Abstract
The interactions between viruses and cellular autophagy have been widely reported. On the one hand, autophagy is an important innate immune response against viral infection. On the other hand, some viruses exploit the autophagy pathway for their survival and proliferation in host cells. Vaccinia virus is a member of the family of Poxviridae which includes the smallpox virus. The biogenesis of vaccinia envelopes, including the core envelope of the immature virus (IV), is not fully understood. In this study we investigated the possible interaction between vaccinia virus and the autophagy membrane biogenesis machinery. Massive LC3 lipidation was observed in mouse fibroblast cells upon vaccinia virus infection. Surprisingly, the vaccinia virus induced LC3 lipidation was shown to be independent of ATG5 and ATG7, as the atg5 and atg7 null mouse embryonic fibroblasts (MEFs) exhibited the same high levels of LC3 lipidation as compared with the wild-type MEFs. Mass spectrometry and immunoblotting analyses revealed that the viral infection led to the direct conjugation of ATG3, which is the E2-like enzyme required for LC3-phosphoethanonamine conjugation, to ATG12, which is a component of the E3-like ATG12–ATG5-ATG16 complex for LC3 lipidation. Consistently, ATG3 was shown to be required for the vaccinia virus induced LC3 lipidation. Strikingly, despite the high levels of LC3 lipidation, subsequent electron microscopy showed that vaccinia virus-infected cells were devoid of autophagosomes, either in normal growth medium or upon serum and amino acid deprivation. In addition, no autophagy flux was observed in virus-infected cells. We further demonstrated that neither ATG3 nor LC3 lipidation is crucial for viral membrane biogenesis or viral proliferation and infection. Together, these results indicated that vaccinia virus does not exploit the cellular autophagic membrane biogenesis machinery for their viral membrane production. Moreover, this study demonstrated that vaccinia virus instead actively disrupts the cellular autophagy through a novel molecular mechanism that is associated with aberrant LC3 lipidation and a direct conjugation between ATG12 and ATG3.
Keywords: :
Introduction
Vaccinia virus is a member of the Orthopoxvirus genus of the family Poxviridae. It is the most well-studied poxvirus and has been successfully implemented as a vaccine to eradicate variola virus, the causative agent of smallpox disease. A unique feature of vaccinia and all poxviruses is that the life cycle is exclusively cytosolic. The morphogenesis of the large, complex, enveloped virus involves several distinct forms of virion.Citation1 The life cycle of the virus begins in sequestered subcellular regions of the infected host cell, referred to as “virus factories,” and is the location where crescent membranes develop and surround dense viroplasm to form spherical immature virus (IV).Citation2 These crescent membranes, containing lipid and protein, are the first visible membrane structures in the host cell post-infection which enclose the core components of the virus and are reported to contain a double-membrane structure.Citation3 In addition, vaccinia requires the procurement of an additional membrane allowing for the nonlytic escape of viral particles from the host cell. The source of the membranes that arise to form the IVs are still controversial despite being intensely scrutinized over the past several decades. It has been suggested that the crescent membranes are not host derived but are instead formed by a de novo synthesis process,Citation4 while opposing researchers have proposed an alternative double-membrane model and argue the membrane originates from the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) of the host cell.Citation3,Citation5,Citation6
Macroautophagy, hereafter referred to as autophagy, is a membrane-trafficking pathway leading to lysosomal degradation of cytosolic components.Citation7-Citation10 The hallmark of this pathway involves the generation of autophagosomes, which are double-membrane structuresCitation9-Citation11 that sequester the cytosol and its constituents and deliver them to the lysosome for degradation. In parallel with the controversy surrounding the crescent membranes of vaccinia, the origin of the double-membrane phagophore assembly sites are also highly debated. The purported concept of de novo synthesis for the crescent membranes which mature to become autophagosomes has been proposed. More recent studies have suggested that the source of the membranes could also be derived from pre-existing organelles, such as the endoplasmic reticulum,Citation12 mitochondria,Citation13 plasma membraneCitation14 or Golgi network.Citation15
The autophagic pathway requires two interlinked ubiquitin-like protein conjugation systems.Citation16-Citation18 In one system, the C-terminal glycine of a ubiquitin-like protein ATG12 is conjugated to a lysine residue of ATG5Citation16 via an isopeptide bond which is catalyzed by ATG7, an E1 equivalent enzymeCitation19 and ATG10, an E2 equivalent enzyme.Citation20 In the other system, ATG8 or, in mammals, MAP-LC3 (microtubule-associated protein 1 light chain 3), is cleaved by the cysteine protease ATG4Citation21 resulting in the cytosolic LC3-I form. Through the E1-like enzyme ATG7 and the E2-like enzyme ATG3, LC3 is conjugated to phosphatidylethanolamine (PE), forming the lipidated LC3-II species. These two systems functionally interact. The ATG12–ATG5 conjugate associates with ATG16L, forming a large multimeric protein complex.Citation22 It is believed that this complex not only serves as a scaffold on which the nascent autophagosome is expandedCitation22 but also acts as an E3-like enzyme for the conjugation of ATG8 or, in mammals MAP-LC3 (microtubule-associated protein 1 light chain 3), to the phospholipids.Citation23
We previously visited the idea that cellular autophagy may play a role in vaccinia virus membrane biogenesis.Citation24 The study showed that inactivation of autophagy by deleting essential autophagy genes, atg5 or beclin 1, affect neither viral production nor viral infectivity, suggesting that cellular autophagy is not required for the vaccinia virus membrane biogenesis. In the present study, however, we found that vaccinia virus does have a tremendous impact on cellular autophagy. Vaccinia virus infection causes massive levels of LC3 lipidation. Surprisingly, the LC3 lipidation induced by vaccinia virus is independent of ATG5 and ATG7, but depends on ATG3. However, neither deletion of atg3 nor lack of LC3 lipidation seems to have an impact on viral production. In search of the molecular mechanism leading to the ATG5- and ATG7- independent LC3 lipidation, we showed that vaccinia virus infection leads to a direct conjugation between ATG12 and ATG3, which is associated with a total absence of autophagosome formation under either basal or amino acid deprivation conditions. Our results showed that vaccinia virus does not use the autophagosome membrane as its source for the core membrane of the immature virus; however, vaccinia virus does actively disrupt the cellular autophagy machinery through a unique molecular mechanism leading to total deficiency of autophagosome formation.
