Abstract
Mutations in the gene for the E3 ubiquitin ligase Parkin are the most prevalent cause of autosomal recessive Parkinson disease (PD), an incurable neurodegenerative disorder. Parkin surveys mitochondrial quality by translocating to depolarized mitochondria and inducing their selective macroautophagic removal (mitophagy). We recently reported that Parkin interacts with Ambra1 (activating molecule in Beclin 1-regulated autophagy), a protein that promotes autophagy in the vertebrate central nervous system. We discovered that prolonged mitochondrial depolarization strongly increases the interaction of Parkin with Ambra1. Ambra1 is recruited in a Parkin-dependent manner to perinuclear clusters of depolarized mitochondria, activates the class III phosphatidylinositol 3-kinase (PtdIns3K) complex around these mitochondria and contributes to their selective autophagic clearance. Here, we discuss these findings and suggest a model where translocated Parkin efficiently triggers mitophagy through combined recruitment of Ambra1 and ubiquitination of outer mitochondrial membrane proteins.
PD is the second most prevalent neurodegenerative disorder, causing a variety of disabling motor and nonmotor problems. Mutations in PARK2, the gene encoding the cytosolic E3 ubiquitin ligase Parkin, are a major cause of autosomal recessive PD, probably through a loss-of-function mechanism. Since 2008, multiple labs have reported that Parkin translocates from the cytosol to depolarized mitochondria and subsequently triggers their elimination through mitophagy. Parkin may thus protect cells by mediating the selective removal of dysfunctional mitochondria that would otherwise induce enhanced oxidative stress and apoptosis. Parkin translocation from the cytosol to depolarized mitochondria is dependent on PINK1, another protein genetically linked to autosomal recessive PD. But how does Parkin, once translocated, trigger the engulfment of mitochondria by autophagosomal membranes?
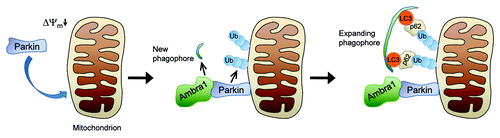
According to one proposed molecular scenario, translocated Parkin ubiquitinates outer mitochondrial membrane proteins such as VDAC1 and mitofusins 1 and 2 (). These ubiquitinated targets may then connect with autophagic membranes via recruitment of receptor proteins that simultaneously bind to ubiquitin and LC3, a protein associated with autophagosomes and expanding phagophores. One such receptor could be p62, although the requirement for p62 in Parkin-mediated mitophagy is controversial. This mechanism would permit recruitment of pre-existing phagophores to mitochondria. However, in this scenario translocated Parkin would not stimulate formation of new phagophores around mitochondria.
Figure 1. Model of Parkin-mediated mitochondrial clearance. Upon loss of mitochondrial membrane potential (Δψm) Parkin translocates to the mitochondria in a PINK1-dependent manner. After translocation, Parkin ubiquitinates outer mitochondrial membrane proteins. In addition, Parkin recruits Ambra1, which induces perimitochondrial nucleation of new phagophores through its effect on class III PtdIns3K. The expanding phagophore then associates with LC3, which tethers the phagophore to the ubiquitinated mitochondria via p62 or other receptor proteins. Ub, ubiquitin.
We recently asked whether Parkin might interact directly with autophagy-regulating proteins to facilitate mitophagy. We used tandem-affinity purification and mass spectrometry to identify binding partners of Parkin. Interestingly, the only autophagy-regulating protein among the identified Parkin interactors was Ambra1. Gian Maria Fimia and colleagues reported in 2007 that Ambra1 promotes autophagy in the central nervous system by activating the class III PtdIns3K complex that is essential for phagophore nucleation. Specifically, Ambra1 promotes the interaction of Beclin 1 and Vps34, two core components of class III PtdIns3K. Fimia and colleagues found that functional deficiency of Ambra1 in mice leads to autophagy impairment and excessive apoptosis in the brain, neuronal accumulation of ubiquitinated proteins and embryonic lethality.
