Abstract
Autophagy is a catabolic process that provides the degradation of altered/damaged organelles through the fusion between autophagosomes and lysosomes. Proper regulation of the autophagic flux is fundamental for the homeostasis of skeletal muscles in physiological conditions and in response to stress. Defective as well as excessive autophagy is detrimental for muscle health and has a pathogenic role in several forms of muscle diseases. Recently, we found that defective activation of the autophagic machinery plays a key role in the pathogenesis of muscular dystrophies linked to collagen VI. Impairment of the autophagic flux in collagen VI null (Col6a1–/–) mice causes accumulation of dysfunctional mitochondria and altered sarcoplasmic reticulum, leading to apoptosis and degeneration of muscle fibers. Here we show that physical exercise activates autophagy in skeletal muscles. Notably, physical training exacerbated the dystrophic phenotype of Col6a1–/– mice, where autophagy flux is compromised. Autophagy was not induced in Col6a1–/– muscles after either acute or prolonged exercise, and this led to a marked increase of muscle wasting and apoptosis. These findings indicate that proper activation of autophagy is important for muscle homeostasis during physical activity.
Introduction
Autophagy can be defined as a process of cytosolic ‘renovation,’ crucial in cell fate decisions.Citation1,Citation2 The autophagic/lysosome system is involved in the degradation of organelles and long-lived proteins and in the clearance of damaged cell components. Furthermore, the autophagic machinery is able to degrade part of the cell material in order to fulfill energetic needs during starvation and stress.Citation3,Citation4 In skeletal muscles a proper autophagic flux is critical to maintain myofiber integrity, and inhibition or perturbation of autophagy contributes to muscle degeneration and wasting.Citation5 The critical function of this process for skeletal muscles is confirmed by the observation that excessive autophagy induces muscle atrophy, while an impairment of the process leads to muscle weakness and myofiber degeneration.Citation6 Therefore, a correct balance between activation and inhibition of autophagy is fundamental for muscle homeostasis.
Collagen VI is a major extracellular matrix protein forming an extended network of beaded microfilaments that is particularly abundant in the endomysium of skeletal muscles.Citation7 Mutations in any of the three genes coding for collagen VI cause several muscle diseases in humans, including Ullrich congenital muscular dystrophy (UCMD), Bethlem myopathy and congenital myosclerosis.Citation8,Citation9 We previously demonstrated that collagen VI null (Col6a1–/–) mice display an early onset myopathic phenotype affecting diaphragm and other skeletal muscles and characterized by myofiber degeneration, spontaneous apoptosis and decreased muscle strength.Citation10 A key pathogenic mechanism involved in the onset of these muscle defects is the impaired activation of the autophagic machinery, leading to accumulation of dysfunctional organelles in Col6a1–/– myofibers.Citation11 The same pathogenic features are present in UCMD patients,Citation11,Citation12 however the clinical features and the progression of the human syndrome are much more severe than the corresponding animal model.
Exercise has been found to be either beneficial or detrimental for muscle function. The beneficial effects of exercise are at least in part mediated by the influence that physical activity has on promoting protein synthesis and reducing protein degradation, thus resulting in an increase of muscle mass and force. The mechanical sensors that can sense the tension and adapt muscle cells are only partially known. One of the most attractive sources is placed in the sarcomere, the basic unit of the contractile machinery of striated muscles, and from this site signals are transmitted to the nucleus to affect gene expression. The giant elastic protein titin, which spans half of the sarcomere extending from the Z-disk to the M-band and interacts with a large number of muscle proteins, provides an exciting example of a sarcomeric activity-dependent signaling complex (signalosome).Citation13 A unique property of titin is the presence in the M-band region of a serine/threonine kinase domain that can be induced to acquire an open active conformation by stretch and contraction.Citation14 In active muscle cells, the titin kinase domain is recruiting a complex formed by the two autophagy-related proteins p62/SQSTM1 and Nbr1 together with the ubiquitin ligases MuRF1 or MuRF2. This complex dissociates during mechanical arrest resulting in nuclear translocation of p62, MuRF1 and MuRF2. Nuclear localization of MuRF2 allows the interaction with the serum response transcription factor SRF, leading to nuclear export of SRF and loss of SRF-dependent gene expression.Citation15 Moreover, since Nbr1 and p62 promote the autophagy of polyubiqitinated proteins by interacting with LC3, their release from the complex may promote the lysosomal-dependent degradation of sarcomeric proteins.Citation16
In this study, we have investigated the effects of acute and chronic physical exercise on autophagy and tissue integrity of limb and respiratory muscles of wild-type mice and in Col6a1–/– animals.
