shows an important role of LC3 in JEV replication independent of its known function in autophagy. We observe that JEV replication complexes extensively overlap with endogenous LC3. The lack of colocalization with LysoTracker Red, LAMP1, and GFP-LC3 indicates that these structures are not acidic, late endosomal or autophagosomal in nature. Furthermore the localization of replication complexes and LC3-I in atg5−/− MEFs indicates that LC3 association with viral replication machinery is independent of its lipidated form and autophagic function. Indeed, LC3 also plays an important role in viral replication as its depletion reduces virus titers. Autophagy-deficient Neuro2a cells and MEFs show elevated levels of LC3-I. Since virus replication complexes are localized to LC3-I structures it is likely that increased LC3-I levels in these cells confer a replication advantage to the virus.
The ERAD is a complex series of events that clears folding-defective polypeptides from the ER membranes for degradation.Citation67,Citation68 To maintain cellular homeostasis, the activity of the ERAD pathway is maintained by a process known as ERAD tuning which involves selective sorting of EDEM1 and other short-lived ER chaperones in 200 to 800 nm vesicles called EDEMosomes.Citation23,Citation68,Citation69 The noncovalent association of LC3-I at the cytosolic face of the membrane distinguishes EDEMosomes from autophagosomes.Citation23 The type-I transmembrane protein SEL1L and LC3-I in combination form a receptor complex which selectively captures EDEM1 and other ER chaperones in the ER lumen for their constitutive clearance from the ER at steady-state.Citation29 Recent studies have demonstrated that the mouse coronavirus and equine arteritis virus can co-opt the ERAD tuning pathway for their replication.Citation27,Citation28,Citation54 These viruses anchor their replication complexes to the membranes of EDEMosomes that are marked by the proteins EDEM1, OS9, SEL1L, and LC3-I.Citation27-Citation29,Citation54 Furthermore, there is defective turnover and accumulation of 2 ERAD factors EDEM1 and OS9 in mouse coronavirus-infected cells.Citation28,Citation54
JEV infection leads to ER stress and the induction of UPR.Citation57,Citation70 As a part of UPR, significant transcriptional upregulation of several genes implicated in ER stress (Bhattacharyya et al., manuscript in preparation), and ERAD is observed (Fig. S11). Our study shows that similar to mouse coronavirus and equine arteritis virus, JEV-infected cells also show extensive overlap between viral replication membranes and EDEMosome like structures that are marked by EDEM1 and LC3-I but not ectopically expressed GFP-LC3. ERAD tuning components LC3-I, EDEM1, and SEL1L play a crucial role in JEV life cycle.
Our study displays the existence of a complex interplay between virus and host-cell. A fine balance exists between autophagy modulation and its effect on outcomes of viral infection. A functional autophagic pathway attempts to control virus spread and exerts prosurvival effects. Interestingly, LC3 a crucial autophagy protein also plays an upstream autophagy independent role in viral replication. Our work gives insight into how viruses target cellular pathways and exploit host proteins for their replication. The role of JEV nonstructural proteins in subverting the ERAD machinery and whether these molecules and pathways are also exploited for replication by other flaviviruses remain interesting avenues to be explored in future studies.
Materials and Methods
Cells and virus
Mouse neuroblastoma (Neuro2a) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Life Technologies, 11960) supplemented with 10% fetal bovine serum (FBS; HyClone, SH 30070). Porcine stable kidney (PS) cells and African green monkey kidney (Vero) cells (National Centre for Cell Science, Pune, India) were grown in Eagle’s minimal essential medium (MEM; Invitrogen, Life Technologies, 11090) with 10% FBS. Wild-type and Atg5-deficient (atg5−/−) MEFs were a kind gift from Prof Noboru Mizushima and obtained through RIKEN Bio-Resource Cell Bank (RCB2710 and RCB2711).Citation35 These cells were grown in DMEM with 10% FBS. All media were additionally supplemented with 100 µg/ml penicillin/streptomycin and 2 mM L-glutamine. For all studies JEV isolate Vellore P20778 generated in PS cells was used.
