Abstract
A large protein complex consisting of Atg5, Atg12 and Atg16L1 has recently been shown to be essential for the elongation of isolation membranes (also called phagophores) during mammalian autophagy. However, the precise function and regulation of the Atg12–5-16L1 complex has largely remained unknown. In this study we identified a novel isoform of mammalian Atg16L, termed Atg16L2, that consists of the same domain structures as Atg16L1. Biochemical analysis revealed that Atg16L2 interacts with Atg5 and self-oligomerizes to form an ~800-kDa complex, the same as Atg16L1 does. A subcellular distribution analysis indicated that, despite forming the Atg12–5-16L2 complex, Atg16L2 is not recruited to phagophores and is mostly present in the cytosol. The results also showed that Atg16L2 is unable to compensate for the function of Atg16L1 in autophagosome formation, and knockdown of endogenous Atg16L2 did not affect autophagosome formation, indicating that Atg16L2 does not possess the ability to mediate canonical autophagy. Moreover, a chimeric analysis between Atg16L1 and Atg16L2 revealed that their difference in function in regard to autophagy is entirely attributable to the difference between their middle regions that contain a coiled-coil domain. Based on the above findings, we propose that formation of the Atg12–5-16L complex is necessary but insufficient to mediate mammalian autophagy and that an additional function of the middle region (especially around amino acid residues 229–242) of Atg16L1 (e.g., interaction with an unidentified binding partner on phagophores) is required for autophagosome formation.
Introduction
Autophagy is a catabolic cellular process in all eukaryotic cells that is responsible for bulk degradation of proteins and organelles, particularly when cells exist under nutrient-deprived conditions (for a review see refs.Citation1–Citation3). One of several types of autophagy, macroautophagy (simply referred to as autophagy below), is the major type that is utilized for many aspects of cellular events, including defense against bacterial intrusion, antigen presentation, and programmed cell death, and dysfunction of autophagy is associated with a variety of human diseases, including cancer, neurodegeneration and microbial infections (for a review see Citationref. 4). Unique dynamic membrane biogenesis in the form of de novo formation of a double-membrane vesicle called the autophagosome occurs in the cytosol during autophagy. In mammals, autophagosomes are formed by elongation and merging of the ends of an isolation membrane (also called the phagophore), and the resulting autophagosomes eventually fuse with lysosomes, which degrade their contents.Citation1-Citation3
Genetic studies of yeast have revealed a set of genes (named ATG for autophagy-related) that are involved in autophagy, and at least 17 ATG genes have been shown to be involved in autophagosome formation.Citation5 Interestingly, most of the ATG genes have been conserved during evolution (from yeasts to humans), and the products of three of the mammalian ATG genes, Atg5, Atg12 (a ubiquitin-like molecule conjugated with Atg5) and Atg16L1 (a WD-repeat-containing molecule that interacts with Atg5), form a tight complex having an apparent molecular mass of ~800-kDa through homo-oligomerization of Atg16L1.Citation6 The resulting Atg12–5-16L1 complex is thought to be essential for elongation of phagophores to occur, because deletion of either Atg5 or Atg16L1 in mice has been found to completely abolish autophagosome formation.Citation7,Citation8 It has recently been suggested that the Atg12–5-16L1 complex is a novel type of E3 ligase that determines the site of lipidation of LC3 (microtubule-associated protein 1 light chain 3)/Atg8,Citation9,Citation10 another ubiquitin-like molecule conjugated to phosphatidylethanolamineCitation11 that mediates membrane tethering and hemifusion in vitro.Citation12 However, the precise function of each component of the Atg12–5-16L1 complex in elongation of phagophores still remains to be elucidated.
Considerable attention has recently been directed toward Atg16L1 for the following two reasons. First, ATG16L1 has been identified as a candidate gene responsible for susceptibility to human Crohn disease, a complex inflammatory disease involving the small intestine,Citation13-Citation15 and second, Atg16L1 (or Atg12–5-16L1 complex) has been identified as a potential effector of small GTPase Rab33,Citation16 one of the membrane-trafficking proteins conserved in all eukaryotes.Citation17-Citation20 Since expression of a coiled-coil (CC) domain of Atg16L1 that binds Rab33, but not of an N-terminal Atg5-binding region or C-terminal WD-repeats, strongly suppresses autophagy,Citation10,Citation16 it has been hypothesized that Rab33-mediated membrane trafficking is involved in autophagosome formation,Citation21 although the precise function of the Rab33-binding ability of Atg16L1 in autophagosome formation remains unknown.
In this study we identified a second isoform of mammalian Atg16L, termed Atg16L2. Biochemical analysis indicated that Atg16L2 retains the biochemical properties of Atg16L1, including its Atg5-binding and ~800-kDa complex forming abilities, although it has weaker Rab33B-binding affinity than Atg16L1. When we investigated the possible involvement of Atg16L2 in autophagy, we discovered that Atg16L2 does not have the ability to mediate autophagy and that its inability to do so is attributable to dysfunction of the CC domain-containing middle region of Atg16L2. Based on these findings, we discuss the distinct functions of Atg16Ls in autophagy.