Results
Long-term voluntary exercise is detrimental for Col6a1–/– muscles
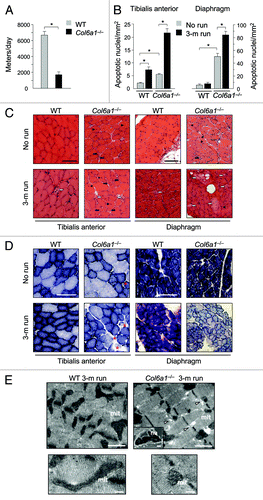
To explore the effects of physical training, we subjected in parallel Col6a1–/– and wild-type mice to muscle training by voluntary exercise on running wheels. Interestingly, Col6a1–/– mice were found to cover significantly shorter distances than wild-type littermates, and they also displayed a gradual decline of their locomotory activity ( and data not shown). Running was continued for three months and at the end of the experiments diaphragm and tibialis anterior (TA) muscles were analyzed and compared with muscles of nonexercised littermates. Long-term exercise had a dramatic impact on Col6a1–/– muscles, leading to myofiber degeneration and muscle wasting (). Indeed, the incidence of myofibers with apoptotic nuclei was markedly increased in the diaphragm and TA of exercised Col6a1–/– mice when compared with nonexercised Col6a1–/– animals (). Histological staining revealed that abnormal myofibers were rare in wild-type muscles even after prolonged physical activity. Conversely, prolonged exercise caused extensive myofiber damage in Col6a1–/– muscles. In particular, Col6a1–/– TA showed a marked increase of centrally nucleated fibers, a marker of myofiber death and regeneration. These fibers, as well as small atrophic fibers, were abundant in exercised Col6a1–/– TA muscles. Col6a1–/– diaphragm showed an increase of necrotic fibers and of inflammation with exercise (). Long-term physical activity induced an increase of mitochondrial content in limb muscles. Indeed, the number of β-oxidative fibers, as revealed by succinate dehydrogenase (SDH) staining, was enhanced in TA. This result was expected given the intense workload of limb muscles during exercise when compared with diaphragm. Col6a1–/– TA showed abnormalities of SDH staining likely reflecting alterations of mitochondrial network. Diaphragm of long-term exercised Col6a1–/– mice displayed a much fainter SDH staining when compared with nonexercised littermates, suggesting a decreased mitochondrial activity (). Electron microscope analysis of the diaphragm confirmed that 3-mo running caused extensive myofiber damage in Col6a1–/– mice, while no detrimental effect was detected in wild-type mice ().
Figure 1. Long-term physical exercise is detrimental for Col6a1–/– muscles. (A) Histogram showing the mean distance covered daily by wild-type and Col6a1–/– mice under voluntary running wheel exercise (*p < 0.001, n = 6 each genotype). Error bars indicate s.e.m. (B) Incidence of apoptosis in TA (left) and diaphragm (right) muscles of 5-mo-old wild-type and Col6a1–/– mice housed either in standard conditions (no run) or after 3-mo running (3-m run) wheel exercise (*p < 0.001, n = 6 each group). Error bars indicate s.e.m. (C) Representative H & E staining of cross-sections of TA and diaphragm muscles isolated from wild-type and Col6a1–/– mice in standard conditions (no run) or after 3-mo run wheel exercise. Under standard conditions, Col6a1–/– muscles display various alterations, with centrally nucleated fibers (black arrowheads) and atrophic myofibers (black arrows) in both diaphragm and TA, and signs of inflammation (white asterisk) in diaphragm. After three months of exercise, only a few degenerating myofibers (white arrows) are detected in wild-type muscles, whereas in Col6a1–/– TA centrally nucleated, atrophic and degenerated myofibers are markedly increased. Three months of exercise leads to extensive myofiber degeneration in Col6a1–/– diaphragm. (D) Representative cross-sections of TA and diaphragm muscles stained for SDH. Histochemical reaction for SDH produces a purple-blue staining with a speckled pattern, which is proportional to the number of mitochondria and their oxidative activity. In resting conditions (no run), most myofibers of TA show scattered staining of variable intensities, due to the predominantly glycolytic metabolic activity of this muscle, while the diaphragm shows intense blue staining in all myofibers, reflecting its oxidative metabolism. After exercise (3-m run), Col6a1–/– mice display several abnormally and poorly stained fibers in TA (red asterisks) and a diffuse pale blue staining of diaphragm. Scale bar, 100 μm. (E) Electron micrographs of diaphragm from wild-type and Col6a1–/– mice after 3 mo of running wheel exercise. Col6a1–/– diaphragm displays several ultrastructural defects, while wild-type diaphragm maintains a normal structure. Col6a1–/– diaphragm presents a marked accumulation of dilated sarcoplasmic reticulum (arrowheads), abnormal mitochondria and apoptotic nuclei (upper panels). Many Col6a1–/– mitochondria appear fragmented and some of them with altered cristae, while wild-type mitochondria maintain elongated shape and normal cristae morphology (lower panels). Scale bars, 1 μm (upper panels) or 200 nm (lower panels). mit, mitochondria; nu, nuclei; WT, wild-type.
