
Abstract
Chloroquine (CQ) is an antimalarial drug and late-stage inhibitor of autophagy currently FDA-approved for use in the treatment of rheumatoid arthritis and other autoimmune diseases. Based primarily on its ability to inhibit autophagy, CQ and its derivative, hydroxychloroquine, are currently being investigated as primary or adjuvant therapy in multiple clinical trials for cancer treatment. Oncogenic RAS has previously been shown to regulate autophagic flux, and cancers with high incidence of RAS mutations, such as pancreatic cancer, have been described in the literature as being particularly susceptible to CQ treatment, leading to the hypothesis that oncogenic RAS makes cancer cells dependent on autophagy. This autophagy “addiction” suggests that the mutation status of RAS in tumors could identify patients who would be more likely to benefit from CQ therapy. Here we show that RAS mutation status itself is unlikely to be beneficial in such a patient selection because oncogenic RAS does not always promote autophagy addiction. Moreover, oncogenic RAS can have opposite effects on both autophagic flux and CQ sensitivity in different cells. Finally, for any given cell type, the positive or negative effect of oncogenic RAS on autophagy does not necessarily predict whether RAS will promote or inhibit CQ-mediated toxicity. Thus, although our results confirm that different tumor cell lines display marked differences in how they respond to autophagy inhibition, these differences can occur irrespective of RAS mutation status and, in different contexts, can either promote or reduce chloroquine sensitivity of tumor cells.
Introduction
Autophagy is an evolutionarily conserved process in which cells recycle macromolecules and organelles by targeting them for lysosomal degradation, thereby allowing components to be used in metabolic and catabolic processes.Citation1 Autophagy is controlled by more than 30 autophagy-related (ATG) genes mostly discovered in yeast and conserved in eukaryotic cells.Citation2 Proteins encoded by these genes direct the nucleation of a small membrane structure called the phagophore, which is elongated by other ATG proteins form a double-membrane vacuole called the autophagosome that encloses organelles, proteins, and cytoplasm. Autophagosomes fuse with lysosomes to form autolysosomes, where the contents are degraded for recycling. Autophagy suppresses anoikis (detachment-induced death)Citation3 and promotes cell survival during nutrient starvation by providing amino acids, fatty acids, and ATP for cell function.Citation4,Citation5 It also promotes resistance to oxidative stress by eliminating oxidized, misfolded, or aggregated proteins, and damaged organelles.Citation1,Citation6 Additionally autophagy plays a role in antigen presentation,Citation7-Citation9 development,Citation10 and differentiation,Citation11 as we have recently shown in macrophages.Citation12
Chloroquine (CQ) is an FDA-approved antimalarial drug, with an established history of generally well-tolerated clinical use,Citation13 and has been used to treat rheumatoid arthritis and other autoimmune diseases due to its mild immunosuppressive properties.Citation13,Citation14 CQ is a weak base with hydrophobic characteristics that diffuses into the lysosomes of cells, where it becomes protonated and trapped, thus leading to a rise in lysosomal pH. These CQ-loaded lysosomes can no longer fuse with autophagosomes, thus blocking autophagy at a late stage.Citation15 Although CQ and its derivative, hydroxychloroquine (HCQ),Citation16 likely have other actions,Citation13 they are currently the only drugs specifically used to inhibit autophagy that are approved for use in human patients.
The effect of the RAS pathway on autophagy has been controversial. Expression of oncogenic RAS has been reported to both induce and repress autophagy. However, pancreatic and other cancers with RAS mutations have been described in the literature as being particularly susceptible to autophagy inhibition and CQ treatment.Citation17-Citation19 This has led to the hypothesis that RAS leads cancer cells to become “addicted” to autophagy,Citation17 and therefore blocking autophagy provides an opportunity for pharmacologic intervention in cancer treatment.Citation19,Citation20 Although autophagy is absolutely critical for the progression of RAS-driven tumors to high-grade cancers,Citation21,Citation22 it is not clear as to whether this requirement for autophagy is maintained once tumor progression has already occurred and the tumor has grown to an appreciable size, such as would be the case in patients receiving therapy. This is important because CQ and HCQ are currently being investigated as primary or adjuvant therapies in more than 30 clinical trials for the treatment of cancer,Citation5,Citation23 based on the premise that their anticancer properties are the result of their inhibition of autophagy.Citation5,Citation16,Citation24-Citation26 Many current trials have been proposed based on the success of inhibiting cancer cell growth in vitro and in mouse models, but they do not involve any selection to identify patients whose tumors are most likely to respond to autophagy inhibition by CQ. If the RAS-driven autophagy-addiction hypothesis is correct, the RAS status of human tumors could potentially provide one such patient selection criteria.
In this study, we sought to determine if an oncogenic RAS mutation necessarily confers autophagy addiction as measured by cancer cell sensitivity to CQ. We found that oncogenic mutation in RAS does not correlate with autophagy addiction or CQ sensitivity. Indeed in different tumor cells, oncogenic RAS may have opposite effects on autophagic flux and the effect of RAS mutation on increasing or decreasing autophagy in a given cell type does not determine whether that cell type will be more or less sensitive to CQ.
Results
RAS has little or no effect on CQ sensitivity in autophagy-deficient DU145 cells
Expression of oncogenic RAS has been proposed to lead to autophagy dependence in cancer cells. We sought to test whether this paradigm is broadly true, because if correct, it may potentially be clinically useful to predict therapeutic outcome. We therefore wanted to examine the affect of RAS in a wide variety of cell lines and contexts. Since CQ and its derivative, HCQ, are the only drugs currently used to deliberately inhibit autophagy in patients, we used cellular sensitivity to CQ as a measurement of autophagy dependence.
RAS not only affects autophagy, but can also influence other survival and death pathways. If RAS affects CQ sensitivity or resistance through its modulation of autophagy, we would expect that RAS would not influence CQ-dependent effects in cells that are genetically autophagy-deficient. We therefore chose to first examine the effect of RAS on CQ sensitivity in previously made DU145 prostate cancer cell lines where the stable expression of HRASG12V confers metastatic ability in previously nonmetastatic cells.Citation27 Importantly, DU145 cells have been reported to be deficient in autophagy due to their lack of full-length ATG5 mRNA transcripts.Citation28 Consistent with this report, we observed little or no LC3-II formation in these cells (Fig. S1A). CQ was not toxic in DU145 cells as measured by MTS and lactate dehydrogenase (LDH) assays, but did have an effect on the cell growth of DU145 as measured by clonogenic assays (Fig. S1B–S1D). However, the expression of oncogenic RAS neither potentiated CQ toxicity nor influenced the CQ-mediated effect on cell growth in these cells. This suggests that oncogenic RAS could not promote CQ toxicity in this autophagy-deficient tumor cell type and that expression of HRASG12V had no effect on the ability of CQ to inhibit cell growth in these cells. Since these particular RAS-transformed cells were apparently not dependent on autophagy, this result also suggested that further investigation into the notion that oncogenic RAS necessarily promotes CQ-mediated toxicity was warranted.