Reagents, antibodies, and plasmids
Antibodies against JEV Envelope (ab41671), JEV NS1 (ab41651), LAMP1 (ab24170), EDEM1 (ab67105), SQSTM1 (ab56416), CALR/calreticulin (ab4), and actin (ab1801) were from Abcam. Two different LC3 antibodies were used for western blots (Cell Signaling Technology, 3868) and immunofluorescence (Abcam, ab51520). GAPDH antibody (2118) was purchased from Cell Signaling Technology. The c-Myc antibody (sc-70463) was obtained from Santa Cruz Biotechnology, Inc. The dsRNA antibody (J2) was purchased from English and Scientific Consulting Bt. Rabbit polyclonals against JEV NS1 and NS3 were generated in the laboratory. Rabbit polyconal against JEV NS5 was a kind gift from Dr Ruey-Yi Chang (National Dong Hwa Univeristy, Taiwan). The Sel1L antibody (S3699), bafilomycin A1 (B1793), pepstatin A (P5318) and MTT assay kit (TOX-1) were purchased from Sigma. Torin1 was purchased from Tocris Bioscience (4247). Fluorescent secondary anti-mouse, anti-rabbit and anti-rat antibodies (A-11004, A-11006, A-11008, A-11011, A-21202, A-21235 and A-31573), LysoTracker Red (L-7528) and ProLong Gold anti-fade reagent with DAPI (P36935) were obtained from Invitrogen, Life Technologies. HRP-coupled secondary antibodies were obtained from Jackson Immunochemicals (711-035-152 and 715-035-150). The following plasmids were obtained from Addgene: Plasmid EGFP-LC3 (11456 deposited by Karla Kirkegaard)Citation6 and BIP-Myc (27164 deposited by Ron Prywes).Citation71 Plasmid HA-NS1 was generated by cloning JEV-NS1 into pSELECT-CHA-zeo (InvivoGen, psetz-cha).
Characterization of JEV antibodies
Polyclonal antisera against JEV NS1, NS3, and NS5 were characterized by western blot analysis of lysates of JEV-infected Neuro2a cells. All antisera reacted with proteins of the expected sizes in JEV-infected but not in mock-infected cells. The staining specificity of rabbit polyclonal antisera and commercially available antibodies was also checked by immunofluorescence in mock and JEV-infected cells. Optimum working dilutions and lack of cross-reactivity with cellular proteins was established for dsRNA and all JEV antibodies. Intracellular distribution of JEV NS1 was analyzed by immunofluorescence using either the rabbit polyclonal or commercial mouse monoclonal (Abcam, ab41651) antibodies.
Virus titration
JEV was titrated by plaque formation on PS monolayers.Citation72 Briefly, PS cells (2 ×105 cells/well) were seeded in 6-well plates to give semi-confluent monolayers in 18 to 24 h. Monolayers were inoculated with 10-fold serial dilutions of virus/experimental culture soup in MEM containing 2% FBS and incubated for 1 h at 37 °C with gentle rocking. The inoculum was removed and 3 washes were given with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.2). The monolayers were overlaid with MEM containing 5% FBS, 1% low-melting point agarose (Agarose Type VII, Sigma, A4018) and appropriate supplements. Plates were incubated at 37 °C for 4 to 5 d until plaques became visible. Cell monolayer was stained with crystal violet after fixing the cells with 10% formaldehyde. Plaque assay results are presented as mean ± standard deviation (SD) of 3 independent experiments.