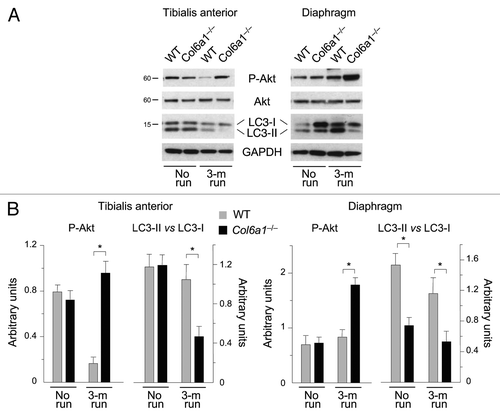
We next investigated autophagy activity in muscles after prolonged exercise. Interestingly, LC3 lipidation was significantly decreased in trained Col6a1–/– mice. Indeed, western blot analysis showed that the LC3-II band was less abundant in both TA and diaphragm of trained collagen VI null animals (). This was particularly evident for TA, where the LC3-II vs. LC3-I ratio was markedly reduced when compared with the corresponding samples of untrained Col6a1–/– animals (). Noticeably, in wild-type muscles the LC3-II vs. LC3-I ratio was not significantly different between trained and untrained animals. Since we previously found that excessive Akt activity inhibits autophagy and contributes to muscle pathology,Citation11,Citation17 we investigated Akt in muscles subjected to long-term wheel exercise. Western blot showed that Akt phosphorylation was increased in both TA and diaphragm of trained Col6a1–/– muscles (). These data indicate that prolonged exercise has a negative impact on the autophagic defect of collagen VI deficient-mice and exacerbates their muscle phenotype.
Figure 2. Autophagy is impaired in Col6a1–/– muscles after long-term physical exercise. (A) Western blot analysis for Akt phosphorylation and LC3 lipidation in TA (left) and diaphragm (right) of 5-mo-old wild-type and Col6a1–/– mice housed either in standard conditions (no run) or after 3-mo run wheel exercise. (B) Densitometric quantifications of P-Akt levels and of LC3-II vs LC3-I ratio, as determined by western blots of TA (left) and diaphragm (right) (*p < 0.05, n = 3).
Treadmill exercise induces autophagy in wild-type but not in Col6a1–/– muscles
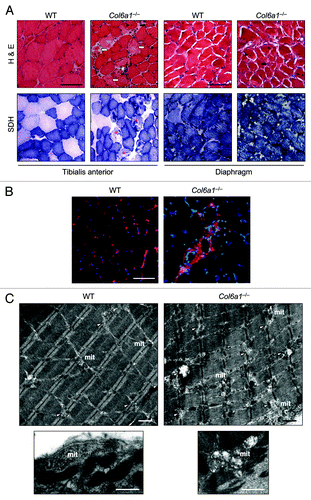
Although the above data indicate that physical activity worsened Col6a1–/– phenotype and concurrently suppressed autophagy, they were obtained by a long-term running protocol where skeletal muscle tissue is likely to produce adaptive responses. Therefore they cannot provide unequivocal evidence for a role of autophagy activation in supporting muscle needs during exercise. In order to address this issue, we performed a different set of experiments where mice were subjected to intense physical activity for short time. We used a treadmill to perform 1 h of training at increasing speed and then muscles were collected and analyzed. Histological analysis indicated that acute exercise caused extensive muscle damage in Col6a1–/– TA (). The severe wasting of Col6a1–/– TA was confirmed by the presence of several degenerating myofibers and of various areas displaying tissue inflammation. SDH staining revealed the presence of myofibers with altered mitochondrial network in Col6a1–/– TA (). One-hour treadmill did not affect tissue morphology of wild-type muscles and showed no evidence of myofiber degeneration or tissue inflammation in wild-type TA (). Electron microscope analysis of TA clearly confirmed that treadmill led to severe myofiber wasting in Col6a1–/– mice (). Almost all myofibers of Col6a1–/– TA showed marked ultrastructural alterations of sarcoplasmic reticulum and mitochondria, without any remarkable difference between oxidative and glycolytic fibers. Sarcoplasmic reticulum appeared highly dilated along all the myofiber length, while mitochondria were fragmented and many of them displayed completely disorganized cristae. Swollen mitochondria were detected in the entire myofiber area, and they were found both among sarcomeres and under the sarcolemma (andFig. S1). Conversely, wild-type TA showed some dilated sarcoplasmic reticulum, but these dilations were much less marked when compared with Col6a1–/– muscles. Some mitochondria of wild-type TA appeared abnormal, but the majority of them had a normal elongated shape with a correct morphology of the cristae ().