Results
Identification of Atg16L2, a novel mammalian Atg16L isoform
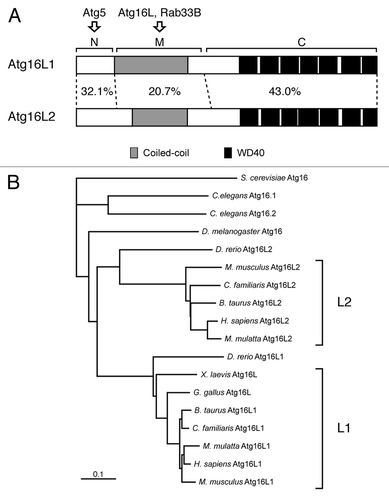
Atg16L1 (formerly called Apg16L) was originally identified as a mammalian homolog of yeast Atg16Citation6 and has been shown to be required for elongation of phagophores in autophagy.Citation8 Both yeast Atg16 and mammalian Atg16L1 share an Atg5-binding domain at the N terminus and an adjacent CC domain that is required for formation of homo-oligomer (most likely homo-dimer),Citation22,Citation23 but, in addition, mammalian Atg16L1 contains WD repeats at the C terminus whose function is unknown ().Citation6 A search of the mouse and human public databases allowed us to identify a second isoform of mammalian Atg16L, designated as Atg16L2, on human chromosome 11q13.4 and on mouse chromosome 7 E3, that is structurally similar to the previously reported Atg16L1 (on human chromosome 2q37.1 and mouse chromosome 1 D) (). The same as Atg16L1, Atg16L2 consists of three structural regions: an N-terminal region, which is homologous to the Atg5-binding region of Atg16L1 (named the N region), a putative CC domain in the middle region (named the M region), and seven WD repeats at the C-terminal region (named the C region) (Fig. S1). Sequence comparison between mouse Atg16L1 and Atg16L2 indicated that the N- and C-terminal regions are well conserved (32.1% amino acid identity in the N region and 43.0% amino acid identity in the C region), but that sequence homology in the M region is relatively low (20.7% amino acid identity). To determine whether Atg16L2 is present in other species, we searched for Atg16L homologs in available vertebrate and invertebrate databases, and as shown in , Atg16L2 was found to form a distinct branch in the phylogenetic tree of Atg16L homologs independent from that of Atg16L1. Interestingly, Atg16L2 homologs were found in all mammals whose databases we searched, but none were found in Xenopus laevis or Gallus gallus, suggesting that Atg16L2 is present only in mammals.
Figure 1. Structural comparison between mouse Atg16L1 and Atg16L2. (A) Schematic representations of mouse Atg16L1 and Atg16L2. Both proteins share a coiled-coil domain (gray boxes) in the middle (M) region and WD repeats (black boxes) at the C terminus. The degree of amino acid identity in each region of Atg16L1 and Atg16L2 (N, M and C) is indicated by percentages. Note that the amino acid identity of the M region of Atg16L1 and Atg16L2 is much lower than that of the N region and C region. (B) The phylogenetic tree of Atg16L homologs from a variety of species was depicted with the CLUSTAL X (1.8) program. Note that the Atg16L2 homologs are conserved mostly in mammals and form a distinct branch from that of the Atg16L1 homologs. Although D. rerio contains two Atg16L homologs, D. rerio Atg16L2 is distantly related to the mammalian Atg16L2 in the phylogenetic tree.
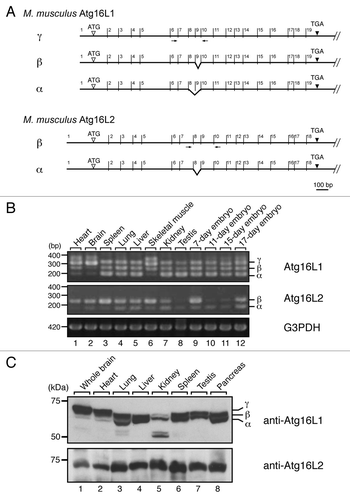
Reverse transcriptase (RT)-PCR analysis indicated that Atg16L2 is expressed in various tissues (i.e., ubiquitous expression) and developmental stages of the mouse, the same as Atg16L1 (). However, we discovered three Atg16L1 alternative splicing isoforms, named α, β and γ,Citation6 and two Atg16L2 alternative splicing isoforms (). The short isoform of Atg16L2 (602 amino acids; designated as Atg16L2α) lacks exon 8 (63 bp), and the long isoform of Atg16L2 (623 amino acids; designated as Atg16L2β) contains all 18 exons (the long isoform was used as the Atg16L2 in all subsequent experiments). Alternative splicing of Atg16L1 occurred in a tissue-specific manner, whereas Atg16L2β expression is dominant in most of the mouse tissues (). Immunoblot analysis with specific antibodies revealed similar ubiquitous expression profiles of Atg16L1 and Atg16L2 at the protein level ().
Figure 2. Alternative splicing variants of Atg16L2. (A) Alternative splicing of Atg16L1 mRNA and Atg16L2 mRNA. The 19 exons of Atg16L1 and 18 exons of Atg16L2 are indicated by numbers above the line representing Atg16L1 and Atg16L2, respectively. Alternatively spliced exons are indicated by broken lines. The positions of the primers (within exons 6 and 10 for Atg16L1 and exons 7 and 10 for Atg16L2) used in B are indicated by arrows. (B) RT-PCR analysis of Atg16L1 and Atg16L2 expression in various mouse tissues and on embryonic day 7, 11, 15 and 17 (upper two panels). RT-PCR analysis of glyceraldehyde-3-phosphate dehydrogenase (G3PDH) expression was also performed (bottom panel) to ensure that equivalent amounts of first-strand cDNA were used. Sequence analysis showed that the additional bands around 400 bp in the top panel were artifacts of Atg16L1γ bands. The size of the molecular weight markers (bp, base pair) is shown at the left. (C) Expression of Atg16L1 and Atg16L2 protein in various mouse tissues. Mouse tissue lysates indicated (50 μg) were subjected to 7.5% SDS-PAGE followed by immunoblotting with anti-Atg16L1 antibody (top panel) and anti-Atg16L2 antibody (bottom panel). Atg16L2 protein was expressed ubiquitously, the same as Atg16L1. The size of the molecular mass markers (in kDa) is shown at the left.
