Abstract
Mitophagy, selective autophagy of mitochondria, has been extensively demonstrated in cultured cell models but has never been described in skeletal muscle in the context of muscle disease. We recently reported the first example of human muscle disease where mitophagy plays a role in the peculiar muscle pathology. This disease is caused by loss-of-function mutations in the CHKB gene encoding choline kinase β. “Patients” and rostrocaudal muscular dystrophy (rmd) mice, spontaneous Chkb mutants, develop congenital muscular dystrophy with a peculiar mitochondrial abnormality—mitochondria are markedly enlarged at the periphery of muscle fibers and absent from the center. Choline kinase is the first enzymatic step in a biosynthetic pathway for phosphatidylcholine, the most abundant phospholipid in eukaryotes. Our discovery demonstrates that a phosphatydilcholine biosynthetic defect leads to mitochondrial dysfunction and increased mitophagy.
Congenital muscular dystrophy with mitochondrial structural abnormalities (CMDmt) (OMIM:602541) is an autosomal recessive disorder. “Patients” have various biallelic mutations—nonsense, missense or splice-site—in the CHKB gene. This is the only human disease caused by a phospholipid de novo synthetic enzyme defect. rmd mice have a 1.6-kb deletion in the Chkb gene, an ortholog of CHKB. Choline kinase activity in muscle is undetectable and the phosphatidylcholine level is decreased in muscles from both “patients” and rmd mice and also in isolated mitochondria from rmd mice ().
Figure 1. In normal muscle fiber, mitochondria distribute uniformly. Choline kinase defect causes a decreased phosphatidylcholine level in the mitochondrial membrane, leading to mitochondrial dysfunction. These mitochondria are eliminated by mitophagy, probably resulting in sparse mitochondria in muscle fiber.
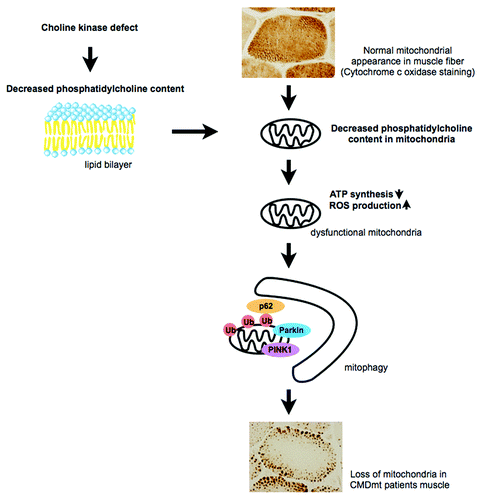
Interestingly, both CMDmt “patients” and rmd mice show distinct mitochondrial morphological abnormalities in muscle fibers: Mitochondria are nearly absent in the center, while they are enlarged at the periphery. This morphological change seems specific to this disease; we tested 15 “patients” with this distinct muscle pathology and identified mutations in CHKB gene in all of them. We hypothesize that altered phospholipid composition in muscle mitochondrial membrane may lead to mitochondrial structural and functional abnormalities.
In rmd mice, muscles are involved with the rostrocaudal gradient of severity; hindlimbs are more severely affected while forelimbs are relatively spared, and mitochondria are more sparse in hindlimbs. Isolated mitochondria from skeletal muscle of rmd mice show impaired respiratory chain enzyme activities, decreased ATP synthesis, and increased superoxide production. Not surprisingly, these mitochondrial bioenergetic dysfunctions are more severe in hindlimbs than in forelimbs, presumably reflecting the different degree of severity in muscle damage.
Mitochondrial bioenergetic abnormalities are also seen in mitochondrial myopathies; genetic diseases of the mitochondrial respiratory chain caused by a variety of defects in mtDNA or nuclear DNA. However, the muscle pathology of mitochondrial myopathies is different from that of CMDmt. In mitochondrial myopathies, mitochondria are commonly increased both in number and size in muscle fibers, showing a ragged red appearance upon modified Gomori trichrome staining. In contrast, in CMDmt, mitochondria are sparse in the center of muscle fibers, while they are enlarged at the periphery. This mitochondrial decrease in number with concomitant increase in size can be confirmed by quantitative analysis in rmd mice. Mitochondrial DNA copy number is also decreased in rmd muscle in an age-dependent manner, most likely due to decreased mitochondrial number, because the pathological change of sparse mitochondria is also progressive with age.
In this disease, mitochondria are eliminated by mitophagy because (1) mitochondria are observed in autophagosomes by EM; (2) mitochondria are polyubiquitinated, and p62 and LC3 are recruited to mitochondria based on immunohistochemistry; and (3) PINK1 and Parkin in isolated mitochondria are increased based on western blotting, suggesting that they are recruited to mitochondria to promote mitophagy. Our data suggest that the decreased number of mitochondria is most likely due to increased mitochondrial clearance in skeletal muscle.
This discovery raises a number of important questions. Is it mitochondrial bioenergetic dysfunction that induces mitophagy in rmd? What will happen if mitophagy is blocked in rmd muscle? Does mitophagy have a protective role, or promote muscle degeneration? In other words, is muscular dystrophy in rmd mice due to impaired mitochondria or excessive clearance of mitochondria?
Another important question is: How does membrane lipid compositional change affect mitochondrial function in vivo? Native PAGE analysis of rmd mitochondria shows that respiratory enzyme activity is decreased, but the respiratory chain protein complexes are properly formed. This finding supports the evidence that proper membrane lipid composition, and thereby lipid-protein interaction, is crucial for the activities of respiratory chain enzymes. Further study is necessary to elucidate the regulation mechanism of respiratory chain enzyme activities by membrane lipid.
CMDmt, due to the choline kinase β defect, should be a good model to investigate the regulation of mitochondrial number by mitophagy and lipid-protein interaction in mitochondrial membrane.
Acknowledgments
This study was supported partly by the Research on Psychiatric and Neurological Diseases and Mental Health of Health and Labour Sciences research grants; partly by Research on Intractable Diseases of Health and Labor Sciences research grants; partly by a Research Grant for Nervous and Mental Disorders (20B-12, 20B-13) from the Ministry of Health, Labour and Welfare; partly by an Intramural Research Grant (23-4, 23-5) for Neurological and Psychiatric Disorders from NCNP; partly by KAKENHI (20390250, 22791019); partly by Research on Publicly Essential Drugs and Medical Devices of Health and Labor Sciences research grants; partly by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO); and partly by a grant from the Japan Foundation for Neuroscience and Mental Health.