Figure 3. Treadmill exercise causes muscle wasting in Col6a1–/– mice. (A) Representative cross-sections of TA and diaphragm muscles stained with H & E or with histochemical reaction for SDH and isolated from 5-mo-old wild-type and Col6a1–/– mice after 1 h treadmill exercise. H & E staining shows normal morphology in both muscles of wild-type animals after treadmill. Instead, TA from Col6a1–/– mice after treadmill displays severe muscle degeneration characterized by inflammation (white asterisk), degenerating myofibers (white arrows), centrally nucleated fibers (black arrowheads), and atrophic myofibers (black arrows). The extent of muscle degeneration in the diaphragm of Col6a1–/– mice after treadmill is similar to that observed in standard conditions. SDH staining of TA and diaphragm is normal in wild-type mice after treadmill. In Col6a1–/– animals subjected to treadmill several abnormal poorly stained fibers (red asterisks) are present in TA, while the diaphragm displays an apparently normal SDH staining. Scale bar, 100 μm. (B) Representative IgG immunolabeling images of TA cross-sections from wild-type and Col6a1–/– mice after 1 h of treadmill exercise, confirming extensive inflammation in Col6a1–/– TA but not in the corresponding wild-type samples. Scale bar, 100 μm. (C) Electron micrographs of TA from wild-type and Col6a1–/– mice after treadmill exercise. Col6a1–/– TA displays a large number of severe ultrastructural alterations, while wild-type TA presents fibers with dilated sarcoplasmic reticulum and some altered mitochondria. Col6a1–/– TA displays a marked accumulation of dilated sarcoplasmic reticulum (arrowheads) and abnormal mitochondria. Almost all Col6a1–/– mitochondria appear fragmented and many of them with completely swollen cristae, while the majority of wild-type mitochondria maintain elongated shape and normal cristae morphology (lower panels). Scale bars, 1 μm. mit, mitochondria; WT, wild-type.
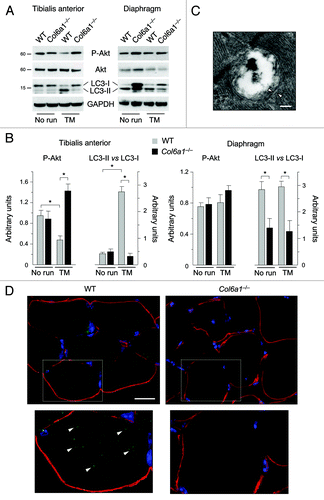
Remarkably, 1 h treadmill had a potent effect on LC3 lipidation in wild-type limb muscles, as indicated by the abundant conversion of LC3-I to LC3-II and the presence of autophagosomes in wild-type TA (). In agreement with the intense workload of limb muscles during this training protocol, it is worthy to point out that autophagy was markedly activated in wild-type TA but not in wild-type diaphragm. Treadmill exercise confirmed an impaired autophagy in Col6a1–/– muscles, as shown by the lower LC3 lipidation which was not different from that of untrained Col6a1–/– muscles. To further investigate the impairment of autophagy in collagen VI null TA, we generated Col6a1–/–;GFP-LC3 mice by breeding Col6a1–/– animals with GFP-LC3 transgenic mice.Citation18 We subjected Col6a1–/–;GFP-LC3 mice and their corresponding wild-type controls to treadmill exercise and then we investigated the presence of GFP-LC3 puncta inside myofibers. In agreement with the LC3 lipidation shown by western blot, many fluorescent autophagic puncta were clearly detectable in wild-type;GFP-LC3 TA. Moreover, GFP-LC3 puncta were found in both glycolytic and oxidative fibers. Interestingly, some autophagic puncta merged with the mitochondrial network suggesting that mitophagy was activated. Conversely, only a few puncta with faint labeling were present in Col6a1–/–;GFP-LC3 muscles (andFig. S2). Finally, 1 h treadmill led to increased Akt phosphorylation in TA of trained Col6a1–/– mice but caused a decrease of Akt activity in wild-type TA (). These data indicate that short-term intense exercise by treadmill activates autophagy in healthy limb muscles. However, strenuous exercise does not restore autophagy in Col6a1–/– muscles and indeed further contributes to myofiber degeneration.
Figure 4. Treadmill exercise induces autophagy in wild-type muscle but not in Col6a1–/– mice. (A) Western blot analysis for Akt phosphorylation and LC3 lipidation in TA (left) and diaphragm (right) muscles from 5-mo-old wild-type and Col6a1–/– mice housed in standard conditions (no run) or after 1 h treadmill exercise (TM). (B) Densitometric quantifications of P-Akt levels and LC3-II vs LC3-I ratio, as determined by western blots of TA (left) and diaphragm (right) (*p < 0.05, n = 3). (C) Electron micrograph of double-membrane (arrowheads) autophagosome in TA of wild-type mice after treadmill exercise. Scale bar, 200 nm. (D) Fluorescence microscope analysis of cryosections of TA from wild-type;GFP-LC3 and Col6a1–/–;GFP-LC3 mice after treadmill exercise. GFP-LC3 puncta (green fluorescence) are abundant in wild-type muscle fibers, but they are scarce and poorly detectable in Col6a1–/– myofibers. Immunohistochemistry for laminin (red fluorescence) was performed to reveal myofiber outline, while Hoechst staining (blue) was used to label nuclei. The lower panels are higher magnifications of the squared areas, clearly showing GFP-LC3 puncta (arrowheads) in wild-type but not in Col6a1–/– myofibers. Scale bar, 25 μm. TM, treadmill; WT, wild-type.