We demonstrated binding of endogenous Parkin and Ambra1 in adult mouse brain. Moreover, prolonged mitochondrial depolarization strongly enhances the interaction of endogenous Parkin and Ambra1 in neuronal cells. We found no evidence for Parkin-mediated ubiquitination of Ambra1. The Ambra1 protein is recruited in a Parkin-dependent manner to perinuclear clusters of depolarized mitochondria, and activates class III PtdIns3K in their immediate vicinity. Knockdown of Ambra1 does not affect Parkin translocation to depolarized mitochondria, but inhibits subsequent mitochondrial clearance. Conversely, Ambra1 overexpression enhances the elimination of depolarized mitochondria, but only in the presence of Parkin.
Our findings suggest a novel mechanism that facilitates Parkin-induced engulfment of mitochondria by autophagosomes (). Rather than merely attracting pre-existing phagophores via ubiquitination of outer mitochondrial membrane proteins and recruitment of receptors such as p62, Parkin actually stimulates the perimitochondrial nucleation of new phagophores through recruitment of Ambra1. After incorporation of LC3, these locally formed new phagophores may then become tethered to ubiquitinated mitochondria via receptors like p62. Ambra1 thus enhances the efficiency of Parkin-mediated mitophagy.
What is the source of the autophagosomal membranes formed around damaged mitochondria? Most previous work attempting to identify the membrane origins of autophagosomes has focused on starvation-induced nonselective autophagy. In starvation-induced autophagy, the ER, the outer mitochondrial membrane, the Golgi and even the plasma membrane have all been implicated as sources of autophagosomal membranes. Recent evidence suggests that mitofusin 2-dependent mitochondria-ER connections are necessary for starvation-induced autophagosome formation. It is possible that the origin of autophagosomal membranes varies depending on the autophagy-triggering conditions. Where the membranes for mitophagy come from, is not known yet. An intriguing and consistent finding is that depolarized mitochondria are trafficked from the cell periphery to the perinuclear region prior to mitophagic degradation. As this area of the cell is enriched in ER membranes, perinuclear mitochondrial clustering could favor the mitochondria-ER crosstalk that may be necessary for autophagosome formation.
A number of other important questions remain to be addressed. What is the biochemical basis for the increased binding of Parkin and Ambra1 after mitochondrial depolarization? Are Parkin and Ambra1 degraded along with the mitochondria, or do they dissociate from mitochondria after setting the mitophagic process in motion? How is the balance between mitophagy and mitochondrial biogenesis regulated? Compelling evidence from Valina and Ted Dawson’s group has recently shown that Parkin ubiquitinates the transcriptional repressor PARIS to target it for proteasomal degradation. PARIS selectively downregulates the levels of PGC1-α, a potent stimulator of mitochondrial biogenesis. Thus, Parkin may control mitochondrial homeostasis by both promoting mitochondrial biogenesis and mediating clearance of dysfunctional mitochondria. How are the effects of Parkin on mitophagy and mitochondrial biogenesis coordinated?
It will be of interest to see whether variations in the AMBRA1 sequence might be associated with the risk of developing PD or other adult-onset brain diseases. Further molecular dissection of the Parkin-PINK1 pathway could also reveal new drug targets for PD. Mitophagy-enhancing compounds could conceivably have a beneficial effect in PD cases caused by deficiency of Parkin or PINK1. However, since autophagy has pleiotropic effects, chronic nonselective autophagy induction is unlikely to be a safe therapeutic strategy in PD. Identification of cofactors specifically involved in Parkin-mediated mitophagy may pave the way for more selective modulation of this pathway.
| Abbreviations: | ||
| Ambra1 | = | activating molecule in Beclin 1-regulated autophagy |
| ER | = | endoplasmic reticulum |
| PARIS | = | Parkin interacting substrate |
| PD | = | Parkinson disease |
| PGC1-α | = | peroxisome proliferator-activated receptor gamma coactivator-1α |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| PINK1 | = | phosphatase and tensin homolog-induced putative kinase 1 |
| VDAC1 | = | voltage-dependent anion channel 1 |
Acknowledgments
C.V.H. is supported by a Ph.D. Fellowship of the Research Foundation Flanders (FWO). W.V. is a Senior Clinical Investigator of the FWO.