Cell treatment and virus infection
Cells were serum-starved, treated with inhibitors, mock- or JEV-infected and processed for western blotting, immunofluorescence, or RNA extraction. For serum-starvation, 60% to 70% confluent cells were given 3 washes in PBS followed by incubation in serum-free media. To determine endocytosed viral load in siRNA-transfected cells (Fig. S2), JEV (5 MOI) was adsorbed on cells at 4 °C for 1 h, followed by one wash with cold PBS and a shift to 37 °C for 1 h. After incubation, cells were washed with chilled PBS and low-pH buffer and lysed in TRIzol reagent (Invitrogen, Life Technologies, 15596026). For infection experiments, JEV was added to cells at the indicated MOI for 1 h at 37 °C. For experiments involving western blotting and immunofluorescence, cells were infected at MOI 5 (24 h) which results in an infection efficiency of ~90%. This ensured that western blots are representative of infection in a majority of the cell population and not a fraction of cells. Similarly for immunofluorescence experiments this ensured uniform or similar expression level of JEV proteins/ dsRNA in all cells. qRT-PCR of JEV RNA, end point plaque assays and cell death assays were done across MOIs 0.1 to 5 and the results showed similar trends. Following infection, cells were washed twice with PBS and complete medium was added. The inhibitors bafilomycin A1, pepstatin A, and Torin1 were dissolved in DMSO. To measure autophagic flux bafilomycin A1, (100 nM) was added to the culture medium for 2 h before fixation. The protease inhibitor pepstatin A (50 µg/ml) was added to cells for 12 h before fixation. For MTOR inhibition, Torin1 (330 nM) was added to cells 2 h after infection and maintained until fixation. After infection or treatment, cells were washed twice with PBS and processed. In all experiments culture supernatants was collected for plaque assays at the indicated time pi. Transfections were performed using Lipofectamine 2000 (Invitrogen, Life Technologies, 11668027) according to the manufacturer’s protocol. Briefly, cells were grown to 60% to 70% confluency on 18-mm coverslips or 35-mm coverslip dishes and transfected with 1 µg DNA. Transfections were typically allowed to proceed for 18 to 24 h before infection and analysis. Transfection efficiency of GFP-LC3 ranged from 30% to 40%.
Western blots
Serum-starved, treated, mock- or JEV-infected cells were washed in PBS and lysed in buffer containing 1% Triton X-100 in 50 mM TRIS-HCl, pH 7.5, 150 mM NaCl and protease inhibitor cocktail (Sigma, P8340). Equal amounts of protein extracts were separated on polyacrylamide gels and transferred to PVDF membranes for immunoblotting. Band intensities were quantified with ImageJ software.
Immunostaining, fluorescence microscopy, and image processing
To mark acidic late endosomal and lysosomal compartments LysoTracker Red (20 µM), was added to cells for 2 h before fixation. Following treatment or infection cells were fixed in 2% paraformaldehyde and permeabilized using 0.1% Triton X-100 or 0.4% Tween-20 for 20 min at room temperature. Blocking was done with 2 mg/ml bovine serum albumin (BSA; Sigma, A7906) in PBS for 1 h prior to incubation with primary antibodies followed by incubation with Alexa 488/568/647 coupled appropriate secondary antibody. Cells were mounted using ProLong Gold anti-fade reagent with DAPI. Images were acquired on an Olympus FV1000 confocal microscope with 20× (NA 1.2) or 60× (NA 1.4) Plan Apo objectives (Olympus, Singapore). Quantification of JEV-infected cells was done as described earlier.Citation73 For colocalization experiments, Z-stacks were acquired at 0.25 µm per slice by sequential scanning with a 60× objective lens. FluoView software was used to generate cross-sectional and maximum intensity projection images. Cellular outlines were traced on images and the Pearson coefficient of colocalization between 2 fluorophores was calculated using the Fluo View software. Colocalization values are represented as mean and standard error of mean from 2 to 5 independent experiments from at least 10 to 15 cells in each experiment.