Discussion
Our recent data have shown that autophagy flux is compromised in collagen VI-deficient muscles. The defective activation of autophagy in Col6a1–/– muscles causes accumulation of abnormal and dysfunctional mitochondria, which in turn leads to apoptotic degeneration of myofibers.Citation11 Physical exercise has an established role in promoting mitochondrial biogenesis and in improving mitochondrial function.Citation19 Most of these effects are mediated by the action of PGC-1α,Citation20 a transcriptional coactivator that regulates genes involved in energy metabolism. PGC-1α binds and enhances the activity of other transcription factors, such as NRF-1, inducing the expression of mitofusins, TFAM and several mitochondrial-related proteins. We and others have shown that PGC-1α overexpression prevents muscle loss during fasting, chronic cardiac failure and aging.Citation21-Citation24 Since PGC-1α expression is strongly modulated by physical activity,Citation25 we explored whether exercise may improve mitochondrial function and therefore might have beneficial effects on muscles of Col6a1–/– mice. Instead our data strongly support the concept that exercise is detrimental for Col6a1–/– muscles. Interestingly, exercise per se is able to induce autophagy in wild-type animals but it is not sufficient to reactivate the compromised autophagy in myofibers of collagen VI-deficient mice.
The connection between muscle contraction and autophagy can be related to the two most important functions of autophagy, i.e., maintaining the cell free of dysfunctional organelles and providing new sources for energy.Citation1-Citation4 It is well established that physical activity and muscle contraction cause an increase in ROS production and oxidative stress.Citation26 Indeed, oxidative stress has an important role in muscle signaling but can also be a damaging factor for contractile proteins and organelles. Therefore, induction of autophagy during acute training is required to remove dangerous unfolded proteins as well as dysfunctional organelles in muscle fibers. Moreover, autophagy is critical to produce alternative sources of energy in order to maintain the basal homeostasis of muscle cells during energetic stress. Since autophagy flux is deficient in Col6a1–/– muscles and is not increased during physical exercise, this defect enhances myofiber instability as a result of accumulation of damaged mitochondria. Further mechanisms may contribute to the aggravated muscle pathology of Col6a1–/– mice after physical exercise. Collagen VI is involved in a broad range of interactions with cell surface molecules, including several integrins and nonintegrin receptors, and with other extracellular matrix components.Citation7 These interactions and the close association of beaded microfilaments with muscle basal lamina suggest that collagen VI provides an important mechanical link of myofibers to the surrounding matrix. Impaired cell-extracellular matrix contacts, failure of activation of membrane-cytoskeleton associated mechanosensors and sarcolemma defects may contribute to the pathomolecular mechanisms underlying muscle damage of Col6a1–/– mice. Indeed, various studies indicate that cell membrane receptors and cytoskeletal proteins integrate a host of signaling functions in muscle mechanotransduction, and distinct signaling pathways can be activated in response to mechanical stress applied to skeletal muscle fibers.Citation27,Citation28 Therefore, absence of autophagy activation by exercise in Col6a1–/– muscles is compatible with defects in membrane- and/or cytoskeleton-associated mechanosensors. Interestingly, recent in vitro studies indicate that extracellular matrix components are able to modulate autophagy in different manners, depending on the specific matrix composition.Citation29,Citation30
Slow- and fast-twitched fibers display distinct mechanical behavior, metabolism and signaling pathways. Differences in loading, mitochondrial content and calcium homeostasis affect the susceptibility or resistance of muscle to damage during contraction. Importantly, both energetic and oxidative stresses are more pronounced in lower limb muscles than in the diaphragm during exercise. In fact the diaphragm is not recruited during limb movement and is not loaded by the body weight. Therefore, the mechanical and metabolic involvement is more pronounced in TA, resulting in a more dramatic increase of apoptotic nuclei in Col6a1–/– TA (about 4 fold) than in Col6a1–/– diaphragm (about 1.5 fold) (see also ). Histological analysis confirms prominent myofiber damage in Col6a1–/– TA after acute exercise. It is worthwhile to underline that myofibers are syncytial cells containing several hundred nuclei, thus the progressive loss of nuclei can trigger instability leading to myofiber degeneration.