Comparison of biochemical properties between Atg16L1 and Atg16L2
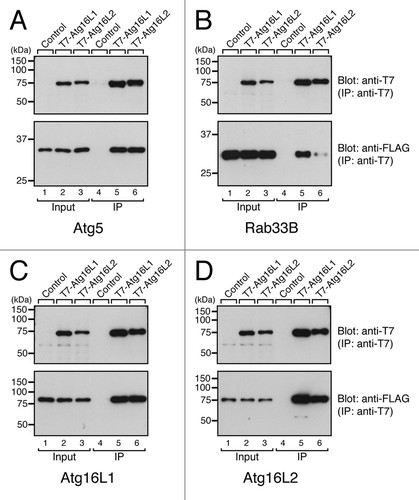
The ubiquitous expression of Atg16L2 and its structural similarity to Atg16L1 prompted us to further investigate the possible involvement of Atg16L2 in autophagy. To do so, we first compared the biochemical properties of Atg16L1 and Atg16L2. Since Atg16L1 has been shown to interact with Atg5 via the N-terminal domain and Rab33B via the CC domainCitation16 and to form a homo-oligomer,Citation6,Citation10,Citation22,Citation23 we attempted to determine whether Atg16L2 interacts with Atg5, Rab33B and Atg16L1/L2 by conventional immunoprecipitation assay in COS-7 cells. Consistent with the high-sequence similarity between the N-terminal domain of Atg16L1 and Atg16L2, Atg16L2 interacted with Atg5, the same as Atg16L1 did (lanes 5 and 6 in the bottom panel of ). In addition, Atg16L1 and Atg16L2 were able to form homo-oligomers and hetero-oligomers (lanes 5 and 6 in the bottom panels of ), despite the low-sequence similarity between the CC domain of Atg16L1 and the CC domain of Atg16L2. Since mouse embryonic fibroblast (MEF) cells endogenously express both Atg16L isoforms (Fig. S2), we also confirmed the presence of endogenous hetero-oligomer between Atg16L1 and Atg16L2 by co-immunoprecipitation assay using specific antibodies (Fig. S3). The only difference we observed was in the Rab33B-binding ability of the two Atg16Ls: Atg16L2 interacted with Rab33B very little, in comparison with Atg16L1 (compare lanes 5 and 6 in the bottom panel of ). To quantitatively analyze the Rab33B-binding ability of Atg16L1 and Atg16L2, we performed in vitro direct binding experiments with purified components, i.e., T7-Atg16Ls and GST (glutathione S-transferase)-Atg5 or GST-Rab33B. In brief, beads coupled with T7-Atg16Ls were incubated with various concentrations of GST-Atg5 or GST-Rab33B, and bound proteins were detected with anti-GST antibody. Consistent with the results of the co-immunoprecipitation assay (), Atg16L2 bound Atg5 with affinity similar to that of Atg16L1 (calculated EC50 value for the Atg16L2·Atg5 interaction and the Atg16L1·Atg5 interaction was 46.2 ± 18.3 nM and 53.5 ± 17.5 nM, respectively, and these values are not significantly different; p > 0.25, Student’s unpaired t-test) (Fig. S4A and S4B), whereas the affinity of the Atg16L2·Rab33B interaction (EC50 = 1.06 ± 0.27 μM) was approximately hundred times lower than that of the Atg16L1·Rab33B interaction (EC50 = 10.2 ± 1.3 nM) (Fig. S4C, S4D and S4E). Despite the weakness of the Atg16L2·Rab33B interaction, the Atg16L2 CC domain specifically interacted with Rab33A/B, but did not specifically react with any of 58 other Rabs, according to the results of a yeast two-hybrid assay (Fig. S4F),Citation16,Citation24 and thus it has the same Rab-binding specificity as Atg16L1.Citation16 The results of the biochemical analyses indicated that Atg16L1 and Atg16L2 basically share the same biochemical properties (e.g., Atg5-binding and oligomerization) but have different Rab33-binding affinity.
Figure 3. Distinct-ligand-binding activities of Atg16L1 and Atg16L2. Atg5-binding activity (A), Rab33B-binding activity (B), and oligomerization activity of Atg16L1 and Atg16L2 (C and D) as determined by co-immunoprecipitation assay in COS-7 cells. pEF-T7-Atg16Ls (or a control pEF-BOS vector) and pEF-FLAG-Atg5 (pEF-FLAG-Rab33B, or pEF-FLAG-Atg16Ls) were co-transfected into COS-7 cells. The proteins expressed were solubilized with 1% Triton X-100, and T7-Atg16Ls were immunoprecipitated with anti-T7 tag antibody-conjugated agarose. Co-immunoprecipitated FLAG-Atg5 (FLAG-Rab33B, or FLAG-Atg16Ls) was detected with HRP-conjugated anti-FLAG tag antibody (lanes 4–6 in the lower panels). To ensure loading of the same amounts of T7-Atg16Ls, immunoprecipitated T7-Atg16Ls were detected with HRP-conjugated anti-T7 tag antibody (lanes 4–6 in the upper panels). Total expressed proteins used for the immunoprecipitation are shown in lanes 1–3. Note that the Rab33B-binding activity of Atg16L2 was weaker than that of Atg16L1 in B but that the Atg5-binding activity and oligomerization activity of Atg16L1 and Atg16L2 were similar in (A, C and D). The size of the molecular mass markers (in kDa) is shown at the left.