Results
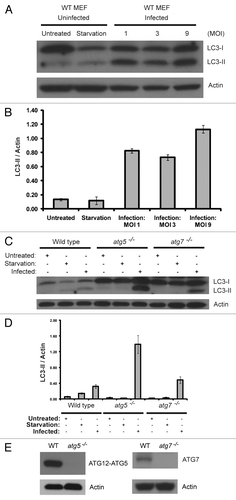
Vaccinia virus infection of fibroblast cells led to massive LC3 lipidation that was independent of (ATG5) and ATG7
A previous study in our lab, employing atg5−/− MEFs and beclin 1−/− embryonic stem (ES) cells, showed that the cellular autophagy machinery is not required for vaccinia virus replication and maturation.Citation24 However, we found that vaccinia virus infection did lead to a massive increase of LC3 lipidation at the various multiplicity of infections (MOI) we tested, as shown in . The ratio of LC3-II over actin is several fold higher than that observed in the cells that underwent amino acid and serum deprivation for 2 h with Hank’s media. Strikingly, when the autophagy-deficient atg5−/− or atg7−/− MEF cell lines were infected with vaccinia virus, they displayed similar levels of LC3 lipidation as observed in the wild-type cells (). As a control, amino acid and serum starvation failed to produce any lipidation of LC3 in these autophagy-deficient cells without vaccinia virus infection. Lack of ATG5 (as indicated by lacking ATG12–ATG5 conjugate) and ATG7 in the respective knockout MEF cells was confirmed with immunoblotting as shown in . This result indicated that vaccinia virus infection generated LC3 lipidation through a unique process that is independent of ATG7 and ATG5.
Figure 1. Vaccinia virus infection increases LC3 lipidation, which is independent of atg5 and atg7. (A) Immunoblotting assay of wild-type cells either grown in normal medium (untreated), or amino acid and serum deprived for 2 h with Hank’s media (starvation), or infected with vaccinia virus at a multiplicity of infection (MOI) of 1, 3 or 9 and harvested 24 h post infection. Cell lysates were subjected to SDS-PAGE and separated proteins were immunoblotted using an anti-LC3 or anti-actin antibodies as indicated. (B) Quantification of LC3-II to actin ratio in (A). (C) Immunoblotting assay of wild-type, atg5−/− and atg7−/− MEF cell lines either grown in normal medium (untreated), or amino acid and serum deprived for 2 h with Hank’s media (starvation), or infected with vaccinia virus at a MOI of 3 and harvested 24 h post infection. Cell lysates were analyzed with an anti-LC3 or anti-actin antibody, as indicated. (D) Quantification of LC3-II to actin ratio in (C). The results in (A) and (C) are representative of three independent experiments. (E) Immunoblotting assay of wild-type, atg5−/− and atg7−/− MEF cell lines grown in normal medium using an anti-ATG12, anti-ATG7 and anti-actin antibodies as indicated.
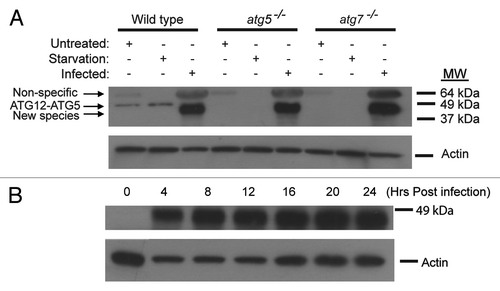
New species of ATG12 conjugate was formed upon vaccinia virus infection
The surprising result that the LC3 lipidation induced by vaccinia virus infection is independent of ATG5 and ATG7 indicated that the virus had exploited a novel molecular mechanism for LC3 lipidation. One possibility is that the viral genome may encode a homolog of ATG5 and ATG7 that is capable of complementing the loss of the mammalian proteins. To explore that possibility, we screened the Addgene (Cambridge, MA) vaccinia virus ORF (open reading frame) library. The library is a collection of all 266 open reading frames from the vaccinia virus genome subcloned into a mammalian expression vector. We transfected each ORF expression plasmid from the library into the atg5−/− cell line to determine if the viral gene could mimic the effect of viral infection in restoring LC3 lipidation. However, no single ORF from the collection could induce the conversion of LC3-I to LC3-II in the atg5−/− cell line (data not shown). The data suggested that the vaccinia virus genome may not encode a homolog of ATG5 or that the complementation of the ATG5 function may require a concerted interaction specifically related to viral life cycle.
In wild-type cells, ATG5 is conjugated to ATG12 in a ubiquitin-like conjugation systemCitation25 and in turn helps facilitate the conjugation of LC3 to PE. Therefore, we decided to employ an alternative approach to identify the functional homolog of ATG5. We reasoned if a functional ATG5 homolog existed, it should form a conjugate with ATG12. We did an immunoblotting assay with an antibody against ATG12 on infected and uninfected wild-type, atg5−/− and atg7−/− MEF cells. As shown in , under untreated and starvation conditions in wild-type cells, a 55-kDa protein band was detected representing the ATG12-ATG5 conjugate. This protein band was not detected in either autophagy deficient cell line as expected. Upon viral infection, a novel protein band that interacts with the anti-ATG12 antibody was detected in wild-type, atg5−/− and atg7−/− MEFs. Under denaturing conditions, this faster migrating protein band was running at approximately 50-kDa. It was interesting to note that the 55-kDa protein band that represents ATG12-ATG5 protein conjugate was no longer observed in the wild-type cells post vaccinia virus infection. We further performed a time course study. As shown in , the novel protein band reached a high level in about 4 h and remained abundant in the infected cells for all the time points tested thereafter. These results suggested that ATG12, whose molecular weight is 16 kDa, might be conjugated to a protein other than ATG5 upon vaccinia virus infection.
Figure 2. Formation of a new ATG12 conjugate upon vaccinia virus infection. (A) Immunoblotting assay of wild-type, atg5−/− and atg7−/− MEF cell lines either untreated, or amino acid and serum deprived for 2 h with Hank’s media (starvation), or infected with vaccinia virus at a MOI of 3 and harvested 24 h post infection. Lysates were subjected to SDS-PAGE and separated proteins were immunoblotted using an anti-ATG12 (upper) or anti-actin (lower) antibody. The results are representative of three independent experiments. (B) Immunoblotting assay of wild-type cells showing the time course of the appearance of the novel protein band after vaccinia virus infection. Wild-type MEFs were infected with vaccinia virus at a MOI of 3, harvested at indicated time points, and analyzed with an anti-ATG12 (upper) or anti-actin (lower) antibody.
Novel ATG12–ATG3 conjugate was generated upon vaccinia virus infection and ATG3 was required for LC3 lipidation
To determine the identity of the unknown protein that was conjugated to ATG12, we generated a stable cell line expressing FLAG and HA epitope tagged mouse ATG12, referred to hereafter as FH-ATG12, in NIH-3T3 cells. The FH-ATG12 stable cell line was subsequently infected with vaccinia virus for 24 h and immunoprecipitation experiments employing M2 agarose beads (the anti-FLAG antibody M2 is crossed-linked to agarose) were performed. Silver stain of the vaccinia virus-infected immunoprecipitation sample revealed a protein band that ran at an approximate size of 50-kDa () corresponding well with western blot results (). Untreated NIH-3T3 and FH-ATG12 immunoprecipitation control samples did not exhibit a protein band that ran at the same molecular weight.