Oncogenic RAS does not correlate with autophagy addiction in lung cancer cells
Therapeutically, if screening for oncogenic RAS mutations were to have a predictive value on which patients would be successfully treated with CQ, it would likely be most successful in cancers that are heterogeneous for RAS mutations. Furthermore, in order for such patient selection criteria to be of use for CQ-mediated therapy, RAS mutation status should largely correlate with CQ-mediated growth suppression and toxicity in such cancers. Consequently, we next examined CQ sensitivity in cells derived from non-small cell lung cancer (NSCLC) tumors, where approximately one-third of tumors display oncogenic mutations in KRAS. Initially, 3 NSCLC cell lines with oncogenic KRAS mutations (H358, G12C; A549, G12S; H2009, G12A) were compared with 3 NSCLC cell lines with wild-type KRAS (H322C, HCC4006 and Calu3). After treatment of the cells for 48 h or 72 h over a large concentration range of CQ in the normal growth media that was typically used to passage these cells, we performed MTS viability assays to measure overall viability and growth effects (; Fig. S2A). Long-term clonogenic assays were used to measure the ability of the cells to grow back after this same treatment (), while LDH release was used to measure acute cytotoxicity (). Of the 6 cell lines tested, only Calu3 cells were susceptible to acute toxicity from CQ in the 30- to 50 μM range (). Though all of the cell types showed at least some growth inhibition in response to CQ exposure (), Calu3 cells also showed the greatest response to CQ in the clonogenic assays followed by the H322C, HCC4006, and H2009 lines, with the A549 and H358 being the least sensitive (), mirroring the data seen in the MTS assay. Surprisingly, cells with mutations in RAS were not more sensitive to autophagy inhibition with CQ, since the 2 most sensitive cell lines had wild-type RAS alleles, with 2 mutant cell lines being the least sensitive. RAS status (Fig. S2B) therefore showed no direct correlation with autophagy dependence in these assays. The amount of autophagic flux in the cell lines as measured by LC3-II accumulation in the presence of CQ did not obviously correlate with CQ toxicity (Fig. S2C). When the activity of RAS was measured in these cells using ELISA (data not shown), RAS activity also failed to correlate with increased CQ sensitivity, since the 2 cell lines with highest RAS activity, H2009 and H358, had an intermediate and resistant phenotype, respectively.
Figure 1. RAS status does not correlate sensitivity to autophagy inhibition in NSCLC lung cell lines. (A–C) H322C, HCC4006, and Calu3 (wt RAS, indicated by filled symbols) and H358, A549, and H2009 (oncogenic KRAS mutant, indicated by unfilled symbols) NSCLC cancer cell lines were treated with varying doses of CQ and assayed by (A) MTS viability assay (72 h), (B) clonogenic growth assay as measured by crystal violet staining several days after removing CQ (72 h treatment) and adding growth media or (C) LDH release cytotoxicity assay (48 h).
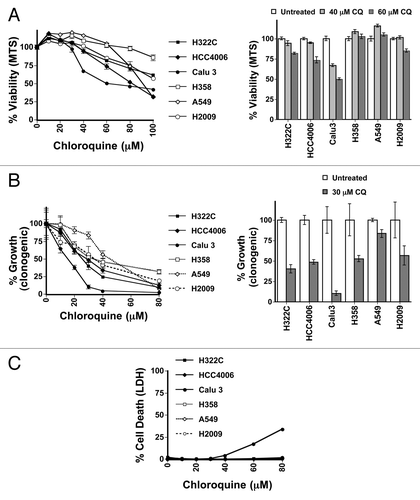
Because our experiments were done in growth media containing additional glutamine, as well as additional nonessential amino acids, which could influence the requirement for autophagy, we repeated our experiments in basal media without these additional factors. We observed slightly greater effects on CQ-mediated growth repression and cytotoxicity in most of the cell lines in the basal media. However, the cells with activating mutations in RAS were not more sensitive than the cells with wild-type RAS to autophagy inhibition with CQ in either viability or cytotoxic assays (Fig. S3A and S3B). To confirm that this result was due to autophagy inhibition, we also treated cells with bafilomycin A1 (BafA1), another late-stage inhibitor of autophagy that reduces lysosomal pH through a different mechanism (it inhibits proton pumping by the lysosomal vacuolar ATPase). While one RAS mutant cell line was among the most sensitive to BafA1 when measured with MTS or LDH assays, 2 of the cell lines having oncogenic RAS mutations were the least sensitive of the 6 cell lines (Fig. S3C and S3D), indicating that BafA1 sensitivity was not correlated with RAS status. Sensitivity to the autophagy inhibitor 3-MA was similar in that it was also not correlated with RAS status (data not shown).
To examine the effect of RAS in specific cell lines, we next expressed oncogenic RAS in NSCLC lines that have wild-type RAS, and conversely knocked down RAS in NSCLC lines that have endogenous oncogenic RAS mutations. Ectopic expression of oncogenic RAS did not sensitize Calu3 or HCC 4006 cells to CQ toxicity, but rather made these cells more resistant to CQ (Fig. S3E and S3F). Similarly, expressing KRASG12V in H322C cells made these cells somewhat more resistant to CQ and BafA1 (Fig. S4A). Knocking down KRAS in H2009 cells that were highly resistant to CQ or BafA1 had little effect on the toxicity of these agents in these cells (Fig. S4B). In H358 cells, KRAS knockdown made these cells slightly more sensitive to CQ and BafA1 (Fig. S4C), while A549 cells became less sensitive to CQ and BafA1 when KRAS was knocked down (Fig. S4D). These data confirm that RAS can indeed contribute CQ and BafA1 toxicity in some cell lines, however oncogenic RAS does not always confer sensitivity to agents that inhibit autophagy. Thus, oncogenic RAS is not sufficient on its own to confer autophagy addiction in NSCLC cell lines and in some cell lines may actually confer CQ resistance rather than sensitivity.