Atg16L2 forms an ~800-kDa complex with Atg12–5 in MEF cells
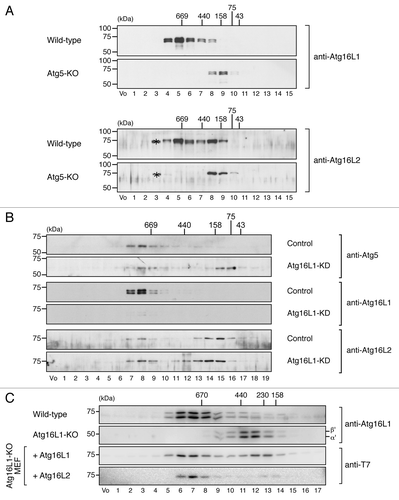
Although Atg16L2 has the ability to bind Atg5 and to form a homo-oligomer in vitro, the same as Atg16L1 does (),Citation6 Atg16L2 may not form ~800-kDa complex with Atg5 and Atg12 in vivo. A gel filtration analysis was performed with MEF cell lysates to determine whether it does, and as expected, the gel filtration analysis clearly demonstrated the presence of an endogenous ~800-kDa complex containing Atg16L2 in MEF cells, although a portion of the Atg16L2 was eluted in a fraction having a low molecular mass (~200-kDa) (fraction 8 in the upper panels of ). By contrast, no ~800-kDa complex was detected in Atg5-knockout (KO) MEF cells (, lower panels). We then attempted to determine whether Atg16L2 alone is able to form an ~800-kDa complex in the absence of Atg16L1 by performing the same gel filtration analysis with lysates of Atg16L1-knockdown (KD) MEF cells. As shown in , a portion of both Atg5 and Atg16L2 was clearly present in fraction 8, which contained the ~800-kDa complex, of Atg16L1-KD MEF cells. Moreover, although the ~800-kDa complex was not formed in the absence of Atg16L1,Citation8 expression of Atg16L2 (or Atg16L1) in Atg16L1-KO MEF cells restored formation of the complex (bottom two panels in ). The absence of the ~800-kDa complex in Atg16L1-KO MEF cells is likely to be attributable to the reduced expression of Atg16L2 protein in comparison with the wild-type MEF cells (data not shown). We therefore concluded that the Atg16L2 is capable of forming an ~800-kDa complex with Atg5 and Atg12 in vivo, the same as Atg16L1.
Figure 4. Atg16L2 forms an ~800-kDa protein complex the same as Atg16L1 does. (A) Cytosolic fractions of the homogenates of wild-type MEF cells or Atg5-KO MEF cells were separated by size exclusion chromatography. Each fraction was subjected to 10% SDS-PAGE followed by immunoblotting with anti-Atg16L1 antibody (top two panels) or anti-Atg16L2 antibody (bottom two panels). The asterisks presumably correspond to nonspecific bands by anti-Atg16L2 antibody. (B) Cytosolic fractions of the homogenates of wild-type MEF cells and Atg16L1-KD MEF cells transfected with specific Atg16L1 siRNA were separated as described in (A). Each fraction was subjected to 10% SDS-PAGE followed by immunoblotting with anti-Atg5 antibody (top two panels), anti-Atg16L1 antibody (middle two panels), or anti-Atg16L2 antibody (bottom two panels). (C) Cytosolic fractions of the homogenates of wild-type MEF cells or Atg16L1-KO MEF cells retrovirally expressing T7-Atg16L-111 (Atg16L1) or T7-Atg16L-222 (Atg16L2) were separated as described in (A). Each fraction was subjected to 10% SDS-PAGE followed by immunoblotting with anti-T7 tag antibody or anti-Atg16L1 antibody. The positions of the molecular mass standards (in kDa) are shown at the top (or left). Vo, void fraction.
Atg16L2-N together with Atg12–5 has E3-like enzyme activity in regard to ectopic LC3 lipidation
It has recently been proposed that the Atg12–5-16L1 complex functions as an E3-like enzyme in regard to LC3 lipidation,Citation9,Citation10 because when Atg16L1-N was fused to the CAAX motif derived from human K-ras (i.e., a plasma membrane localization signal), it promoted targeting of Atg12–5 to the plasma membrane and facilitated ectopic LC3 lipidation on the plasma membrane. Consistent with the fact that Atg16L2 binds Atg5 (), a similar Atg16L2-NKras-CAAX mutant also promoted ectopic LC3 lipidation on the plasma membrane (Fig. S5), indicating that the Atg12–5-16L2-N complex has the ability to function as an E3-like enzyme, the same as the Atg12–5-16L1-N complex does.
Atg16L2 is unable to localize on phagophores
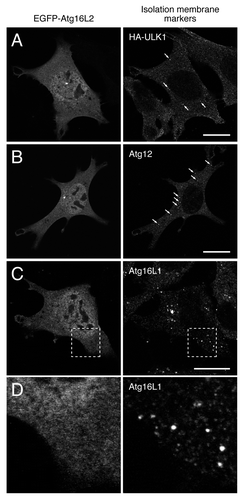
Since Atg16L1 and Atg16L2 share fundamental biochemical properties (i.e., Atg5-binding, ~800-kDa complex formation, and E3-like activity in regard to LC3 lipidation), we initially hypothesized that Atg16L2 participates in autophagosome formation, the same as Atg16L1 does. To test our hypothesis, we first determined whether Atg16L2 is capable of localizing on phagophores, the same as Atg16L1 does. Since our specific anti-Atg16L2 antibody could not be used for an immunofluorescence analysis, the intracellular localization of EGFP (enhanced green fluorescent protein)-tagged Atg16L2 was determined by confocal fluorescence microscopy. Unexpectedly, however, EGFP-Atg16L2 was mainly localized in the cytosol, and it did not colocalize with any of three phagophore markers (i.e., ULK1 (Unc-51-like kinase 1), Atg12 and Atg16L1) (). EGFP-Atg16L2 also appeared to be present in the nucleus, but its apparent nuclear localization of EGFP-Atg16L2 may have been an artifact of EGFP-tagging, because HA-tagged Atg16L2 showed clear cytosolic localization and did not accumulate in the nucleus (data not shown). The absence of Atg16L2 on phagophores suggests that Atg16L2 is not involved in autophagosome formation.