Quantitative real-time (qRT) PCR
Cells were given the appropriate treatment or infection and RNA was extracted following lysis in Trizol reagent. The cDNA was prepared using random hexamers. The qRT-PCR of autophagy and ERAD pathway genes was performed with SYBR green reagents and normalized to the endogenous house keeping control GAPDH. Primers for all the genes were designed based on sequences available from the Harvard qPCR primer bank. To determine JEV RNA copies in infected cells, qPCR was done using Taqman probes and Gapdh was used as an internal control. JEV was amplified using the following probes—Taqman probe: CCACGCCACT CGACCCATAG ACTG (5′ end FAM, 3′end TAMRA), 5′ primer: AGAGCACCAA GGGAATGAAA TAGT, 3′ primer: AATAAGTTGT AGTTGGGCAC TCTG. Gapdh was used as an internal control-Taqman probe sequence: ACAACCTGGT CCTCAGTGTA GC (5′ end FAM, 3′end TAMRA), 5′ primer: CCTGCCAAGT ATGATGAC, 3′ primer: GGAGTTGCTG TTGAAGTC. The PCR conditions were as follows: 94 °C for 2 min (1 cycle), 94 °C for 15 s, 55 °C for 30 s and 72 °C for 1 min (40 cycles). The qPCR was done on Applied Biosystems ABI 7500 instrument (Applied Biosystems, Life Technologies, Singapore). The fold change in the expression level of genes and JEV RNA levels is represented as mean ± SD of 3 independent experiments.
siRNA depletion experiments
Mouse specific Atg7-, Edem1-, Sel1l-, Lc3a-, and Lc3b-targeting siRNA was purchased from Dharmacon (ON-TARGET plus SMART pool). Cells were transfected with siRNA’s according to manufacturer recommendations and harvested at 24 and 48 h to check for protein knockdown. Cell lysates were run on SDS-PAGE and western blotting was done for ATG7, EDEM1, SEL1L, LC3, and GAPDH (loading control). For infection experiments, cells were transfected with siRNAs for 48 h following which they were infected with JEV.
Cell fractionation by Optiprep density gradients
JEV-infected (MOI 5) Neuro2a cells were washed with ice-cold PBS, resuspended in subcellular fractionation buffer (250 mM Sucrose, 20 mM HEPES, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, protease inhibitor cocktail) and scraped with a rubber policeman. Cells were broken by Dounce homogenization to release intracellular compartments. The nuclear fraction was removed by centrifugation at 500 g for 10 min. The resulting postnuclear supernatant fraction was mixed with OptiPrep (Sigma, D1556) to make a final concentration of 15% OptiPrep, and was loaded on top of a discontinuous OptiPrep density gradient (17%, 20%, 23%, 27%, and 30%). Separation was done by ultracentrifugation in a SW55Ti rotor (45,000 rpm/100,000 g for 2 h, at 4 °C) on a Beckman Coulter OptimaL-100K ultracentrifuge (Beckman Coulter Inc., St Indianapolis, IN, USA). Fractions were collected from the top and separated on SDS-PAGE gels for further analysis.
Cell death assays
Apoptosis in JEV-infected cells was assayed using the ANXA5/Annexin V-FITC Apoptosis kit (KA0714) from Abnova. Briefly, cells were stained with ANXA5-FITC and propidium iodide and were analyzed by flow cytometry using a Becton Dickinson (BD) FACSCantoII flow cytometer. Percentage of cell death is represented as mean and standard error of mean from 3 independent experiments. The MTT assay was performed using the In Vitro Toxicology Assay Kit (Sigma, TOX-1) as per the manufacturer’s instructions.