TP53 contribution to CQ-mediated toxicity and growth inhibition in mutant RAS cells varies depending on the tumor cell line
The previous data suggests that oncogenic RAS may not be sufficient on its own to confer autophagy addiction and CQ sensitivity in the lung cell lines. RAS may therefore mediate autophagy addiction in combination with other factors. In the case of KRAS-driven tumors, tumor progression to high-grade cancer requires autophagy.Citation21,Citation22 However, when TP53 (Trp53 in murine models) is deleted in such tumors, this requirement could change. A recent report suggests that when TP53 is deleted, the requirement for autophagy is somewhat reduced, but TP53-independent mechanisms may eventually cause growth arrest.Citation21 A second report suggests that in the absence of TP53, autophagy is no longer required, and the absence of autophagy may actually accelerate tumor growth and progression.Citation22 To test if elimination of TP53 affects CQ-mediated growth arrest and toxicity in oncogenic KRAS mutant cell lines, we examined CQ-mediated effects in HCT116 cells in which TP53 has been deleted by homologous recombination.Citation29 Treatment of TP53−/− cells with CQ caused a reduction in growth as well as cell death; however, both of these effects were reduced when compared with the effect of CQ on the wild-type HCT116 cell line (Fig. S5A–S5C). We did not observe substantive differences in the amount of autophagy between the wild-type or TP53−/− cell lines (Fig. S5D). The difference in reduction of viable cells upon CQ treatment was similar to the differences seen when treating the same cell lines with the DNA damage-inducing damage drug doxorubicin (Fig. S5E). Although we did not observe a perfect correlation between TP53 status and CQ/ BafA1-mediated effects in the RAS mutant NSCLC lines, the most sensitive of these lines, A549, has wild-type TP53.
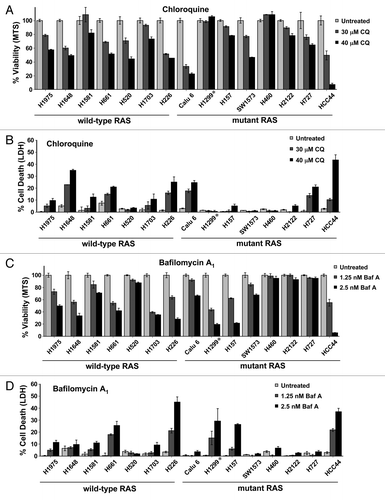
To further investigate whether TP53 coupled with RAS status could predict whether tumor cell lines would be sensitive to autophagy inhibition, we expanded our cell line analysis to 15 additional NSCLC lines, 8 of which possess oncogenic RAS mutations (one of these cell lines, H1299, has a mutation in N-RAS, the remainder have activating mutations in KRAS). Upon treatment of these cell lines with CQ, we observed that somewhat more of the wild-type RAS cell lines were sensitive to CQ compared with those possessing oncogenic RAS mutations (). This was true whether observing cell growth (MTS, ) or cytotoxicity (LDH, ). A similar amount of wild-type RAS cell lines were also sensitive to BafA1 compared with RAS mutant cells (), though at very high concentrations of BafA1 (> 5 nM) almost all cell lines had some sensitivity, with the exception of 2 mutant RAS cell lines (Fig. S6A and S6B). This confirms that oncogenic RAS status does not generally make cells particularly sensitive to these autophagy inhibitors. Additionally, we found no correlation between TP53 status and sensitivity to either CQ or BafA1 in the mutant RAS cell lines (Fig. S6C). In fact, the cell line with oncogenic RAS that was most sensitive to both inhibitors was the cell line, HCC44, which has a TP53 mutation(s).
Figure 2. Neither RAS status nor TP53 status correlates with sensitivity to autophagy inhibition in NSCLC lung cell lines. (A–D) Multiple NSCLC cancer cell lines with wild-type or oncogenic RAS status as designated were treated with the indicated doses of (A and B) CQ or (C and D) BafA1 and then assayed by (A and C) MTS viability assay (72 h) or (B and D) LDH release cytotoxicity assay (72 h). *H1299 has an oncogenic mutation in NRAS instead of KRAS.
Thus, these data suggest that while oncogenic RAS and wild-type TP53 are capable of contributing to autophagy dependence (and resultant CQ and BafA1 sensitivity) in some contexts, they are not sufficient to always do so. Additionally the sensitivity of a majority of NCSLC cell lines to autophagy inhibitors is not wholly dependent on these factors.
RAS protects against autophagy inhibition by CQ in HOSE cells
Our previous data in the NSCLC cell lines dealt primarily with oncogenic KRAS. Although HRAS activates similar pathways as KRAS, they are not equivalent and have different localization within the cell and traffic differently.Citation30 To examine the effect of HRAS activity on CQ sensitivity, we chose a system where HRAS is specifically known to have effects on autophagy. Therefore, we used a tetracycline-inducible HRASG12V human ovarian surface epithelial (HOSE) cell line. Recent data indicates that doxycycline-dependent induction of oncogenic RAS in these cells leads to increased autophagy, which over a several-week time course results in an autophagy-dependent death.Citation31 Consistent with what was previously reported, we observed that autophagic flux increased over the first 3 to 8 d after doxycycline induction of HRASG12V as measured by LC3-II formation in the presence of CQ or BafA1 (; Fig. S7A). Increased autophagy upon RAS expression was confirmed by quantitation of LC3 puncta (). Additionally, when these cells were infected with a tandem mCherry-GFP-LC3 construct, they had a greater red/green ratio 3 to 6 d after RAS induction, indicating a higher autophagic flux, while in control cells treated with CQ treatment overnight the red/green ratio decreased (). After about 2 to 3 wk, some RAS-expressing cells had distinct morphological changes, some grew more slowly, and some cells began to die (Fig. S7B–S7D, data not shown). Although ATG7-knockdown HOSE cells grew slower compared with the nonsilencing control cells, they were protected from long-term RASG12V-induced cell death (Fig. S7E), consistent with the effect observed by Elgendy et al.Citation31 However, late autophagy inhibition using CQ did not protect cells from cell death, as CQ caused death by itself in these cells, even in the absence of RAS induction (data not shown).