Figure 5. Atg16L2 is unable to localize on phagophores even under starvation conditions. (A–D) EGFP-Atg16L2 was transiently expressed in MEF cells stably expressing HA-ULK1 and in wild-type MEF cells. The starved cells were fixed and stained with anti-HA antibody (A), anti-Atg12 antibody (B), and anti-Atg16L1 antibody (C and D). Arrows indicate phagophores. A magnified view of the boxed area in (C) is shown in (D). Note that EGFP-Atg16L2 did not form any dots and that it did not colocalize with any of three phagophore markers, ULK1, Atg12 and Atg16L1. Scale bars, 20 μm.
Atg16L2 is not required for starvation-induced autophagy
The inability of Atg16L2 to recruit phagophores prompted us to investigate a direct involvement of Atg16L2 in starvation-induced autophagy by performing rescue experiments with Atg16L1-KO MEF cells.Citation8 In brief, Atg16L1-KO MEF cells retrovirally expressing T7-tagged Atg16L1/L2 or no Atg16L molecules were incubated with HBSS (Hanks' Balanced Salt Solutions) for 1 h, and autophagosomes were then visualized by immunofluorescence staining with anti-LC3 antibody. Consistent with the previous finding that Atg16L1 is an essential factor for autophagy to occur,Citation8 no LC3-positive dots were observed in the control Atg16L1-KO MEF cells even under the starvation conditions (), and re-expression of Atg16L1 (designated as Atg16L-111 in ) in the Atg16L1-KO MEF cells fully restored the formation of LC3-positive dots (). By contrast, expression of Atg16L2 (designated as Atg16L-222 in ) in Atg16L1-KO MEF cells did not restore any formation of either LC3-positive dots or Atg12-positive dots at all (), strongly indicating that Atg16L2 is unable to compensate for the function of Atg16L1 in autophagy.
Figure 6. Atg16L2 is unable to restore autophagosome formation in Atg16L1-deficient (KO) MEF cells. (A) Schematic representation of Atg16L1 (open boxes) and Atg16L2 (shaded boxes) swapping mutants. The M region containing a CC domain was swapped between Atg16L1 and Atg16L2, and the resulting swapping mutants were designated as Atg16L-121 and Atg16L-212. (B) Atg16L2 is unable to restore LC3-dot formation in Atg16L1-KO MEF cells. Atg16L1-KO MEF cells expressing either no Atg16L (a), T7-Atg16L-111 (virtually equivalent to Atg16L1) (b), T7-Atg16L-222 (virtually equivalent to Atg16L2) (c), T7-Atg16L-212 (d), or T7-Atg16L-121 (e), were cultured in HBSS for 1 h and fixed with 4% paraformaldehyde. The cells were stained with anti-LC3 antibody. The right panels show magnified views of the boxed area. LC3-dot formation in the Atg16L1-KO MEF cells was restored by expression of proteins containing the Atg16L1-M region (Atg16L-111 and Atg16L-212), but not by expression of proteins containing the Atg16L2-M region (Atg16L-121 and Atg16L-222). (C) Atg16L2 mutant containing the L1-M region (Atg16L-212) restores LC3 lipidation and p62 degradation in Atg16L1-KO MEF cells. MEF cells expressing T7-Atg16L-swapping mutants (or control cells) were cultured under nutrient-rich or starvation conditions. The cell lysates were analyzed by immunoblotting with anti-T7 tag antibody, anti-Atg16L1 antibody, anti-LC3 antibody, anti-p62 antibody and anti-α-tubulin antibody. The bottom panel shows expression of α-tubulin, a loading control. Consistent with the results in (B), expression of proteins containing Atg16L1-M region (Atg16L-111 and -212) induced LC3 lipidation and p62 degradation (lanes 4 and 8), whereas expression of proteins containing the Atg16L2-M region (Atg16L-121 and Atg16L-222) did not restore autophagic activity (lanes 6 and 10). Although for some unknown reason the protein expression levels of Atg16L-222 and Atg16L-121 were always lower than those of Atg16L-111 and Atg16L-212 under our experimental conditions, the absence of any rescue effect by Atg16L-222 or Atg16L-121 is unlikely to be attributable to an insufficient protein expression level in Atg16L1-KO MEF cells by the following reasons. First, it has recently been shown that autophagy occurs normally in Atg5 tetracycline-off cell lines even in the presence of a level of Atg5 protein that is undetectable by immunoblotting.Citation26 Second, mice in which there was complete knockout of Atg16L1 died soon after birth because of a neonatal deficiency of autophagy,Citation8 whereas mice with reduced expression of Atg16L1 were born and survived to adulthood.Citation15 Therefore, a level of Atg16L protein that is detectable under normal immunoblotting conditions is presumably sufficient to support autophagy. α’ and β’, indicate Atg16L1α and β lacking the CC domain, respectively, in Atg16L1-KO MEF cells. The asterisk indicates a nonspecific band.