Figure 3. Immunoprecipitation and mass spectrometry analysis of the novel ATG12 conjugate. (A) NIH-3T3 and NIH-3T3 stable cell line expressing epitope tagged ATG12 (FH-ATG12) were left untreated or infected with vaccinia virus at a MOI of 3 and harvested 24 h post infection. Samples were immunoprecipitated with α-FLAG M2 affinity gel beads and eluted with FLAG peptide. The final eluate was separated by SDS-PAGE and visualized by silver stain. Labeled protein bands were identified through mass spectrometry analysis; double asterisk denotes cellular protein not fully characterized. (B) ATG3 peptide sequences identified by mass spectrometry. The numbers indicate the relative amino acid position in full-length mouse ATG3 sequence.
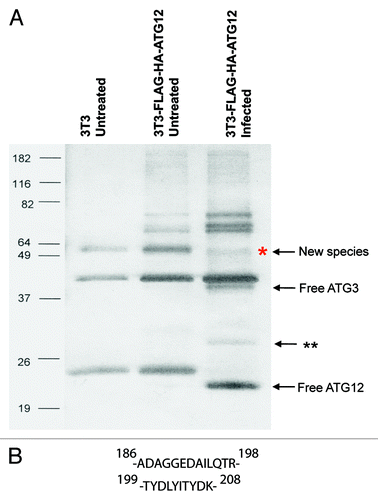
Mass spectrometry analysis was performed to determine the identity of the novel fusion protein. Screening of the mass spectrometry amino acid sequence data against the protein databanks that are encoded by the Western Reserve and Copenhagen vaccinia virus strains yielded no matches with high confidence levels; however, screening against the murine protein database identified ATG3 as the most plausible protein conjugating to ATG12 ().
To confirm the mass spectrometry findings, we performed immunoblotting analysis on the immunoprecipitated proteins. As shown in , indeed, the immunoprecipitated protein band was recognized by both the ATG12 and the ATG3 antibodies. We further tested if the ATG12 interacting protein band shown in can be recognized by the ATG3 antibody. Cell lysates were re-examined with an anti-ATG3 antibody. As shown in , upon vaccinia virus infection, wild-type, atg5−/− and atg7−/− cell lines all exhibited an intense 50-kDa protein band that was in the position previously identified with the ATG12 antibody. Again, as in the case when probed with anti-ATG12 antibody (), this protein band was not detected under untreated or starvation conditions in wild-type cells. To determine that the 50-kDa protein was truly reactive to both ATG3 and ATG12 antibodies, the band was verified in several ways. First, fresh membranes were exposed to each individual antibody; second, after probing and stripping of one antibody, the membrane was then reprobed with the second antibody; third, the order of probing antibodies were reversed. All these analyses indicated that the protein that is reactive to the ATG3 and ATG12 antibodies is the same protein. Based on predicted molecular weight of ATG12 (16-kDa) and ATG3 (36-kDa), the detection of an ATG12–ATG3 protein conjugate would be expected to run at an approximate molecular weight of 52-kDa, in-line with the empirical western blot data.
Figure 4. Novel ATG12-ATG3 conjugate and ATG3 dependent LC3 lipidation. (A) Immunoprecipitation and immunoblotting analysis of the new ATG12 conjugate. NIH-3T3 and NIH-3T3 cells stably expressing FH-ATG12 were left untreated or infected with vaccinia virus at a MOI of 3 and harvested 8 h post infection. Lysates were immunoprecipitated with α-FLAG. The protein complexes were separated using SDS-PAGE and immunoblotted with anti-ATG12 or anti-ATG3 antibodies, as indicated. (B) Immunoblotting assay of wild-type, atg5−/−, and atg7−/− MEF cells with an anti-ATG3 antibody. Indicated cell lines were either untreated, amino acid and serum deprived for 2 h with Hank’s media or infected with vaccinia virus at a MOI of 3 and harvested 24 h after infection. Actin levels serve as a loading control. (C) Immunoblotting of wild-type, atg5−/− and atg3−/− MEF cells with a LC3 antibody. Indicated cell lines were either untreated, amino acid and serum deprived for 2 h with Hank’s media or infected with vaccinia virus at a MOI of 3 and harvested 24 h post infection. Lysates were probed with anti-LC3 and anti-actin antibodies. The results are representative of three independent experiments. (D) Quantification of LC3-II to actin ratio in (C). The data are the means ± SD from three independent experiments.
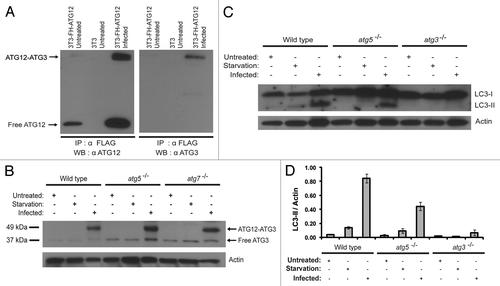
We then investigated the functional significance of the ATG12–ATG3 conjugation and determined if the LC3 lipidation depends upon ATG3. Wild-type, atg5−/− and atg3−/− cell lines were infected with vaccinia virus (). Consistent with our previous observations (), wild-type and atg5−/− cells exhibit the conversion of LC3-I to LC3-II post vaccinia virus infection. However, in the atg3−/− cell line, no LC3 lipidation was detected upon viral infection, indicating LC3 lipidation is dependent upon ATG3 during viral infection. Together, our results indicated that vaccinia virus leads to the production of ATG12–ATG3 conjugate and ATG3 is required for the virus-induced LC3 lipidation. It is noteworthy that viral infection in the atg3−/− MEFs still led to production of a residual conjugate product (Fig. S1A). This is likely due to the fact that the atg3−/− MEFs express an alternative spliced form of atg3 transcript (Fig. S1B). In this transcript, the neo cassette that replaces atg3 exon 10 encoding the catalytic domain of ATG3 is spliced out (Fig. S1C). The residual conjugate might be the conjugation product between ATG12 and the nonfunctional ATG3Δexon10 (Fig. S1A).