In our previous work we have shown that Akt blocks both autophagy-lysosome and ubiquitin-proteasome systems.Citation11,Citation17 In this study we could also confirm the inverse correlation between Akt activity and autophagy induction. Indeed Akt is hyperactivated in Col6a1–/– muscles during both chronic and acute exercise, blocking autophagosome formation. The fact the Akt is dephosphorylated in wild-type muscles after acute exercise may seem unexpected. We and others have shown that the IGF-1/Akt pathway is important for muscle hypertrophyCitation31,Citation32 and therefore should be activated by exercise. However, the IGF-1 pathway contains several feedback and feed-forward loops that regulate the activity of critical components, such as Akt, and that can explain our data. For instance, activation of IGF-1 signaling leads to a negative feedback mechanism via S6K that reduces IRS1 activity, leading to decreased Akt phosphorylation.Citation33 Another regulatory mechanism involves TRB3, a pseudokinase that inhibits Akt.Citation34 TRB3 is repressed by FoxO transcription factors that are negatively regulated by Akt.Citation35,Citation36 Therefore, persistent Akt activation would finally induce expression of TRB3, which in turn blocks Akt. Both these mechanisms can explain why Akt is dephosphorylated in treadmill training. It has been reported that an acute bout of contractions leads to Akt activation and increased protein synthesis.Citation37 However, when contraction is prolonged, the sustained activation of the pathway results in Akt inhibition via the negative feedback of S6K and/or via the upregulation of TRB3, consequent to FoxO inhibition.Citation33,Citation34,Citation37 These mechanisms would be important for reducing Akt activity in order to reactivate the degradation pathways and facilitate the removal of damaged proteins and organelles.
In humans, mutations of collagen VI genes cause various inherited disorders of skeletal muscle. The total absence of collagen VI in Col6a1–/– mice highly resembles the marked collagen VI deficiency of UCMD patients.Citation7,Citation38 In spite of this, under standard housing conditions, the muscle pathology and the progression of disease displayed by the murine model are much milder that those of UCMD patients. Remarkably, exercise causes a striking worsening of the phenotype and an increase of myofiber degeneration and death of collagen VI null mice. In particular, running leads to accumulation of damaged organelles in Col6a1–/– muscles. Therefore, physical exercise has a detrimental effect on Col6a1–/– animals and leads to a muscle pathology which is closer to the human UCMD condition, thus representing a useful experimental set-up for understanding the pathophysiological mechanisms and testing and/or validating therapeutic approaches. One key feature of muscles of both Col6a1–/– model and UCMD/BM is defective autophagy. This is due to an impaired activation of the autophagic machinery with defective formation of autophagosomes in Col6a1–/– mice, which becomes unmistakably evident under food starvation, a well-known autophagic stimulus. Interestingly, the autophagic flux is not fully compromised in Col6a1–/– muscles and it can be reactivated by different genetic, pharmacological or dietary means, allowing for removal of dysfunctional organelles and amelioration of muscle pathology with beneficial effects on Col6a1–/– mice.Citation11,Citation39 The results of the present study show that physical exercise stimulates autophagy in muscles of wild-type mice, however this stimulus is not able to trigger autophagy in collagen VI-deficient mice and it has detrimental effects on Col6a1–/– muscles. Thus, the inability to properly activate autophagy in diseased muscles under conditions of intense workload and increased need for energy sources leads to deterioration of muscle fiber defects.
In conclusion, here we reported that physical activity stimulates autophagy in normal muscles but is detrimental for collagen VI deficient muscles, where the autophagic flux is defective. Our data strongly support a view where proper activation of the autophagic machinery in skeletal muscles is crucial for maintaining tissue homeostasis during physical activity.
Materials and Methods
Mice
Col6a1+/– mice were backcrossed in the inbred C57BL/6J strain for eight generations.Citation10 All data were obtained in 16- to 24-week-old mice, by comparing age-matched Col6a1–/– (collagen VI null) and Col6a1+/+ (wild-type) animals. GFP-LC3 miceCitation18 were provided by RIKEN BRC (GFP-LC3#53 strain, RBRC00806). Col6a1–/–;GFP-LC3 mice were generated crossing Col6a1–/– with GFP-LC3 mice. Mice were housed in individual cages in an environmentally controlled room (23°C, 12 h light/12 h dark cycle) and provided food and water ad libitum. For long-term voluntary exercise, 2-mo-old mice were housed individually in cages containing a wheel connected to a sensor able to monitor the distance run by each animal. Before starting the measurements, mice were left to train in the wheels for one month in order to adapt for individual variations in baseline activities. After three months, mice were sacrificed by cervical dislocation and muscles dissected. Treadmill exercise was performed by subjecting mice to forced run in a treadmill device for 1 h. Briefly, mice were first left to adapt to treadmill for 5 min at a speed of 5 cm/sec. After this, exercise was performed by starting with a speed of 10 cm/sec that was increased of 2 cm/sec every 2 min until a maximum of 40 cm/sec. After 1 h, mice were sacrificed by cervical dislocation and muscles dissected.