Statistical analysis
Statistical evaluations were performed using the Student t test. Differences were considered significant at values of P < 0.05.
| Abbreviations: | ||
| ATG | = | autophagy-related |
| CNS | = | central nervous system |
| dsRNA | = | double-stranded RNA |
| EDEM1 | = | ER-degradation enhancer, mannosidase alpha-like 1 |
| ER | = | endoplasmic reticulum |
| ERAD | = | ER-associated degradation |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| JEV | = | Japanese encephalitis virus |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| MAP1LC3 (LC3) | = | microtubule-associated protein 1 light chain 3 |
| MEF | = | mouse embryonic fibroblast |
| MOI | = | multiplicity of infection |
| MTOR | = | mechanistic target of rapamycin |
| NS1 | = | nonstructural protein 1 |
| pi | = | post-infection |
| ROS | = | reactive oxygen species |
| SQSTM1 | = | sequestosome 1 |
| UPR | = | unfolded protein response |
Additional material
Download Zip (930.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Department of Biotechnology (DBT), Government of India grant—BT/MB/01/VIDRC/08. MS is supported by UGC-JRF fellowship, MN is supported by CSIR-JRF fellowship and RK is supported by ICMR-SRF fellowship. We thank Arup Banerjee, Shahid Jameel, and Rohan Dhiman for critical inputs on the manuscript, Deepak Sharma for technical assistance, and all Virology lab members for constant support and encouragement.
Related Research Data
References
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009; 5:527 - 49; http://dx.doi.org/10.1016/j.chom.2009.05.016; PMID: 19527881
- Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol 2004; 2:301 - 14; http://dx.doi.org/10.1038/nrmicro865; PMID: 15031729
- Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle 2010; 9:1295 - 307; http://dx.doi.org/10.4161/cc.9.7.11109; PMID: 20305376
- Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367 - 80; http://dx.doi.org/10.1016/j.chom.2009.09.005; PMID: 19837376
- Jackson WT, Giddings TH Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005; 3:e156; http://dx.doi.org/10.1371/journal.pbio.0030156; PMID: 15884975
- Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol 2009; 19:359 - 78; http://dx.doi.org/10.1002/rmv.630; PMID: 19750559
- Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 2006; 116:2161 - 72; http://dx.doi.org/10.1172/JCI26185; PMID: 16886061
- Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, Azocar O, Vidalain PO, Vidal M, Lotteau V, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009; 6:354 - 66; http://dx.doi.org/10.1016/j.chom.2009.09.006; PMID: 19837375
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009; 30:588 - 98; http://dx.doi.org/10.1016/j.immuni.2009.02.009; PMID: 19362021
- Carpenter JE, Jackson W, Benetti L, Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J Virol 2011; 85:9414 - 24; http://dx.doi.org/10.1128/JVI.00281-11; PMID: 21752906
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74:739 - 89; http://dx.doi.org/10.1146/annurev.biochem.73.011303.074134; PMID: 15952902
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J 2008; 27:1110 - 21; http://dx.doi.org/10.1038/emboj.2008.31; PMID: 18337753
- Tallóczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 2002; 99:190 - 5; http://dx.doi.org/10.1073/pnas.012485299; PMID: 11756670
- Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255 - 68; http://dx.doi.org/10.1083/jcb.200903070; PMID: 19635843
- Taguwa S, Kambara H, Fujita N, Noda T, Yoshimori T, Koike K, Moriishi K, Matsuura Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol 2011; 85:13185 - 94; http://dx.doi.org/10.1128/JVI.06099-11; PMID: 21994453
- Schmid D, Dengjel J, Schoor O, Stevanovic S, Münz C. Autophagy in innate and adaptive immunity against intracellular pathogens. J Mol Med (Berl) 2006; 84:194 - 202; http://dx.doi.org/10.1007/s00109-005-0014-4; PMID: 16501849