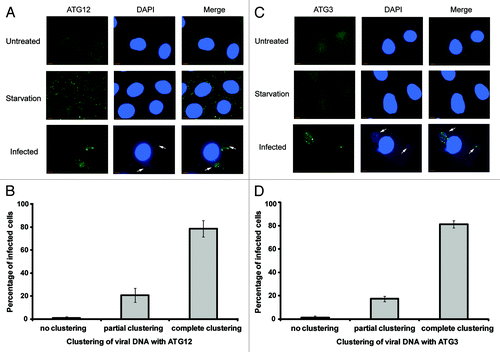
Viral DNA and ATG12/ATG3 proteins were clustered together in vaccinia-infected cells
An immunofluorescence study was performed to determine changes in subcellular localization of ATG12 and ATG3 post-vaccinia virus infection. When stained for ATG12 or ATG3, wild-type cells under normal growth conditions exhibited a diffuse pattern throughout the cell (, upper). However, upon viral infection, a new pattern was observed in which ATG12 or ATG3 was clustered in the cytoplasm. To determine if viral activity colocalized with the ATG12 or ATG3 clustering, DAPI staining was performed in wild-type cells to visualize viral DNA. After 8 h of viral infection, viral DNA was clearly observed. In almost all cells, the location where ATG3 or ATG12 was clustered in near proximity to the cytoplasmic viral DNA (, lower). Quantification of the immunofluorescence results show that ATG12 and ATG3 consistently localize near the subcellular regions of the cell where viral DNA is concentrated (). Due to the unavailability of commercial antibodies of different species origins against ATG3 and ATG12 that are suitable for immunofluorescent study, it was not possible to directly analyze the colocalization between ATG12 and ATG3 upon viral infection. It was important to note that while the ATG12 or ATG3 clusters seemed to interdigitate with the viral DNA clusters, ATG12/ATG3 proteins did not overlap with the viral DNA. Together, our results indicated that vaccinia virus led to ATG12/ATG3 clustering near the viral factory in the infected cells, supporting that ATG12–ATG3 conjugation is a result of viral activities.
Figure 5. Clustering of ATG12 and ATG3 with viral DNA. (A) Wild-type MEF cells were untreated, amino acid and serum deprived for 2 h with Hank’s media or infected with vaccinia virus at a MOI of 1 for 8 h. Cells were fixed, immunostained for endogenous ATG12 and co-stained with DAPI. Arrows denote areas of viral activity. (B) Quantification of clustering between viral DNA and ATG12. A minimum of 50 cells was counted and scored for the localization of viral activity with ATG12. (C) Wild-type MEF cells were untreated, amino acid and serum deprived for 2 h with Hank’s media or infected with vaccinia virus at a MOI of 1 for 8 h. Cells were fixed, immunostained for endogenous ATG3 and stained with DAPI. Arrows denote areas of viral activity. (D) Quantification of clustering between viral DNA and ATG3. A minimum of 50 cells was counted and scored for the localization of viral activity with ATG3. Complete clustering was defined as all cellular ATG3/ATG12 localizing with viral DNA (cytoplasmic DAPI staining), and vice versa. Cells with partial clustering pattern were defined as those in which all cellular ATG3/ATG12 is localized with viral DNA, with a fraction of viral DNA not localized with cellular ATG3/ATG12.
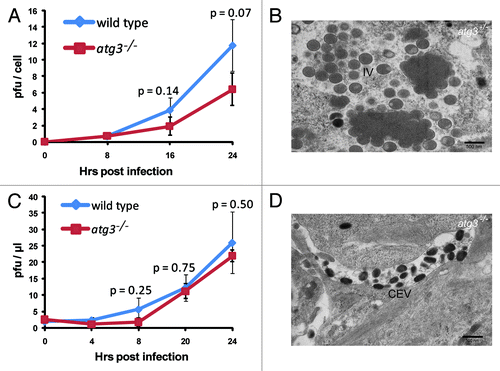
Viral proliferation was not dependent upon ATG3
Our observation that no LC3 lipidation was formed in atg3−/− cells allowed us to further determine a possible role of ATG3 and the ATG3-dependent LC3 lipidation in viral proliferation. A viral growth assay was completed comparing the viral production in wild-type and in atg3−/− cells. Growth curve analysis of vaccinia virus shows that there was no significant difference in levels of intracellular virus when comparing wild-type and atg3−/− cell lines at the observed time points (). Consistently, the electron microscopy pictures showed that there was no defect in viral crescent formation and viral maturation of the intracellular viral particles (). In addition to the viral crescent membrane, the intracellular mature virus (IMV) acquires an outer wrapping membrane forming the intracellular enveloped virus (IEV) during the viral morphogenesis process. The proper formation and function of the outer membrane is required for nonlytic release from mammalian cells. To test if ATG3 affects the proper outer membrane formation, the extracellular viral particles were collected. The viral growth curve of these particles was determined by plaque assay and the morphology of those viral particles was analyzed by electron microscopy (). Again, growth curve analysis of vaccinia virus shows that there was little difference in levels of extracellular viral particle production between wild-type and atg3−/− cell lines at the observed time points () and seemingly normal extracellular viral particles were observed under electron microscopy (). Together, these results indicated that ATG3 and the ATG3-dependent LC3 lipidation are neither essential for the formation of IV membrane, nor required for outer membrane maturation required for the nonlytic release from the host cells.
Figure 6. Vaccinia virus maturation and replication is independent of ATG3. (A) Growth curves of vaccinia virus were determined to compare levels of intracellular virus production in wild-type and atg3−/− MEF cells infected at a MOI of 3. Error bars indicate SD and p values are given at indicated time points. p values were calculated by Student’s t test. (B) Representative electron microscopy image of vaccinia virus infected atg3−/− MEF. Cells were analyzed 24 h post infection at a MOI of 3. (C) Growth curves of vaccinia virus were determined to compare levels of extracellular virus production in wild-type and atg3−/− MEF cells infected at a MOI of 3. (D) Representative electron microscopy image of vaccinia virus infected atg3−/− MEF. Cells were analyzed 24 h post infection at a MOI of 3. Error bars indicate SD and p values are given at indicated time points. Scale bars are shown; IV, immature virus; IMV, intracellular mature virus. p values were calculated by Student’s t test.
Vaccinia virus infection abrogated basal and starvation-induced autophagosome formation and abolished autophagy flux
Our results clearly indicated that the vaccinia virus manipulated the cellular autophagy machinery, yet inactivation of cellular autophagy had no impact on viral proliferation. This prompted us to speculate that the function of the viral-induced ATG12–ATG3 conjugation and LC3 lipidation may not be to support viral replication or maturation but rather to disrupt cellular autophagy, which often represents a strong innate immunity response toward viral infection. To test this hypothesis, we analyzed autophagy activity in the wild-type cells before and after vaccinia virus infection with electron microscopy. As shown in , without vaccinia virus infection, basal autophagy activity was easily detectable. The average number of autophagosomes per cell was approximately two. Very strikingly, upon vaccinia virus infection, any cell that showed signs of viral infection, such as formation of IV and IMV, no autophagosomes were observed (). We further tested if vaccinia virus infection blocked amino acid and serum deprivation induced autophagy activation. As shown in , after cells were cultured in Hanks’ medium for 2 h, autophagy activity increased 4.5 fold, as manifested by the increased number of autophagosomes in cells (comparing untreated and starvation alone). However, in the vaccinia virus-infected cells, further amino acid and serum deprivation did not lead to any autophagosome formation (). Interestingly, despite the fact that no autophagosomes or other nonviral membrane structures were visible with electron microscopy study, vaccinia virus infection led to the formation of GFP-LC3 puncta (). The morphology of these puncta is not exactly the same as the puncta formed in response to amino acid deprivation (). These puncta likely reflect the massive accumulation of GFP-LC3-II in the form of aggregates.