Western blotting
Muscles were pulverized by grinding in liquid nitrogen, lysed and immunoblotted as previously described.Citation26 When needed, membranes were stripped using a stripping buffer (25 mM glycine, 1% SDS, pH 2.0) and reprobed. The following antibodies from Cell Signaling Technologies were used: rabbit polyclonal anti-Akt (9272); rabbit monoclonal (clone 193H12) anti-phospho-Akt (Ser473) (4058); rabbit polyclonal anti-LC3B (2775). Mouse monoclonal anti-GAPDH (MAB374) was from Chemicon International. Western blots were performed in three independent experiments. Densitometric quantification was performed by the ImageJ software.
TUNEL assay
Sections (7-µm-thick) were prepared from diaphragm, after fixation with 4% paraformaldehyde and paraffin embedding, and from TA frozen in 2-methylbutane. TUNEL was performed with the ApopTag peroxidase in situ apoptosis detection system (Chemicon, S7100). Visualization of all nuclei was obtained by staining with Hoechst 33258 (Sigma-Aldrich, B1155). The number of total and TUNEL-positive nuclei was determined in randomly selected fields by using a Zeiss Axioplan microscope equipped with a digital camera.
Histology and fluorescence microscopy
H & E and SDH (Nitro Blue Tetrazolium, Sigma-Aldrich, N5514) stainings were performed on 7-μm-thick cryosections of diaphragms and TA following standard protocols. For immunoglobulin detection, muscle sections were first incubated for 20 min at RT in a blocking solution containing 0.5% bovine serum albumine and then incubated for 30 min at RT with an anti-mouse Cy3-conjugated secondary antibody (Sigma-Aldrich, C21B1). Nuclei were stained with DAPI (Sigma-Aldrich, F6057). GFP-LC3 puncta were analyzed in cryosections of TA from wild-type;GFP-LC3 and Col6a1–/–;GFP-LC3 mice. TA muscles were fixed in 4% paraformaldehyde in 0.1 M Na phosphate buffer pH 7.4 for 4 h at 4°C and than incubated overnight in 30% sucrose. Finally, muscles were frozen in liquid nitrogen and 7-μm-thick cryosections were cut. Sections were stained overnight with a polyclonal antibodies against laminin (Sigma-Aldrich, L9393) or against TOM20 (Santa Cruz Biotechnology, sc-11415) and 1 h with secondary antibody IRIS 5-goat anti rabbit IgG (Cyanine Technologies, 5WS-08). Nuclei were marked with Hoechst (Sigma-Aldrich, B1155). GFP-LC3 puncta were detected using Leica SP5 confocal microscope.
Transmission electron microscopy
Stretched diaphragms and TA muscles were fixed with 2.5% glutaraldehyde in phosphate buffer 0.1 M (pH 7.4) and embedded in Epon E812 resin (Sigma-Aldrich, 45345). Ultrathin sections were stained with uranyl acetate and lead citrate, and observed in a Tecnai-G2 F12 (FEI) electron microscope.
Statistical analysis
Data are expressed as mean ± s.e.m. Statistical significance was determined by unequal variance Student’s t-test.
Additional material
Download Zip (192.8 KB)Acknowledgments
We thank Walter Giuriati for help with electron microscopy, Bert Blaauw for help with treadmill experiments and Tania Tiepolo for her involvement in the initial TUNEL analysis. We are grateful to Prof. Noboru Mizushima for supplying the GFP-LC3 mice. This work was supported by grants from the Telethon Foundation (GGP10225 and TCP04009), European Union Seventh Framework Programme (BIO-NMD and MYOAGE), the Association Francaise contre les Myopathies, and the Italian Ministry of University and Research. Paolo Grumati was supported by the University of Padova (Progetto Giovani Studiosi).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008; 15:344 - 57; http://dx.doi.org/10.1016/j.devcel.2008.08.012; PMID: 18804433
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol 2010; 298:C1291 - 7; http://dx.doi.org/10.1152/ajpcell.00531.2009; PMID: 20089936
- Bernardi P, Bonaldo P. Dysfunction of mitochondria and sarcoplasmic reticulum in the pathogenesis of collagen VI muscular dystrophies. Ann N Y Acad Sci 2008; 1147:303 - 11; http://dx.doi.org/10.1196/annals.1427.009; PMID: 19076452
- Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet 2005; 42:673 - 85; http://dx.doi.org/10.1136/jmg.2002.002311; PMID: 16141002
- Merlini L, Martoni E, Grumati P, Sabatelli P, Squarzoni S, Urciuolo A, et al. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008; 71:1245 - 53; http://dx.doi.org/10.1212/01.wnl.0000327611.01687.5e; PMID: 18852439
- Irwin WA, Bergamin N, Sabatelli P, Merlini L, Megighian A, Reggiani C, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 2003; 35:367 - 71; http://dx.doi.org/10.1038/ng1270; PMID: 14625552