- Orvedahl A, Levine B. Viral evasion of autophagy. Autophagy 2008; 4:280 - 5; PMID: 18059171
- Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 2012; 209:1029 - 47; http://dx.doi.org/10.1084/jem.20110996; PMID: 22508836
- Lang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J 1998; 17:3597 - 607; http://dx.doi.org/10.1093/emboj/17.13.3597; PMID: 9649430
- Tamura N, Oku M, Sakai Y. Atg8 regulates vacuolar membrane dynamics in a lipidation-independent manner in Pichia pastoris. J Cell Sci 2010; 123:4107 - 16; http://dx.doi.org/10.1242/jcs.070045; PMID: 21045113
- Al-Younes HM, Al-Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, Brinkmann V, Braun PR, Meyer TF. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy 2011; 7:814 - 28; http://dx.doi.org/10.4161/auto.7.8.15597; PMID: 21464618
- Calì T, Galli C, Olivari S, Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 2008; 371:405 - 10; http://dx.doi.org/10.1016/j.bbrc.2008.04.098; PMID: 18452703
- Le Fourn V, Gaplovska-Kysela K, Guhl B, Santimaria R, Zuber C, Roth J. Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell Mol Life Sci 2009; 66:1434 - 45; http://dx.doi.org/10.1007/s00018-009-9038-1; PMID: 19266160
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003; 299:1397 - 400; http://dx.doi.org/10.1126/science.1079474; PMID: 12610306
- Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 2003; 299:1394 - 7; http://dx.doi.org/10.1126/science.1079181; PMID: 12610305
- Monastyrska I, Ulasli M, Rottier PJ, Guan JL, Reggiori F, de Haan CA. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy 2013; 9:164 - 74; http://dx.doi.org/10.4161/auto.22743; PMID: 23182945
- Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010; 7:500 - 8; http://dx.doi.org/10.1016/j.chom.2010.05.013; PMID: 20542253
- Bernasconi R, Galli C, Noack J, Bianchi S, de Haan CA, Reggiori F, Molinari M. Role of the SEL1L:LC3-I complex as an ERAD tuning receptor in the mammalian ER. Mol Cell 2012; 46:809 - 19; http://dx.doi.org/10.1016/j.molcel.2012.04.017; PMID: 22633958
- Sips GJ, Wilschut J, Smit JM. Neuroinvasive flavivirus infections. Rev Med Virol 2012; 22:69 - 87; http://dx.doi.org/10.1002/rmv.712; PMID: 22086854
- Durán RV, Hall MN. Regulation of TOR by small GTPases. EMBO Rep 2012; 13:121 - 8; http://dx.doi.org/10.1038/embor.2011.257; PMID: 22240970
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21 - 35; http://dx.doi.org/10.1038/nrm3025; PMID: 21157483
- Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One 2013; 8:e52909; http://dx.doi.org/10.1371/journal.pone.0052909; PMID: 23320079
- Li JK, Liang JJ, Liao CL, Lin YL. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 2012; 14:159 - 68; http://dx.doi.org/10.1016/j.micinf.2011.09.001; PMID: 21946213
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell 1999; 10:1367 - 79; http://dx.doi.org/10.1091/mbc.10.5.1367; PMID: 10233150
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 2009; 7:e38; http://dx.doi.org/10.1371/journal.pbio.1000038; PMID: 19209957
- García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J 2009; 421:29 - 42; http://dx.doi.org/10.1042/BJ20090489; PMID: 19402821
- Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007; 3:154 - 7; http://dx.doi.org/10.4161/auto.3634; PMID: 17204842
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Mackenzie JM, Kenney MT, Westaway EG. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J Gen Virol 2007; 88:1163 - 8; http://dx.doi.org/10.1099/vir.0.82552-0; PMID: 17374759
- Muller DA, Young PR. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res 2013; 98:192 - 208; http://dx.doi.org/10.1016/j.antiviral.2013.03.008; PMID: 23523765
- Salonen A, Ahola T, Kääriäinen L. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 2005; 285:139 - 73; http://dx.doi.org/10.1007/3-540-26764-6_5; PMID: 15609503