Figure 7. Absence of autophagosomes or autophagosome-like structures post vaccinia virus infection. Representative electron microscopy images of (A) untreated, (B) starved, (C) vaccinia virus infected for 24 h at a MOI of 3, and (D) vaccinia virus infected for 18 h at a MOI of 3 followed by 2 h of starvation in wild-type MEF cells. (E) Number of autophagosomes per cell under indicated conditions with cell counts (n). Arrows denote autophagosomes. Scale bars are shown; N, nucleus; IV, immature virus; IMV, intracellular mature virus. P values were calculated by Student’s t-test.
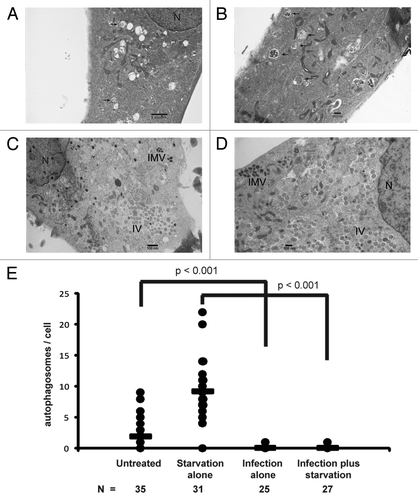
Figure 8. Analyses of the effect of vaccinia virus infection on GFP-LC3 distribution and autophagy flux. (A) Distribution of GFP-LC3 in uninfected and vaccinia virus infected cells. NIH 3T3 cells that stably express GFP-LC3 were either untreated, or underwent amino acid and serum starvation for 2 h, or infected with vaccinia virus, as indicated. Fluorescent microscopy was performed to visualize the distribution of GFP-LC3. (B) Quantification of (A). (C–E) Immunoblotting assay of wild-type or atg5−/− MEFs either uninfected or infected with vaccinia virus (MOI of 3) for 8 h followed by treatment with 100 nM of bafilomycin A1 for indicated time. Lysates were subjected to SDS-PAGE and separated proteins were immunoblotted using an anti-LC3 and anti-actin antibodies as indicated.
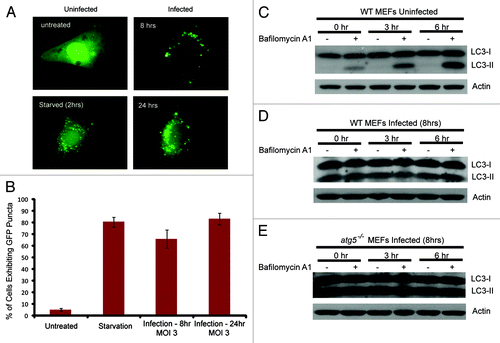
The lack of autophagosomes in the virus-infected cells may be due to either lack of autophagosome biogenesis or, alternatively, due to extremely high efficiency of autophagy flux. To differentiate these two possibilities, we determined the effect of vaccinia virus infection on autophagy flux. As shown in , treating uninfected wild-type MEFs with bafilomycin A1, which inhibits the fusion between autophagosome and lysosome, led to increasing accumulation of LC3-II due to lack of lysosomal degradation. In contrast, when the wild-type cells were infected with vaccinia virus, bafilomycin A1 could no longer increase LC3-II accumulation (), similar to what was observed in the atg5−/− MEFs where no autophagy flux was expected (). Taken together, these results strongly support that vaccinia virus actively disrupts the normal cellular autophagy machinery that is required for autophagosome formation.
Discussion
The interactions between cellular autophagy and viruses and consequently the co-evolution of the virus and cellular autophagy machinery is a fascinating area of research.Citation26-Citation30 On the one hand, studies involving RNA and DNA viruses, including the enveloped single stranded RNA Sindbis virus,Citation31,Citation32 double stranded DNA Herpes simplex virus (HSV-1),Citation33,Citation34 and the single stranded RNA plant tobacco mosaic virus,Citation35 have demonstrated the key role autophagy plays as an antiviral immune defense mechanism. On the other hand, for other viruses, co-evolution has not only allowed for viral escape of autophagy, but also the subversion of the autophagic pathway as a possible integral component of the viral life cycle.Citation36-Citation44 Our current study of the interaction between vaccinia virus and cellular autophagy has led to a number of interesting discoveries. We found that vaccinia virus infection leads to massive lipidation of LC3, which is, surprisingly, atg5- and atg7-independent. In searching for the molecular mechanism underlying the LC3 lipidation, we found the autophagy protein ATG12 is directly conjugated to ATG3 upon viral infection. Moreover, we showed that ATG3 is essential for the viral-induced LC3 lipidation. These results suggest that vaccinia virus may selectively alter the cellular autophagy machinery by inducing ATG12–ATG3 conjugation causing massive LC3 lipidation. We further investigated the functional consequence of the LC3 lipidation. Interestingly, neither lack of ATG3 nor lack of LC3 lipidation has any significant impact on viral morphogenesis, viral proliferation or viral infectivity. On the other hand, vaccinia virus infection has a drastic impact on autophagosome formation in the cells. Paradoxically, despite the high levels of LC3 lipidation, the viral-infected cells are deficient in autophagosome formation and have no autophagy flux. Taken together, these results indicate that vaccinia virus does not require cellular autophagy membrane biogenesis machinery for its morphogenesis and proliferation. Instead, the data support that vaccinia virus selectively inactivates cellular autophagy machinery through a novel molecular mechanism, i.e., the conjugation of ATG12 to ATG3 and consequent aberrant LC3 lipidation.
Autophagy has been clearly established as a key component of all branches of immunity, including intrinsic, innate and adaptive systems.Citation26,Citation28,Citation45,Citation46 Our study indicates that vaccinia virus selectively inactivates cellular autophagy machinery through a novel mechanism. These discoveries suggest that the activation of cellular autophagy may impose a strong negative selection pressure on the survival of vaccinia virus. It would be extremely interesting to identify the viral proteins that trigger the autophagy inactivation events. By studying the behavior of the mutant vaccinia virus lacking the autophagy inactivating genes in both wild-type and the autophagy-deficient cells, one may be able to determine the exact nature of the antiviral activity of autophagy against vaccinia virus. These studies can provide further insight into our understanding of the interactions between virus and the host cells, which may provide a novel venue for therapies against viral infection.