Figure 3. Acute RAS expression activates autophagy and promotes resistance to CQ in a human ovarian surface epithelial (HOSE) cell line. (A) HOSE cells with a tetracycline-inducible HRASG12V transgene (HOSE-HRASG12V) were left untreated or were treated with doxycycline (100 ng/mL) for 8 d. CQ (30 μM) was added for increasing lengths of time and then cells were lysed and immunoblotted for RAS, LC3 and ACTB to measure basal autophagy. (B) GFP-tagged LC3-expressing HOSE-HRASG12V cells that were treated or untreated with doxycyline for 6 d to induce RAS expression were analyzed for puncta formation using fluorescence microscopy. (C) HOSE-HRASG12V cells infected with tandem mCherry/GFP-LC3 retrovirus were left untreated or were treated with doxycycline (100 ng/mL) for 3 or 6 d and analyzed by flow cytometry. Some cells were treated with CQ (30 μM) overnight as a control for no flux. A rightward shift in the peak indicates increasing mCherry/GFP fluorescence ratio and thus an increase in autophagic flux. (C–F) HOSE-HRASG12V cells were left untreated or were treated with doxycycline (100 ng/mL) for 5 to 7 d. Cells were then replated and treated with doses of CQ with or without further doxycycline treatment (100 ng/mL) and cell viability and toxicity was measured by (C) MTS viability assay, (D) LDH release or (C) viability using propidium iodide exclusion as counted under a fluorescence microscope. **P < 0.01, *P < 0.05
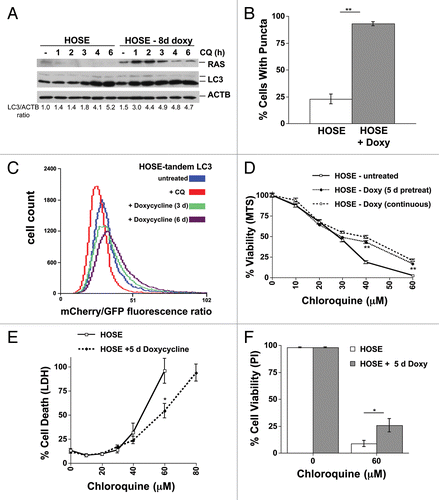
Since RASG12V-induced toxicity was only achieved by long-term RASG12V induction, we examined whether RASG12V affected CQ toxicity in short-term treated cells where RASG12V-induction had effects on autophagy, but where toxicity had not yet occurred. Correspondingly, we treated cells for 5 d with doxycycline, and then with CQ for 48 h. Short-term HRASG12V induction protected the HOSE cells from the higher concentrations of CQ that fully suppress autophagy (). Expression of oncogenic KRASG12V similarly protected (Fig. S7F). These data confirm that HRAS activity leads to increased autophagy in these cells, but does not itself confer autophagy dependence or CQ sensitization in this context, and oncogenic HRAS actually has a protective effect from autophagy inhibition by CQ.
RAS has opposite effects on autophagy and CQ sensitivity in genetically defined isogenic cells
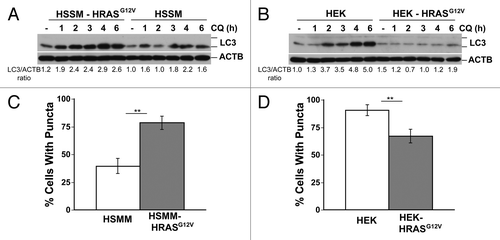
Cancer cells have many genetic aberrations and thus divergent results obtained in the previous experiments could have been influenced by secondary effects unrelated to RAS signaling itself. We therefore sought to examine the effect of RAS expression in cells that were immortalized in a defined manner and then transformed specifically by RAS expression. To do this we compared immortalized human skeletal muscle myoblasts (HSMM) and human embryonic kidney cells (HEK) with isogenic derivatives that differ only in the stable integration of HRASG12V.Citation32,Citation33 Additionally, both sets of cells inactivate TP53 by the same mechanism (expression of SV40 large T-antigen) ensuring that TP53 should not be a factor in any differences in sensitivity to autophagy inhibition. Surprisingly, oncogenic HRASG12V expression sensitized HSMM cells to CQ () as measured by MTS assay, but protected HEK cells from CQ sensitivity (). Clonogenic assays also reflected RASG12V-induced sensitization of HSMM and a RASG12V-mediated protective effect in HEK cells (). CQ caused direct cytotoxicity in these cell lines as shown by both LDH release () and propidium iodide uptake (). Similar results were seen in oncogenic KRASG12V-expressing cells (Fig. S8A and S8B). When basal autophagic flux was examined in HSMM and HEK cells, RASG12V induced autophagic flux in HSMM cells (), but repressed flux in HEK cells (), as measured by time dependent increases in LC3-II in the presence of autophagy inhibition using western blots. These differences in autophagic flux were confirmed by analyzing puncta formation (). HRASG12V also had similar effects on stimulated autophagy by trehalose or Earle’s balanced salt solution (EBSS) in these 2 cell lines in that RASG12V potentiated LC3-II formation in HSMM cells, but repressed LC3-II formation in HEK cells (Fig. S8C and S8D).