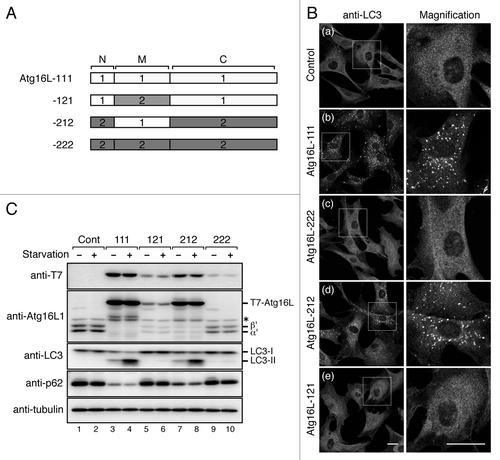
To identify the region of Atg16L2 that is responsible for its inability to rescue autophagy-deficiency in Atg16L1-KO MEF cells, we prepared two chimera mutants (named Atg16L-121 and Atg16L-212) in which the CC domain-containing M region of Atg16L1 and Atg16L2 is swapped (). It should be noted that expressing Atg16L-212, which contains the Atg16L1-M region, in Atg16L1-KO MEF cells fully restored the formation of LC3-positive dots (). Conversely, Atg16L-121, which contains the Atg16L2-M region, had no ability to restore autophagic activity in Atg16L1-KO MEF cells (). The autophagic activity of Atg16L1-KO MEF cells expressing Atg16L-111, Atg16L-222, Atg16L-121, or Arg16L-212 was further evaluated by two independent approaches, i.e., LC3 lipidation and degradation of p62.Citation25 As shown in , Atg16L-111 and Atg16L-212 were able to restore LC3 lipidation and p62 degradation in Atg16L1-KO MEF cells (lanes 3, 4, 7 and 8), but Atg16L-222 and Atg16L-121 were not (lanes 5, 6, 9 and 10), consistent with the results of the immunofluorescence analysis in . These results collectively indicated that the M region of Atg16L1 and Atg16L2 must have different functions in autophagy.
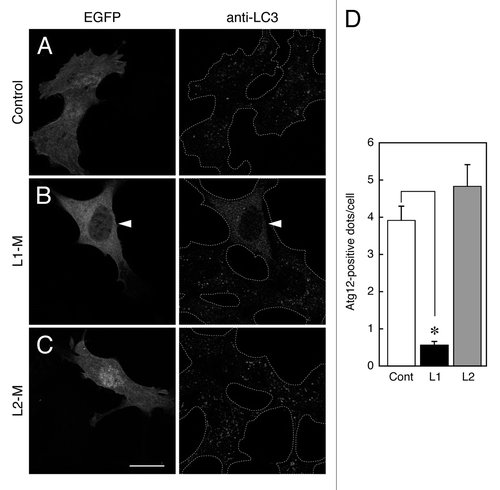
We recently showed that expression of the Atg16L1 M region strongly suppresses starvation-induced autophagy,Citation10,Citation16 indicating that the Atg16L1 M region plays a crucial role in autophagosome formation. We therefore wondered whether the Atg16L2 M region has an inhibitory effect on autophagy. To determine whether it does, we investigated the effect of expression of Atg16L2-M on autophagy by monitoring the formation of Atg12-positive dots and LC3-positive dots. Consistent with the results on the chimeric analysis between Atg16L1 and Atg16L2 (), MEF cells transiently expressing EGFP-tagged Atg16L2-M contained normal numbers of Atg12-positive dots () and LC3-positive dots (), the same as the control EGFP-expressing cells (), indicating that Atg16L2-M is unable to inhibit phagophore formation or autophagosome formation. By contrast, the numbers of Atg12-positive dots and LC3-positive dots were dramatically reduced in MEF cells expressing EGFP-Atg16L1-M () under the same experimental conditions, consistent with the results of our previous studies.Citation10,Citation16 Some EGFP-Atg16L2-M also appeared to be present in the nucleus, but its apparent nuclear localization may have been an artifact of EGFP-tagging, because myc-tagged Atg16L2-M showed clear cytosolic localization and did not affect LC3-dot formation (data not shown).
Figure 7. Effect of overexpression of Atg16Ls-M on phagophore formation. (A–C) MEF cells transiently expressing EGFP-Atg16L1-M, EGFP-Atg16L2-M, or EGFP alone were cultured in HBSS for 1.5 h and fixed in 4% paraformaldehyde. The cells were stained with anti-LC3 antibody. The arrowheads indicate EGFP-Atg16L1-M expressing MEF cells showing inhibition of autophagosome formation. Scale bar, 20 μm. (D) Decreased number of Atg12-positive dots in cells expressing Atg16L1-M, but not Atg16L2-M, under starvation conditions. MEF cells cultured in HBSS for 1.5 h were fixed and stained with anti-Atg12 antibody. The number of Atg12-positive dots in the cells expressing Atg16Ls-M was counted. Bars represent the means and SE of one representative experiment (n > 100). *p < 0.001 (Mann-Whitney test). Similar results were obtained in an independent experiment.