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010; 16:1313 - 20; http://dx.doi.org/10.1038/nm.2247; PMID: 21037586
- Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci USA 2007; 104:991 - 6; http://dx.doi.org/10.1073/pnas.0610270104; PMID: 17215366
- Lange S, Ehler E, Gautel M. From A to Z and back? Multicompartment proteins in the sarcome. Trends Cell Biol 2006; 16:11 - 8; http://dx.doi.org/10.1016/j.tcb.2005.11.007; PMID: 16337382
- Gräter F, Shen J, Jiang H, Gautel M, Grubmuller H. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys J 2005; 88:790 - 804; http://dx.doi.org/10.1529/biophysj.104.052423; PMID: 15531631
- Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005; 308:1599 - 603; http://dx.doi.org/10.1126/science.1110463; PMID: 15802564
- Waters S, Marchbank K, Solomon E, Whitehouse C, Gautel M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett 2009; 583:1846 - 52; http://dx.doi.org/10.1016/j.febslet.2009.04.049; PMID: 19427866
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458 - 71; http://dx.doi.org/10.1016/j.cmet.2007.11.001; PMID: 18054315
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, LeBrasseur NK, et al. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 2007; 282:30014 - 21; http://dx.doi.org/10.1074/jbc.M704817200; PMID: 17702743
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, et al. Transcription co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002; 418:797 - 801; http://dx.doi.org/10.1038/nature00904; PMID: 12181572
- Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep 2010; 12:433 - 9; http://dx.doi.org/10.1007/s11906-010-0157-8; PMID: 20967516
- Sandri M, Lin J, Handschin C, Yang CW, Arany ZP, Lecker SH, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 2006; 103:16260 - 5; http://dx.doi.org/10.1073/pnas.0607795103; PMID: 17053067
- Geng T, Li P, Yin X, Yan Z. PGC-1α Promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am J Pathol 2010; 178:1738 - 48; http://dx.doi.org/10.1016/j.ajpath.2011.01.005; PMID: 21435455
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 2009; 106:20405 - 10; http://dx.doi.org/10.1073/pnas.0911570106; PMID: 19918075
- Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor γ coactivator 1 α or 1 β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem 2010; 285:19460 - 71; http://dx.doi.org/10.1074/jbc.M110.113092; PMID: 20404331
- Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007; 35:411 - 29; http://dx.doi.org/10.1002/mus.20743; PMID: 17266144
- Gautel M. The sarcomeric cytoskeleton: who picks up the strain?. Curr Opin Cell Biol 2011; 23:39 - 46; http://dx.doi.org/10.1016/j.ceb.2010.12.001; PMID: 21190822
- Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol 2011; 12:349 - 61; http://dx.doi.org/10.1038/nrm3118; PMID: 21602905
- Lock R, Debnath J. Extracellular matrix regulation of autophagy. Curr Opin Cell Biol 2008; 20:583 - 8; http://dx.doi.org/10.1016/j.ceb.2008.05.002; PMID: 18573652
- Tuloup-Minguez V, Greffard A, Codogno P, Botti J. Regulation of autophagy by extracellular matrix glycoproteins in HeLa cells. Autophagy 2011; 7:27 - 39; http://dx.doi.org/10.4161/auto.7.1.13851; PMID: 20980830
- Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J 2009; 23:3896 - 905; http://dx.doi.org/10.1096/fj.09-131870; PMID: 19661286
- Glass DJ. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol 2010; 346:267 - 78; http://dx.doi.org/10.1007/82_2010_78; PMID: 20593312
- Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 2006; 27:411 - 25; http://dx.doi.org/10.1016/j.mam.2006.08.002; PMID: 16973212
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003; 300:1574 - 7; http://dx.doi.org/10.1126/science.1079817; PMID: 12791994
- Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest 2006; 116:2464 - 72; PMID: 16906224
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. FoxO transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117:399 - 412; http://dx.doi.org/10.1016/S0092-8674(04)00400-3; PMID: 15109499
- Sakamoto K, Hirshman MF, Aschenbach WG, Goodyear LJ. Contraction regulation of Akt in rat skeletal muscle. J Biol Chem 2002; 277:11910 - 7; http://dx.doi.org/10.1074/jbc.M112410200; PMID: 11809761
- Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol 2002; 6:193 - 8; http://dx.doi.org/10.1053/ejpn.2002.0593; PMID: 12374585
- Grumati P, Coletto L, Sandri M, Bonaldo P. Autophagy induction rescues muscular dystrophy. Autophagy 2011; 7:426 - 8; http://dx.doi.org/10.4161/auto.7.4.14392; PMID: 21543891