Figure 4. RAS has opposite effects on CQ sensitivity in genetically defined immortalized human skeletal muscle myoblasts (HSMM) and human embryonic kidney cells (HEK). (A–H) Immortalized HSMM (A, C, E, and G) or HEK cells (B, D, F, and H) with or without stable expression of HRASG12V were treated with CQ for 48 h and assayed by (A and B) MTS viability assay, (C and D) clonogenic growth assay as measured by crystal violet staining several days after removing the CQ and adding growth media, (E and F) LDH release cytotoxicity assay, or (G and H) propidium iodide exclusion as counted under a fluorescence microscope. **P < 0.01, *P < 0.05, #P < 0.08
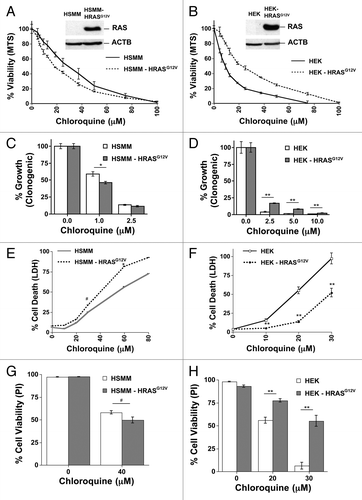
Figure 5. RAS has opposite effects on autophagy in genetically defined immortalized human skeletal muscle myoblasts (HSMM) and human embryonic kidney cells (HEK). (A and B) Immortalized HSMM (A) or HEK cells (B) with or without stable expression of HRASG12V were treated with CQ (30 μM) for the indicated time points and then cells were lysed and immunoblotted for LC3 and ACTB. (C and D) GFP tagged LC3-expressing HSMM (C) or HEK cells (D) cells with or without HRASG12V were analyzed for puncta formation using fluorescence microscopy. **P < 0.01
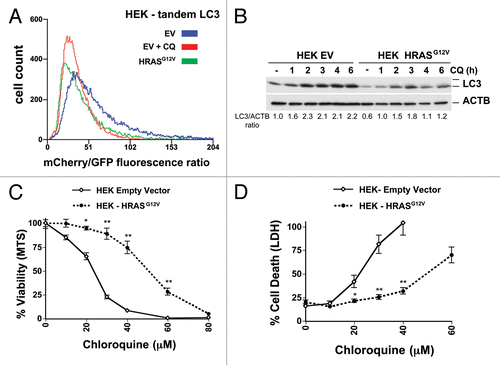
To confirm that the repressive effect of RAS on autophagy in the HEK cells was not an artifact of long-term selection of the cells, we created new HRASG12V-expressing HEK- cells by acutely expressing HRASG12V in immortalized HEK cells that already stably expressed Tandem mCherry-GFP LC3. Unlike in the HOSE cells () where HRASG12V induced autophagic flux, a decreased mCherry/GFP fluorescence ratio in the HRASG12V-expressing cells when compared with the empty vector control cells indicated that oncogenic HRAS reduced autophagic flux in the HEK cells (). This decrease in autophagic flux in the new HRAS HEK cells was further verified using western blotting of LC3 (). In these same cells, HRASG12V substantially decreased CQ toxicity as shown by MTS and LDH assays (). Thus, oncogenic HRAS, similar to oncogenic KRAS, can differentially potentiate or inhibit CQ-induced toxicity, depending on the cell type, and HRAS can inhibit or repress autophagy in different cell types.
Figure 6. Oncogenic RAS expression inhibits both autophagy and CQ sensitivity human embryonic kidney cells (HEK). (A) Immortalized HEK cells infected with tandem mCherry/GFP-LC3 retrovirus, stably selected for antibiotic resistance, and sorted for high fluorescence population. Cells were then infected with a HRASG12V retrovirus or control empty vector (EV), and after growth, analyzed by flow cytometry. Some cells were treated with CQ (30 μM) overnight as a control for no flux. A leftward shift in the peak indicates decreasing mCherry/GFP fluorescence ratio and thus a decrease in autophagic flux compared with the empty vector control cells. (B) Cells from (A) were treated with CQ (30 μM) for the indicated time points and then cells were lysed and immunoblotted for LC3 and (C and D) Cells from (A) were treated with CQ for 48 h and assayed by (C) MTS viability assay and (D) LDH release. **P < 0.01, *P < 0.05
One cannot predict whether RAS will potentiate or inhibit CQ sensitivity in a given cell type based solely on whether RAS inhibits or stimulates autophagy in that cell line
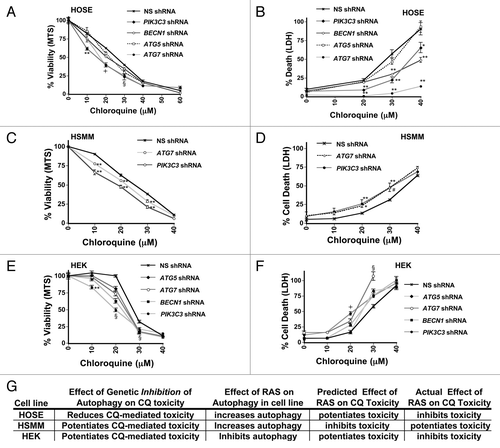
CQ inhibits autophagy, but it is a late stage inhibitor of the autophagic process (it blocks fusion of the autophagosome with the lysosome by altering the lysosomal pH) and is therefore fundamentally different from genetically inhibiting autophagy at the early stages by using ATG protein-targeted shRNAs that prevent the formation of the autophagosome. Thus, CQ toxicity could be the result of either inhibiting autophagy in cells that require it for survival, or by blocking the autophagic process at a late stage, leading to intermediates that are toxic to the cells. If CQ toxicity results from the first scenario, further reduction of autophagy by genetically reducing autophagosome formation should increase CQ toxicity. If CQ toxicity results from the latter scenario, then inhibiting autophagosome formation should inhibit CQ toxicity by reducing toxic intermediates. To distinguish between these possibilities, we infected cell lines displaying different RAS-mediated effects on CQ toxicity (HOSE, HSMM, and HEK cells) with lentiviruses expressing shRNAs that target mRNAs encoding PIK3C3/VPS34, BECN1, ATG5, or ATG7 (Fig. S9A–S9C). Interestingly, genetically inhibiting autophagosome formation slowed the growth of HOSE cells, but protected them from CQ-toxicity especially at higher concentrations (). In contrast, genetically inhibiting autophagosome formation sensitized the HSMM and HEK cell lines to CQ, especially at lower concentrations (). These data suggest that CQ toxicity in HOSE cells could be due to interrupted autophagy, while CQ toxicity in HSMM and HEK cells likely occurs because these cell lines need autophagy for survival. When considering the effect of genetic reduction in autophagy has on CQ-mediated toxicity in each of these cell lines in combination with the observed effect of RAS on autophagy in each cell line, the expected effect of RAS on CQ toxicity if it were solely acting on autophagy does not match the observed effect of RAS on CQ-mediated toxicity in these cell lines (summarized in ). Taken as a whole with the rest of our data, these data indicate that not only can oncogenic RAS have cell-type specific differential effects on autophagic flux and on CQ-sensitivity but also that the effect of RAS on autophagy is not correlated either positively or negatively with its effects on CQ sensitivity.