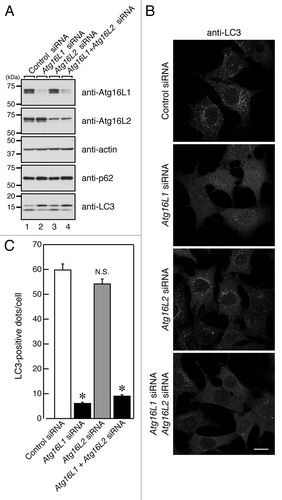
To rule out the possibility that Atg16L2 is required for a different step in autophagy, i.e., a step in which Atg16L1 is not involved, we used small interfering RNA (siRNA) to knock down endogenous Atg16L2, and evaluated the effect of the knockdown on starvation-induced autophagy by assessing p62 degradation, LC3 lipidation and LC3-dot formation. However, comparison with the control cells showed that knockdown of endogenous Atg16L2 had no effect on p62 degradation, LC3 lipidation, or LC3-dot formation in Atg16L2-KD MEF cells (), whereas autophagy was dramatically suppressed in Atg16L1-KD MEF cells under the same experimental conditions (). Moreover, simultaneous knockdown of Atg16L1 and Atg16L2 did not suppress autophagy further (). We also investigated whether knockdown of Atg16L2 would affect autophagy flux under nutrient-rich or starvation conditions, but no effect of Atg16L2-KD on autophagy flux in MEF cells was detected under nutrient-rich or starvation conditions either (Fig. S7). Based on these results, we concluded that Atg16L2 is not required for canonical autophagy, despite forming an ~800-kDa complex with Atg12–5 and having E3-like activity. In contrast to our findings, however, Lipinski et al. very recently reported that knockdown of Atg16L2 by a genome-wide siRNA screen resulted in increased autophagy flux in human neuroblastoma H4 cells,Citation27 suggesting that Atg16L2 functions as a negative regulator in autophagy. This discrepancy may be attributable to the difference in the cell lines used, i.e., fibroblasts cells in our study and neuroblastoma cells in theirs. Alternatively, since protein level analyses are not generally performed and off-target effects by siRNAs are not sufficiently evaluated in genome-wild siRNA screen experiments, Atg16L2 may not be directly involved in basal autophagy in H4 cells. Further work will be necessary to investigate these possibilities.
Figure 8. Effect of knockdown of endogenous Atg16Ls on autophagosome formation. (A) MEF cells treated with Atg16Ls siRNA (or control siRNA) were cultured in HBSS for 1.5 h and harvested. The cells were solubilized with 1% Triton X-100, and their lysates were subjected to 10% SDS-PAGE followed by immunoblotting with the specific antibodies indicated. The size of the molecular mass markers (in kDa) is shown at the left. (B) MEF cells prepared as described in (A) were fixed in 4% paraformaldehyde and stained with anti-LC3 antibody. Scale bar, 20 μm. (C) The number of LC3-positive dots in (B) was counted. Bars represent the means and SE of one representative experiment (n > 150). *p < 0.001 (Tukey’s Multiple Comparison Test).
Discussion
The present study identified Atg16L2, a novel isoform of mammalian-specific Atg16L that contains the same domain structures as Atg16L1 (). Biochemical analyses demonstrated that Atg16L2 binds Atg5 and forms an ~800-kDa complex in MEF cells, the same as Atg16L1 does, but that it interacts with Rab33B with approximately 100 times less affinity than Atg16L1 does ( and Fig. S4). The most unexpected findings in this study were that Atg16L2 is not recruited to phagophores () and that it cannot restore autophagosome formation in Atg16L1-KO MEF cells (), despite forming the ~800-kDa complex with Atg12–5 and having E3-like activity in regard to LC3 lipidation ( and Fig. S5), strongly indicating that formation of an ~800-kDa complex is necessary but insufficient to promote autophagosome formation. We further demonstrated by chimeric analysis between Atg16L1 and Atg16L2 that the nonessential role of Atg16L2 in autophagy is attributable to the CC domain-containing M region of Atg16L2 (). In addition, expression of the Atg16L1-M region, but not the Atg16L2-M region, strongly attenuated LC3-dot (or Atg12-dot) formation ().Citation10,Citation16 We therefore speculate that a key factor that associates with the Atg16L1 M region, but not with the Atg16L2 M region, must be present within the cell. Small GTPase Rab33 is a likely candidate for such a key factor, because the only difference found between Atg16L1 and Atg16L2 is the reduced Rab33B-binding activity of the M region of Atg16L2, and expression of a GTPase-deficient mutant of Rab33B (Rab33B-QL) attenuated autophagosome maturation.Citation28 However, since, unlike knockdown of Atg16L1 (), knockdown of Rab33B had no effect on autophagosome formation,Citation16 Rab33B may not be the key factor that recruits the Atg12–5-16L1 complex to phagophores. Consistent with this notion, our detailed deletion analyses indicated that the minimal essential region of Atg16L1 for autophagosome formation (Fig. S8B) and for colocalization with LC3 dots (Fig. S9) (i.e., amino acid residues 1–242 of Atg16L1) was clearly different from the minimal essential region for Rab33B binding (Fig. S8A) (i.e., amino acid residues 1–214 of Atg16L1). Therefore, we speculate that amino acid residues 229–242 of Atg16L1 contribute to autophagosome formation and its recruitment to phagophores, presumably by binding to the above-mentioned key factor (e.g., lipid or protein on phagophores). It should be noted that amino acid residues 229–242 of Atg16L1 are not conserved in the corresponding region of Atg16L2 (i.e., only one amino acid is conserved, and two amino acid deletions are found in Atg16L2; Fig. S1, yellow), suggesting that Atg16L2 is unable to interact with such a key factor. Identification of a key factor that specifically associates with the Atg16L1 M region in the future would clarify the function of Atg16L1 in autophagosome formation.