- Uchil PD, Satchidanandam V. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J Biol Chem 2003; 278:24388 - 98; http://dx.doi.org/10.1074/jbc.M301717200; PMID: 12700232
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009; 5:365 - 75; http://dx.doi.org/10.1016/j.chom.2009.03.007; PMID: 19380115
- Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol 1997; 71:6650 - 61; PMID: 9261387
- Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 2012; 86:7360 - 71; http://dx.doi.org/10.1128/JVI.00157-12; PMID: 22553322
- Gillespie LK, Hoenen A, Morgan G, Mackenzie JM. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J Virol 2010; 84:10438 - 47; http://dx.doi.org/10.1128/JVI.00986-10; PMID: 20686019
- Miorin L, Romero-Brey I, Maiuri P, Hoppe S, Krijnse-Locker J, Bartenschlager R, Marcello A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J Virol 2013; 87:6469 - 81; http://dx.doi.org/10.1128/JVI.03456-12; PMID: 23552408
- Westaway EG, Mackenzie JM, Khromykh AA. Replication and gene function in Kunjin virus. Curr Top Microbiol Immunol 2002; 267:323 - 51; http://dx.doi.org/10.1007/978-3-642-59403-8_16; PMID: 12082996
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Reggiori F, de Haan CA, Molinari M. Unconventional use of LC3 by coronaviruses through the alleged subversion of the ERAD tuning pathway. Viruses 2011; 3:1610 - 23; http://dx.doi.org/10.3390/v3091610; PMID: 21994798
- Lorenz IC, Kartenbeck J, Mezzacasa A, Allison SL, Heinz FX, Helenius A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J Virol 2003; 77:4370 - 82; http://dx.doi.org/10.1128/JVI.77.7.4370-4382.2003; PMID: 12634393
- Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol 2001; 75:10787 - 99; http://dx.doi.org/10.1128/JVI.75.22.10787-10799.2001; PMID: 11602720
- Bhattacharyya S, Sen U, Vrati S. Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J Gen Virol 2014; 95:71 - 9; http://dx.doi.org/10.1099/vir.0.057265-0; PMID: 24114795
- Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity 2014; 40:51 - 65; http://dx.doi.org/10.1016/j.immuni.2013.10.020; PMID: 24374193
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012; 12:334 - 45; http://dx.doi.org/10.1016/j.chom.2012.07.013; PMID: 22980330
- Orvedahl A, Levine B. Autophagy and viral neurovirulence. Cell Microbiol 2008; 10:1747 - 56; http://dx.doi.org/10.1111/j.1462-5822.2008.01175.x; PMID: 18503639
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Jordan TX, Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect 2012; 14:126 - 39; http://dx.doi.org/10.1016/j.micinf.2011.09.007; PMID: 22051604
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007; 1:23 - 35; http://dx.doi.org/10.1016/j.chom.2006.12.001; PMID: 18005679
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell 2012; 151:1163 - 7; http://dx.doi.org/10.1016/j.cell.2012.11.012; PMID: 23217703
- Merulla J, Fasana E, Soldà T, Molinari M. Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic 2013; 14:767 - 77; http://dx.doi.org/10.1111/tra.12068; PMID: 23521725
- Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol 2011; 23:176 - 83; http://dx.doi.org/10.1016/j.ceb.2010.10.002; PMID: 21075612
- Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 2006; 80:11868 - 80; http://dx.doi.org/10.1128/JVI.00879-06; PMID: 16987981
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 2002; 3:99 - 111; http://dx.doi.org/10.1016/S1534-5807(02)00203-4; PMID: 12110171
- Vrati S, Agarwal V, Malik P, Wani SA, Saini M. Molecular characterization of an Indian isolate of Japanese encephalitis virus that shows an extended lag phase during growth. J Gen Virol 1999; 80:1665 - 71; PMID: 10423134
- Kalia M, Khasa R, Sharma M, Nain M, Vrati S. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 2013; 87:148 - 62; http://dx.doi.org/10.1128/JVI.01399-12; PMID: 23055570