Figure 7. Genetic inhibition of autophagosome formation can potentiate or inhibit CQ- induced toxicity. HOSE (A and B), Immortalized HSMM (C and D) or HEK (E and F) cells were infected with shRNAs targeting PIK3C3, BECN1, ATG5, or ATG7, or a nonsilencing (NS) shRNA control, as indicated. These cells were treated with CQ for 48 h and assayed by MTS viability assay (left panels: A, C, and E) or LDH release cytotoxicity assay (right panels: B, D, and F). (G) Summary of the effects of RAS on autophagy and CQ toxicity are compared with the effect on CQ toxicity after inhibiting autophagosome formation using genetic means. RAS has different effects on autophagic flux and on CQ-sensitivity, but the direction of effect of RAS on autophagy does not predict whether RAS will potentiate or inhibit CQ toxicity. **P < 0.01, *P < 0.05, #P < 0.08, § for all knockdowns, P < 0.025 vs. nonsilencing control, l for all knockdowns except ATG5, P < 0.01 vs. nonsilencing control.
Discussion
Previous studies have demonstrated conflicting results concerning the effect of RAS on autophagy, with a few studies suggesting that oncogenic RAS inhibits autophagy, while others have claimed that oncogenic RAS induces, or supports the induction of autophagy. Our work confirms that oncogenic RAS can have both stimulating and repressive effects on autophagy, and that these differing effects are tumor cell specific and context dependent. The consequences of RAS activation on CQ sensitivity also varied depending on the cell type, with some cell lines being sensitized to CQ by oncogenic RAS and others becoming more resistant. Thus, oncogenic RAS is not sufficient to confer autophagy dependence or increased sensitivity of a cell to CQ.
A recent study published after we had originally submitted this manuscript similarly suggests that oncogenic RAS-driven autophagy dependence is context dependent, since in the absence of TP53, autophagy is no longer required for RAS-mediated tumor growth and progression, and the absence of autophagy may actually accelerate these in RAS-driven tumors where TP53 has been deleted.Citation22 In our experiments, TP53 status did not correlate with CQ or BafA1 sensitivity, however our in vitro experiments also differ from the previously mentioned study in vivo in 2 important ways. First, they were observing effects in the context of tumor formation and progression, while we observed effects on cells that had already come from fully developed tumors. Second, their data was based on TP53 deletion, while a majority of our cell lines had mutant TP53, and since TP53 can have effects on autophagy and other pathways in the cytoplasmCitation34 that do not depend on DNA binding,Citation35 it is possible that in some of our TP53 mutant cell lines important TP53 activities may be retained.
Our study shows that there are multiple ways in which a tumor cell can respond to CQ treatment. Some cancer cells require autophagy for proper growth, and therefore CQ reduces the number of actively proliferating cells but does not necessarily kill them. In other cells, CQ treatment results in cytotoxicity because the cell requires autophagy for cell survival. Additionally some cancer cells appear to be dependent on autophagy in terms of both growth and survival, and thus low levels of CQ prevent cell growth, while higher levels of CQ are toxic. In addition, chloroquine can be cytotoxic not because the cell requires autophagy, but because the cell is sensitive to cytotoxic intermediates created by late stage inhibition of autophagy. In this case, greater autophagosome formation can lead to increased CQ toxicity while reduced autophagosome formation can alleviate CQ toxicity. Lastly, in some cells CQ has minimal effect on either growth or cytotoxicity at concentrations that completely inhibit autophagy, indicating that they have no substantial need of the autophagic process for cell growth or viability. In some cases, especially at high concentrations, CQ may also act through mechanisms of action that are independent of autophagy. With the exception of some our clonogenic assays (), the range of CQ concentrations that we have primarily observed effects in our short-term experiments in this study are often substantially higher than the maximal concentrations of CQ typically achieved in the blood. (The maximal peak blood concentration of CQ and its main metabolites observed in rheumatoid arthritis patients in one study was just over 7 μM,Citation36 and the more typical peak concentration of CQ in the blood ranges between 1 to 3 μM.Citation36,Citation37) However, since chloroquine preferentially accumulates over time in lysosomes, the maximal concentration of CQ in tissues is much higher than this, and can approach 100 μM in the liver, bone marrow, spleen, and leukocytes.Citation38 Therefore, it is not implausible that CQ could accumulate within tumors at the doses used in these short-term experiments. Additionally, when exposing some of our cells for long periods of CQ treatment (> 7 d), we observed CQ effects on growth and toxicity for some of our cell types at concentrations well below 5 μM (data not shown), indicating that physiologically attainable concentrations of CQ are sufficient to observe effects on growth and toxicity in cancer cells.
In genetically defined transformed and immortalized isogenic HSSM and HEK cells, the stable ectopic expression of HRASG12V had opposing effects on autophagy. In HSMM cells, HRASG12V activated basal autophagic flux, while in the HEK cells, HRASG12V repressed basal autophagy. In both these cases, the cells with higher basal levels of autophagy were more sensitive to CQ consistent with autophagy intermediates being toxic to these cells. However, further experimentation showed that this was not the case since a reduction in autophagy in both cell types using shRNA to genes required for autophagosome formation actually sensitized to CQ-toxicity. Thus, RAS can positively or negatively influence autophagy and also potentiate or inhibit CQ-toxicity in different cells lines in a context dependent manner. In addition, our data indicate that RAS can stimulate other survival pathways that inhibit CQ-mediated toxicity that are independent from its effect on autophagy. This means that even knowing if oncogenic RAS inhibits or potentiates autophagy in a given tumor cell would not necessarily allow one to predict how RAS status will affect CQ sensitivity in that cell. Therefore we suggest that the RAS status alone even in combination with TP53 status, would not provide a suitable selection basis for CQ-based cancer therapy designed to inhibit autophagy and it will be necessary to identify other biomarkers that more accurately predict tumor response to autophagy inhibition to maximize the utility of autophagy inhibition therapy with CQ or other agents in the clinic.