When studied by electron microscopy, the lack of autophagosome formation in the vaccinia virus-infected cells is astounding (). The deficiency in autophagy in the virus infected cells was also confirmed by autophagy flux analysis (). The exact mechanism by which the vaccinia virus causes autophagy deficiency is not clear. Based on the known functions of the autophagy proteins, we propose a working model which is illustrated in . In the case of normal cellular autophagy activation (), the ATG12 ubiquitin-like protein conjugation system generates an ATG12–ATG5 conjugate which complexes with ATG16L. Together they form a large multimeric protein complex (~800 kDa) where autophagosomes elongate and mature.Citation23 The ATG12–ATG5-ATG16L complex serves at least two related purposes: first, it serves as a scaffold that brings the molecular components required for autophagosome elongation; second, it functions as a ubiquitin E3-like enzyme that facilitates the conjugation of LC3 to phosphatidylethanolamine (PE), which is required for autophagosome elongation and/or closing. In particular, ATG12 in this ATG12–ATG5-ATG16L complex is directly related to its E3-like enzyme activity. During cellular autophagy activation, ATG12 directly interacts with the ATG3-LC3 intermediate and is responsible for bringing LC3 into proximity with the membrane embedded PE, thereby leading to the conjugation between the substrates LC3 and PE.Citation23 Under the condition of vaccinia virus infection (), we speculate that the viral-induced ATG12–ATG3 conjugation disrupts autophagosome formation in the following way. The novel ATG12–ATG3 conjugate covalently links the E2-like enzyme to the E3-like enzyme component, causing the “short-circuiting” of the LC3 lipidation regulatory circuitry and leading to massive LC3 lipidation (). In the meantime, lack of ATG12–ATG5 conjugation fails to generate an ATG12–ATG5-ATG16L complex that brings other components required for autophagosome elongation. Thus this spatial dislocation of LC3 lipidation from other molecular components required for autophagosome elongation might have caused the defect in autophagosome formation in the viral-infected cells.
Figure 9. A possible mechanism by which vaccinia virus disrupts autophagy machinery. (A) The functions of the ATG12 and LC3 ubiquitin-like protein conjugation systems are required for normal autophagosome formation. The ATG12 ubiquitin-like conjugation system leads to formation of the ATG12–ATG5–ATG16L multimeric complex, which serves as an E3-like enzyme for lipidation of LC3 at phagophore, facilitating autophagosome maturation. (B) Vaccinia virus infection causes ATG12–ATG3 conjugation, which may have both E2- and E3-like enzyme activity. The ATG12–ATG3 complex results in massive aberrant LC3 lipidation which is dislocated from phagophore, causing failure in autophagosome formation.
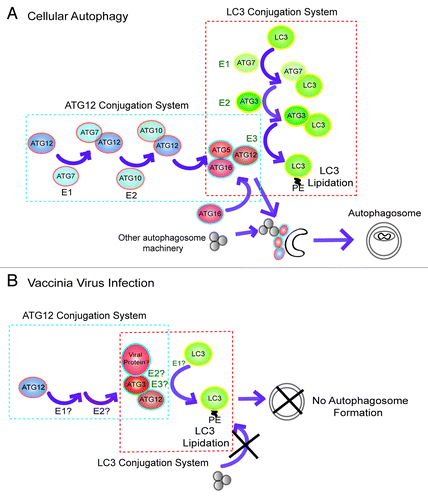
The conjugation between ATG12 and ATG3 has been recently identified by Jayanta Debnath’s group and it was found to be functionally involved in regulation of mitochondrial homeostasis.Citation47 It is therefore possible that vaccinia virus might have exploited the endogenous molecular system. It must be noted that the transient expression of an ATG12–ATG3 fusion protein by itself failed to increase LC3 lipidation in either wild-type or atg5−/− cells (Data not shown). Thus, it appears that the function of the novel ATG12–ATG3 conjugate depends on cellular content and additional viral components must have contributed to the aberrant LC3 lipidation and autophagy deficiency. Importantly, the virus-induced LC3 lipidation is independent of ATG7, the E1-like enzyme for normal cellular autophagosome formation. Alternative E1-like enzyme is therefore required for the completion of the chemical reaction. Further studies are necessary to dissect the molecular interactions and their functional consequences between vaccinia virus and cellular autophagy.
Materials and Methods
Cells and viruses
Wild-type, atg3−/−,Citation48 atg5−/− Citation49 and atg7−/− Citation50 mouse embryonic fibroblast (MEF) cells were generous gifts from Drs. Komatsu and Mizushima. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 100 units of penicillin/ml, 100 µg/ml of streptomycin and 0.29 mg/ml of L-glutamine at 37°C and 5% CO2. A modified version of wild-type vaccinia virus was employed as previously described.Citation22
Antibodies
The primary antibodies used are as follows: goat polyclonal anti-actin (Santa Cruz Biotechnology, sc1616), rabbit polyclonal anti-Atg12 (Cell Signaling Technology, 2011S), rabbit polyclonal anti-Atg7 (Cell Signaling Technology, 2631S) and rabbit polyclonal anti-Atg3 (Abgent, AP1807a). The antibody against LC3 was raised in rabbits using full-length LC3 as antigen.Citation51 The horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology with the exception of goat anti-rabbit secondary antibody (Bio-Rad, 170-6515). The Alexa Fluor 488 goat anti-rabbit fluorescent dye conjugated secondary antibody was purchased from Invitrogen (A-11008).
Construction of vectors and cell lines
The mouse full-length Atg12 cDNA was purchased from Open Biosystems (MMM1013-9498108). The Atg12 PCR product was subcloned into the multicloning site of pIRES1-FHneo under transcriptional control of the CMV IE promoter. The pIRES1neo had previously been modified to contain the coding sequences of FLAG and HA epitope tags and the resulting plasmid is referred to as pIRES1-FHneo.Citation52 The construct was transfected into NIH-3T3 cells employing Lipofectamine reagent (Invitrogen, 18324-020). Selection in media supplemented with 500 µg/ml geneticin (Invitrogen, 11811-031) began 48 h following transfection and colonies were picked after 2 weeks of selection. Colonies were expanded to a 12-well plate and tested for the ability to express FLAG-HA-ATG12 by western blot probing with HA antibody. The clones that express the tagged version of ATG12 were selected.
Transfections and infections
Cells were seeded in six-well plates (35-mm) at 2.5 × 105 cells per well or in 100-mm plates at 2.0 × 106 cells per plate for transfection the next day. Transfections were completed with 2, 5 or 10 µg of plasmid DNA using Lipofectamine or Lipofectamine 2000 reagent (Invitrogen, 11668-019) and incubated for 5 h at 37°C. Cells were allowed to recover overnight and treated as indicated prior to harvesting. Cells were seeded in six-well plates at approximately 2.5 × 105 cells per well for vaccinia virus infection the next day. Cells were inoculated with virus at a multiplicity of infection (MOI) as indicated and allowed to incubate at 37°C for the times indicated.
Protein extraction and immunoblotting
Under the conditions and indicated time points, cells were harvested with lysis buffer containing 10 mM TRIS-HCl [pH 7.9], 10% glycerol, 0.1 mM EDTA, 100 mM KCl, 0.2% NP-40, 0.5 mM PMSF, 1 mM DTT and mini-complete protease inhibitor cocktail (Roche, 11836153001). Lysates were syringed with 23 gauge ¾ inch needles on ice and nuclei and insoluble debris were pelleted in an Eppendorf microcentrifuge at 10,000 rpm for 2 min at 4°C. Cell extracts were then stored at -20°C or immediately subjected to sodium dodecyl sulfate-PAGE (SDS-PAGE). Cell extracts were mixed with 2× Laemmli loading buffer and heated at 95°C for 5 min prior to electrophoresis on 16% SDS-polyacrylamide gels. For immunoblotting, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, IPVH00010) for 2.5 h or overnight. Prior to incubating with primary antibody, membranes were blocked with 5% milk in phosphate-buffered saline (PBS) supplemented with 0.1% (vol/vol) Tween-20 (PBST) for one h at room temperature. Chemiluminescent detection was completed with ECL western blotting reagents (Amersham, 95038-566). Quantifying LC3-II and actin was determined by measuring band intensity using ImageJ software and used to calculate the ratio of LC3-II to actin.
Mass spectrometry
Lysates were immunoprecipitated with α-FLAG M2 affinity gel beads (Sigma, A-2220) and eluted with FLAG peptide (Sigma, F3290). The final eluate was separated by SDS-PAGE and visualized with SilverSNAP stain for mass spectrometry kit (Thermo Scientific, 24600). Bands of interest were in-gel tryptically digested and subject to mass spectrometry performed by the Biological Mass Spectrometry Facility at UMDNJ-Robert Wood Johnson Medical School and Rutgers, The State University of New Jersey.
RNA purification
Total RNA was isolated from wild-type and atg3−/− MEF cells using Absolutely RNA Miniprep Kit (Agilent Technologies, 400800). Reverse transcription was performed with 2 μg of total RNA using the AffinityScript QPCR cDNA Synthesis Kit (Agilent Technologies, 600559) employing atg3 specific primers: 5′-ATGCAGAATGTGATCAACACG-3′ and 5′-CTACATTGTGAAGTGTCTTGTG-3′.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for 15 min, permeabilized in 0.2% Triton X-100 for 10 min and subject to blocking buffer (10% BSA in PBST) for 1 h. Cells were stained overnight with primary antibody diluted in 1% BSA in PBST followed by a fluorescent dye-conjugated secondary antibody diluted in 1% BSA in PBST for 1 h. Cells were counterstained with 50 ng/ml of DAPI (Invitrogen, D9542) diluted in 1% BSA in PBST. Cells were mounted using Citifluor and analyzed by fluorescent microscopy. Imaging was performed using a 100× objective of a Zeiss Axioplan 2 microscope equipped with a Zeiss AxioCam camera and mercury lamp. Images were acquired using Openlab software.
Viral growth analysis
Human osteosarcoma cells, 143B, were added to six-well plates at a concentration of 8 × 105 per well and incubated overnight at 37°C in DMEM containing 5% FBS. The following day, when the cells had reached confluency, the medium was removed and replaced with 1.5 ml of IMDM containing 0.1% BSA. The virus inoculum was then added to the wells and allowed to infect for 2 h before the supernatant was removed and the cells overlaid with 3 ml of a 1:1 mixture of 2× DMEM / 5% FBS and 2% agarose. After a 2-d incubation, an additional 1 ml of 2× DMEM / 2% agarose containing 200 µg X-gal was added to each well. The following day the blue plaques were counted and the virus titer calculated. Virus inoculum was adjusted, by trial and error, to give between 5 and 30 plaques per well and the virus titer was determined from a series of inoculum dilutions and/or volumes generated in triplicate.
To determine the levels of intracellular virus, virus-infected cells were harvested at indicated time points by first removing the supernatant, then collecting the cells by scraping into PBS containing 0.1% BSA. The virus was released from the cells by three freeze-thaw cycles followed by sonication. The inoculum used for the plaque assay was prepared by low-speed centrifugation to remove cellular debris and PBS / 0.1% BSA was used to make serial dilutions.
To determine the levels of extracellular virus, the supernatant was collected from virus-infected cells at the indicated time points and the inoculum was prepared by low speed centrifugation of the supernatant to remove detached cells and debris. Serial dilutions of this inoculum were made using PBS / 0.1% BSA.
Electron microscopy
Electron microscopy was performed by the University of Medicine and Dentistry of New Jersey (UMDNJ) electron microscopy core facility. Wild-type and atg3−/− MEFs were treated under the conditions indicated, fixed and embedded. Sections were cut and examined with a JOEL 1200EX transmission electron microscope. Cells were counted and scored for the presence of autophagosomes and the number of autophagosomes per cell. P values were calculated by Student’s t test.
Analysis of GFP-LC3
NIH-3T3 fibroblasts stably expressing GFP-LC3 (rat) were grown on coverslips prior to Hanks’ media starvation or vaccinia virus infection. Cells were fixed with 4% paraformaldehyde, washed with PBS, mounted using Citifluor (Ted Pella, Inc.) and analyzed by fluorescent microscopy. At least 200 cells from three independent experiments were scored.
Autophagy flux analysis with bafilomycin A1 treatment
Fibroblast cells either untreated or infected with vaccinia virus (8 h post infection) were treated with 100 ng/ml bafilomycin A1. Cells were harvested at various time points, cell lysates were separated by SDS-PAGE, immunoblotted with anti-serum against LC3.
Vaccinia virus ORF library
The vaccinia virus ORF library was purchased from Addgene. The library is a collection of open reading frames from the vaccinia virus genome and the individual ORFs were subcloned into pcDNA 3.1D V5-His-TOPO mammalian expression vectors. The kit was shipped as bacterial glycerol stocks in a 96-well format. All plasmids were subsequently purified via a miniprep kit purchased from Qiagen (27106).