Materials and Methods
Cell culture
Unless otherwise noted, all cells were grown in DMEM (Cellgro, 10-013-CV) supplemented with 10% fetal bovine serum (Sigma, F6178), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Cellgro, 30-002-Cl) at 37 °C and 5% CO2. Non-small cell lung cancer lines were obtained from the Protein Production, Monoclonal Antibody and Tissue Culture Shared Resource of the University of Colorado Cancer center. This facility previously validated all cell lines. H322C, HCC4006, Calu3, H358, A549, and H2009 cell lines were initially cultured in RPMI 1640 (Cellgro, 10-040-CV) with 10% fetal bovine serum, 2 mM glutamine, 1% HEPES (Cellgro, 25-060-Cl), 1% nonessential amino acids, and penicillin/streptomycin. Beginning with , as mentioned in the text, all NSCLC cell lines were grown in RPMI 1640 with 10% FBS without additional additives. The tetracycline-inducible HRASG12V human ovarian surface epithelial (HOSE) cell line was obtained from Seamus Martin (Trinity College, Dublin),Citation31 and grown in DMEM with 10% tetracycline-tree fetal bovine serum (Hyclone, SH30070.03T). Genetically defined immortalized human skeletal muscle myoblasts (HSMM T-H, or here denoted HSMM) and human embryonic kidney (HEK-HT, or here denoted HEK) with or without stable integration of HRASG12V were obtained from Chris Counter (Duke University School of Medicine), and have been described previously.Citation32,Citation33 DU145 cells with ectopic expression of HRASG12V or empty vector were obtained from JuanJuan (Ivy) Yin of the Kathleen Kelly Laboratory (National Cancer Institute, National Institutes of Health, Bethesda),Citation27 and were cultured in RPMI with 10% FBS and 1% nonessential amino acids and penicillin/streptomycin.
Reagents
Chloroquine (C6628), EBSS (E2888), doxorubicin (D1515), puromycin (P8833), and bafilomycin A1 (B1793) were obtained from Sigma, as well as the anti-ACTB/β-actin antibody (A5441). Doxycycline was obtained from Clontech (8634-1). Anti-LC3 was from Novus-Biologicals (NB100-2220). Anti-SQSTM1/p62 was from Abnova (H00008878-M01). Anti-RAS antibody (#3965), and anti-mouse (#7076) and anti-rabbit (#7074) HRP-conjugated secondary antibodies were from Cell Signaling Technology. The RAS GTPase Activation ELISA Kit was from Millipore (17-424).
Viability and toxicity assays
For short-term MTS assays, cells were plated in 48-well plates and 24 h later were treated with or without CQ and incubated for 48 or 72 h. MTS reagent (Promega, G3581) was added according to the manufacturer’s instructions and the absorbance was read at 490 nm.
For clonogenic assays, small numbers of cells were plated in 12-well plates (short-term, 5 to 7 d) and 24 h later, they were treated with CQ or left untreated for 48 or 72 h. Cells were washed several times and media was replaced with normal media. Cells were allowed to recover and grow for 5 to 7 d, fixed, and stained with crystal violet (Becton Dickinson and Company, 212525). Plates were scanned and then stain was solubilized with 30% acetic acid and absorbance was measured at 540 nm. For the HOSE-RAS long-term assays, cells were plated in 6-well plates and allowed to grow for 16 to 21 d, with the media changed every 2 to 3 d. Where treated, doxycycline was also replenished.
For LDH assays, cells were plated in 250 μL in 48-well plates and 24 h later, they were treated with or without CQ and incubated for 48 h. Fifty microliters of media was assayed from each well with the CytoscanTM-LDH Cytotoxicity Assay Kit (G-Biosciences, 786-210) according to the manufacturer’s instructions and the absorbance was read at 490 nm. Cell death percentages were calculated as compared with identically treated wells in which cells were lysed to indicate the total amount of LDH present.
For some assays, cell viability was determined by counting the percentage of propidium iodide stained cells (1μg/mL) as counted under a fluorescence microscope in the presence of Hoescht 33342 nuclear counterstain as an indicator of the total cell number. Typically > 20 fields were counted.
Western blotting
After treatment, cells were washed with PBS (Cellgro, 21-040-CV) and lysed in stringent RIPA buffer. Equal amounts of cell extracts were resolved by 12% or 15% SDS-PAGE and analyzed by western blot and visualized by enhanced chemiluminescence (ECL, Pierce, 32106; or Immobilon Western Chemiluminescent HRP Substrate, Millipore, WBKLS0500). Density analysis was performed using ImageJ 1.43u (NIH, USA).
Autophagic flux assay
Flow cytometric analysis of autophagy was done as previously described.Citation39 Briefly, all cells were infected with pBABE retroviruses encoding mCherry-GFP-LC3Citation31 and selected for either puromycin or hygromycin resistance. Flow cytometry was done using 488 and 561 nM lasers for red and green fluorophore excitation, respectively. Fluorescent cell populations were originally enriched after selection by sorting the cells for red and green double positive cells on a Moflo XDP 100 machine (Beckman Coulter, University of Colorado Cancer Center Anschutz Medical Campus Flow Cytometry Shared Resource). Analysis was done on either this machine or a Gallios 561 (Beckman Coulter, University of Colorado Cancer Center Anschutz Medical Campus Flow Cytometry Shared Resource). Nonviable cells were excluded from analysis by gating on the appropriate forward/side scatter profile. The mCherry/GFP signal ratio was determined and then plotted as a histogram by Summit 5.1 (Beckman Coulter). Since GFP is rapidly quenched by the low pH of the lysosome when autophagosomes merge with them, while mCherry is more stable to pH fluctuations, cells with high autophagy flux should be less green and thus have a higher mCherry/GFP ratio than cells with lower autophagic flux. Overnight treatment of cells with chloroquine or bafilomycin A1 was done as a control to show that the mCherry/GFP fluorescence ratio properly decreased in the absence of autophagic flux, while treatment with EBSS (not shown) was done to show that the ratio properly increased.
For quantification of puncta, cells were infected with a GFP-tagged LC3 virus. Cells were plated on coverslips and then fixed in 4% formaldehyde and mounted on slides. Using a fluorescence microscope, green fluorescent pictures were taken of multiple fields of cells and then the number of puncta for the cells were counted. The percentage of cells having high levels of puncta was quantified based on > 10 puncta/cell.
Statistical analysis
Statistical analysis was performed using the 2-tailed Student t test.
| Abbreviations: | ||
| ATG | = | autophagy-related |
| BafA1 | = | bafilomycin A1 |
| CQ | = | chloroquine |
| HCQ | = | hydroxychloroquine |
| HEK | = | immortalized embryonic kidney cells |
| HSMM | = | human skeletal muscle myoblasts |
| HOSE | = | human ovarian surface epithelial cell line |
| HRAS | = | Harvey rat sarcoma viral oncogene homolog |
| HRASG12V | = | HRAS containing the valine 12 oncogenic mutation |
| KRAS | = | Kirsten rat sarcoma viral oncogene homolog |
| KRASG12V | = | KRAS containing the valine 12 oncogenic mutation |
| LC3-II | = | phosphatidylethanolamine conjugate of microtubule-associated protein 1 light chain 3 (MAP1LC3) |
| LDH | = | lactate dehydrogenase |
| MTS | = | (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) |
| NSCLC | = | non-small cell lung cancer |
| RAS | = | rat sarcoma viral oncogene homolog (unspecified) |
| TP53 | = | tumor protein p53 (TRP53, transformation related protein 53 in murine models) |
Additional material
Download Zip (7.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH grants CA111421 and CA 124354 (AT) and by a Cancer League of Colorado Cancer Research Grant (MJM) from the Cancer League of Colorado, Inc. in association with the University of Colorado Cancer Center and also supported by ACS IRG #57-001-53 from the American Cancer Society, as well as Shared Resources from the Cancer Center Support Grant P30CA046934.