| Abbreviations: | ||
| ATG | = | autophagy related |
| IV | = | immature virus |
| EEV | = | extracellular enveloped virus |
| MOI | = | multiplicity of infection |
| ORF | = | open reading frame |
| pfu | = | plaque forming unit |
Additional material
Download Zip (629.2 KB)Acknowledgments
The authors thank Dr. Komatsu for the atg3−/− and atg7−/− cell lines, Dr. Mizushima for the atg5−/− cell line, Rajesh Patel for technical support on electron microscopy, and Haiyan Zheng for technical support on mass spectrometry. The research is supported by NIH grants: 1R01CA116088 (J.G.M. and S.J.), 1R01AG030081 (H.T. and S.J.), and R01CA42908 (E.C.L.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol 2002; 83:2915 - 31; PMID: 12466468
- Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. Protein composition of the vaccinia virus mature virion. Virology 2007; 358:233 - 47; http://dx.doi.org/10.1016/j.virol.2006.08.025; PMID: 17005230
- Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van 't Hof W, et al. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol 1993; 121:521 - 41; http://dx.doi.org/10.1083/jcb.121.3.521; PMID: 8486734
- Dales S, Mosbach EH. Vaccinia as a model for membrane biogenesis. Virology 1968; 35:564 - 83; http://dx.doi.org/10.1016/0042-6822(68)90286-9; PMID: 5677800
- Risco C, Rodriguez JR, Lopez-Iglesias C, Carrascosa JL, Esteban M, Rodriguez D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J Virol 2002; 76:1839 - 55; http://dx.doi.org/10.1128/JVI.76.4.1839-1855.2002; PMID: 11799179
- Rodriguez D, Barcena M, Mobius W, Schleich S, Esteban M, Geerts WJ, et al. A vaccinia virus lacking A10L: viral core proteins accumulate on structures derived from the endoplasmic reticulum. Cell Microbiol 2006; 8:427 - 37; http://dx.doi.org/10.1111/j.1462-5822.2005.00632.x; PMID: 16469055
- Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ 2009; 16:956 - 65; http://dx.doi.org/10.1038/cdd.2009.39; PMID: 19373247
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1 - 32; http://dx.doi.org/10.1007/978-3-642-00302-8_1; PMID: 19802558
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol 1994; 124:903 - 13; http://dx.doi.org/10.1083/jcb.124.6.903; PMID: 8132712
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433 - 7; http://dx.doi.org/10.1038/ncb1991; PMID: 19898463
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 67; http://dx.doi.org/10.1016/j.cell.2010.04.009; PMID: 20478256
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747 - 57; http://dx.doi.org/10.1038/ncb2078; PMID: 20639872
- van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell 2010; 21:2270 - 84; http://dx.doi.org/10.1091/mbc.E09-04-0345; PMID: 20444982
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature 1998; 395:395 - 8; http://dx.doi.org/10.1038/26506; PMID: 9759731
- Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 2004; 15:231 - 6; http://dx.doi.org/10.1016/j.semcdb.2003.12.004; PMID: 15209383
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. 'Protein modifications: beyond the usual suspects' review series. EMBO Rep 2008; 9:859 - 64; http://dx.doi.org/10.1038/embor.2008.163; PMID: 18704115
- Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem 2001; 276:1701 - 6; PMID: 11096062
- Mizushima N, Yoshimori T, Ohsumi Y. Mouse Apg10 as an Apg12-conjugating enzyme: analysis by the conjugation-mediated yeast two-hybrid method. FEBS Lett 2002; 532:450 - 4; http://dx.doi.org/10.1016/S0014-5793(02)03739-0; PMID: 12482611
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem 2004; 279:36268 - 76; http://dx.doi.org/10.1074/jbc.M401461200; PMID: 15187094
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679 - 88; http://dx.doi.org/10.1242/jcs.00381; PMID: 12665549
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092 - 100; http://dx.doi.org/10.1091/mbc.E07-12-1257; PMID: 18321988
- Zhang H, Monken CE, Zhang Y, Lenard J, Mizushima N, Lattime EC, et al. Cellular autophagy machinery is not required for vaccinia virus replication and maturation. Autophagy 2006; 2:91 - 5; PMID: 16874104
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 1998; 273:33889 - 92; http://dx.doi.org/10.1074/jbc.273.51.33889; PMID: 9852036
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009; 10:461 - 70; http://dx.doi.org/10.1038/ni.1726; PMID: 19381141
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. Virgin HWt. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259 - 63; http://dx.doi.org/10.1038/nature07416; PMID: 18849966
- Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle 2010; 9:1295 - 307; http://dx.doi.org/10.4161/cc.9.7.11109; PMID: 20305376
- Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 2009; 335:323 - 33; http://dx.doi.org/10.1007/978-3-642-00302-8_16; PMID: 19802573
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Orvedahl A, MacPherson S, Sumpter R Jr., Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010; 7:115 - 27; http://dx.doi.org/10.1016/j.chom.2010.01.007; PMID: 20159618
- Tallóczy Z, Jiang W. Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 2002; 99:190 - 5; http://dx.doi.org/10.1073/pnas.012485299; PMID: 11756670
- Tallóczy Z. Virgin HWt, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006; 2:24 - 9; PMID: 16874088
- Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005; 121:567 - 77; http://dx.doi.org/10.1016/j.cell.2005.03.007; PMID: 15907470
- Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol 2009; 81:1241 - 52; http://dx.doi.org/10.1002/jmv.21502; PMID: 19475621
- Jackson WT, Giddings TH Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005; 3:e156; http://dx.doi.org/10.1371/journal.pbio.0030156; PMID: 15884975
- Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255 - 68; http://dx.doi.org/10.1083/jcb.200903070; PMID: 19635843
- Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008; 374:240 - 8; http://dx.doi.org/10.1016/j.virol.2008.02.016; PMID: 18353420
- O'Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011; 410:142 - 50; http://dx.doi.org/10.1016/j.virol.2010.10.042; PMID: 21112602
- Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004; 279:10136 - 41; http://dx.doi.org/10.1074/jbc.M306124200; PMID: 14699140
- Schiøtz BL, Roos N, Rishovd AL, Gjoen T. Formation of autophagosomes and redistribution of LC3 upon in vitro infection with infectious salmon anemia virus. Virus Res 2010; 151:104 - 7; http://dx.doi.org/10.1016/j.virusres.2010.03.013; PMID: 20350574
- Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol 2007; 81:12543 - 53; http://dx.doi.org/10.1128/JVI.00755-07; PMID: 17804493
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, et al. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 2008; 82:9143 - 53; http://dx.doi.org/10.1128/JVI.00641-08; PMID: 18596087
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7:767 - 77; http://dx.doi.org/10.1038/nri2161; PMID: 17767194
- Sumpter R Jr., Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin Cell Dev Biol 2010; 21:699 - 711; http://dx.doi.org/10.1016/j.semcdb.2010.04.003; PMID: 20403453
- Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010; 142:590 - 600; http://dx.doi.org/10.1016/j.cell.2010.07.018; PMID: 20723759
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008; 19:4762 - 75; http://dx.doi.org/10.1091/mbc.E08-03-0309; PMID: 18768753
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Jin S, Kalkum M, Overholtzer M, Stoffel A, Chait BT, Levine AJ. CIAP1 and the serine protease HTRA2 are involved in a novel p53-dependent apoptosis pathway in mammals. Genes Dev 2003; 17:359 - 67; http://dx.doi.org/10.1101/gad.1047003; PMID: 12569127