We wish to thank Chris Counter for the HSMM T-H and HEK-HT cells, JuanJuan (Ivy) Yin from the Kathleen Kelly Laboratory for the DU145 cells expressing HRAS, and Seamus Martin for the HOSE cells with a tetracycline-inducible HRASG12V transgene. This work was supported by Cancer League of Colorado Cancer Research Grant to MJ Morgan from the Cancer League of Colorado, Inc. in association with the University of Colorado Cancer Center, and also supported by ACS IRG #57-001-53 from the American Cancer Society (MJM). In addition, this work was supported by NIH grants CA111421 and CA 124354 (AT).
References
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 7; http://dx.doi.org/10.1038/nrm2245; PMID: 17712358
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med 2008; 8:78 - 91; http://dx.doi.org/10.2174/156652408783769616; PMID: 18336289
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533 - 41; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047; PMID: 21878654
- Metcalf DJ, García-Arencibia M, Hochfeld WE, Rubinsztein DC. Autophagy and misfolded proteins in neurodegeneration. Exp Neurol 2012; 238:22 - 8; http://dx.doi.org/10.1016/j.expneurol.2010.11.003; PMID: 21095248
- Chemali M, Radtke K, Desjardins M, English L. Alternative pathways for MHC class I presentation: a new function for autophagy. Cell Mol Life Sci 2011; 68:1533 - 41; http://dx.doi.org/10.1007/s00018-011-0660-3; PMID: 21390546
- Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res 2008; 68:6889 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-08-0161; PMID: 18757401
- Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ 2009; 16:991 - 1005; http://dx.doi.org/10.1038/cdd.2009.8; PMID: 19229247
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728 - 41; http://dx.doi.org/10.1016/j.cell.2011.10.026; PMID: 22078875
- Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012; 119:2895 - 905; http://dx.doi.org/10.1182/blood-2011-08-372383; PMID: 22223827
- Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009; 625:220 - 33; http://dx.doi.org/10.1016/j.ejphar.2009.06.063; PMID: 19836374
- Katz SJ, Russell AS. Re-evaluation of antimalarials in treating rheumatic diseases: re-appreciation and insights into new mechanisms of action. Curr Opin Rheumatol 2011; 23:278 - 81; http://dx.doi.org/10.1097/BOR.0b013e32834456bf; PMID: 21448012
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; http://dx.doi.org/10.4161/auto.5338; PMID: 18188003
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460 - 70; http://dx.doi.org/10.1101/gad.2016311; PMID: 21317241
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 78; http://dx.doi.org/10.1091/mbc.E10-06-0500; PMID: 21119005
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478 - 89; http://dx.doi.org/10.1158/1078-0432.CCR-10-2372; PMID: 21325076
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447 - 61; http://dx.doi.org/10.1101/gad.219642.113; PMID: 23824538
- Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504:296 - 300; http://dx.doi.org/10.1038/nature12865; PMID: 24305049
- http://www.cancer.gov/clinicaltrials/search/results?protocolsearchid=9855259.
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528 - 39; http://dx.doi.org/10.1038/nrclinonc.2011.71; PMID: 21587219
- Turcotte S, Giaccia AJ. Targeting cancer cells through autophagy for anticancer therapy. Curr Opin Cell Biol 2010; 22:246 - 51; http://dx.doi.org/10.1016/j.ceb.2009.12.007; PMID: 20056398
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Yin J, Pollock C, Tracy K, Chock M, Martin P, Oberst M, Kelly K. Activation of the RalGEF/Ral pathway promotes prostate cancer metastasis to bone. Mol Cell Biol 2007; 27:7538 - 50; http://dx.doi.org/10.1128/MCB.00955-07; PMID: 17709381
- Ouyang DY, Xu LH, He XH, Zhang YT, Zeng LH, Cai JY, Ren S. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy 2013; 9:20 - 32; http://dx.doi.org/10.4161/auto.22397; PMID: 23075929
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 501; http://dx.doi.org/10.1126/science.282.5393.1497; PMID: 9822382
- Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 2003; 4:373 - 84; http://dx.doi.org/10.1038/nrm1105; PMID: 12728271
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42:23 - 35; http://dx.doi.org/10.1016/j.molcel.2011.02.009; PMID: 21353614
- Linardic CM, Counter CM. Genetic modeling of Ras-induced human rhabdomyosarcoma. Methods Enzymol 2008; 438:419 - 27; http://dx.doi.org/10.1016/S0076-6879(07)38028-2; PMID: 18413264
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, Counter CM. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev 2002; 16:2045 - 57; http://dx.doi.org/10.1101/gad.993902; PMID: 12183360
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676 - 87; http://dx.doi.org/10.1038/ncb1730; PMID: 18454141
- Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008; 7:3056 - 61; http://dx.doi.org/10.4161/cc.7.19.6751; PMID: 18818522
- Augustijns P, Geusens P, Verbeke N. Chloroquine levels in blood during chronic treatment of patients with rheumatoid arthritis. Eur J Clin Pharmacol 1992; 42:429 - 33; PMID: 1307690
- Mzayek F, Deng H, Mather FJ, Wasilevich EC, Liu H, Hadi CM, Chansolme DH, Murphy HA, Melek BH, Tenaglia AN, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials 2007; 2:e6; http://dx.doi.org/10.1371/journal.pctr.0020006; PMID: 17213921
- Grundmann M, Mikulíková I, Vrublovský P. Tissue distribution of subcutaneously administered chloroquine in the rat. Arzneimittelforschung 1971; 21:573 - 4; PMID: 5108164
- Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DWH, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 2014; 16:47 - 54; http://dx.doi.org/10.1038/ncb2886; PMID: 24316673