At this stage, the physiological function of Atg16L2 is completely unknown. However, since Atg16L2 is capable of homo-oligomer and hetero-oligomer with Atg16L1, one possible function of Atg16L2 might be to modulate the efficiency of autophagy by replacing Atg16L1 in the ~800-kDa complex. The ratio between Atg16L1 and Atg16L2 in the ~800-kDa complex would then to be expected to determine the efficiency of mammalian autophagy. However, since protein expression level of Atg16L2 is almost ten times smaller than that of Atg16L1 in MEF cells (Fig. S2), contribution of Atg16L2 in the control of autophagy seemed to be limited at least in MEF cells. Another possibility is that mammalian-specific Atg16L2 may be involved in a specialized type of autophagy (e.g., exclusion of infectious bacteria, antigen presentation on major histocompatibility complex class II molecules, and clearance of insoluble aggregates in neural cells),Citation29 rather than starvation-induced autophagy. Further study is necessary to determine whether any of these possibilities are actual functions of Atg16L2.
In summary, we identified Atg16L2, a second mammalian Atg16L isoform with the ability to form an ~800-kDa complex with Atg12-conjugated Atg5 in vivo and that has E3-like activity in regard to LC3 lipidation in vitro. Nevertheless, Atg16L2 was found to be dispensable for starvation-induced autophagy because of dysfunction of the CC domain-containing M region of Atg16L2. Thus, MEF cells contain at least two different ~800-kDa complexes, an Atg12–5-16L1 complex, which is capable of mediating autophagosome formation, and an Atg12–5-16L2 complex, which is incapable of facilitating canonical autophagy. On the basis of these findings, we propose that formation of the Atg12–5-16L1 complex alone is necessary but insufficient to promote autophagosome formation and that an additional key factor that associates with the Atg16L1 M region is required for mammalian autophagy to occur.
Materials and Methods
Materials
Horseradish peroxidase (HRP)-conjugated anti-FLAG tag (M2) mouse monoclonal antibody, anti-FLAG tag antibody-conjugated agarose and anti-α-tubulin mouse monoclonal antibody (clone B5-1-2) were obtained from Sigma-Aldrich Corp. (A8592, A2220 and T6074, respectively). HRP-conjugated anti-T7 tag mouse monoclonal antibody, anti-T7 tag mouse monoclonal antibody and anti-T7 tag antibody-conjugated agarose were purchased from Merck Biosciences Novagen (69048, 69522 and 69026, respectively). Anti-p62/sequestosome 1 (SQSTM1) rabbit polyclonal antibody and anti-Atg12 (or Apg12) rabbit polyclonal antibody were from BIOMOL Research Laboratories (BML-PW9860) and Zymed Laboratories (36-6400), respectively. Anti-LC3 rabbit polyclonal antibody against GST-LC3, anti-Atg16L1 rabbit polyclonal antibody against GST-Atg16L1-M (amino acid residues 80–265 of Atg16L1),Citation16 and anti-Atg16L2 rabbit polyclonal antibody against GST-Atg16L2-M (amino acid residues 85–280 of Atg16L2) were prepared as described previously.Citation30 Modified siRNA duplexes (Stealth RNA) against mouse Atg16L1, Atg16L2 and a nontargeting scramble sequence (control siRNA) were obtained from Invitrogen Corp. (MSS293862, MSS232103 and 12935-113, respectively). All other reagents used in this study were analytical grade or the highest grade commercially available.
Molecular cloning of mouse Atg16L2 cDNAs
cDNAs encoding the mouse Atg16L2β (a longer splicing isoform of Atg16L2; simply referred to as Atg16L2 throughout the text) were amplified from Marathon-Ready adult mouse testis cDNA (BD Biosciences Clontech, 639405) by PCR using the following pairs of oligonucleotides with a restriction enzyme site (underlined: BamHI or KpnI) or with a stop codon (in bold), essentially as described previously:Citation31 5′-GGA TCC ATG GCA GGA CCT GGC GCC-3′ (Atg16L2-Met primer, sense) and 5′-AGC ATT GAC CTC AGA GAGA-3′ (Atg16L2-C1 primer, antisense); and 5′-CGG TAC CAA AGC ATC CCT GTG-3′ (Atg16L2-N1 primer, sense) and 5′-TCA GTG CCA GAG CAC CAC CT-3′ (Atg16L2-stop primer, antisense). Purified PCR products were directly inserted into the pGEM-T Easy vector (Promega, A1360) and verified with an automated sequencer. The Atg16L2 cDNA containing a full open reading frame was constructed by using appropriate restriction enzyme sites and then subcloned into the pEF-T7 and pEF-FLAG tag mammalian expression vectors modified from pEF-BOS as described previously.Citation31
Construction of an Atg16L2 truncation mutant, Atg16L swapping mutants and Atg16L1 deletion mutants
A deletion mutant of Atg16L2 (M, amino acid residues 85–280) was constructed by conventional PCR techniques using the following pairs of oligonucleotides with a BamHI site (underlined) or a stop codon (in bold): 5′-GGA TCC GAC CAA GTC TCA TCA CCA-3′ and 5′-TCA AGA CCT GAA AGG CCT CTT-3′. The resulting cDNAs were transferred to the pEGFP-C1 vector (BD Biosciences Clontech, 6084-1). For construction of Atg16L swapping mutants (see for details), three cDNA fragments of Atg16L1/L2 (N, M and C) were separately amplified by PCR using the following pairs of mutagenic oligonucleotides with an artificial MunI or HindIII site (underlined) or a stop codon (in bold) as described previously:Citation32 5′-GGA TCC ATG TCG TCG GGC CTG CGC GC-3′ and 5′-CAA TTG GGC CAT TTC ATG-3′ for Atg16L1-N (amino acid residues 1–79); 5′-CAA TTG AGG ATC AAA CAC-3′ and 5′-AAG CTT CTT AGT AGC TGC TCT-3′ for Atg16L1-M (amino acid residues 80–265); 5′-AAG CTT TCG CAG CCT GCT GGA-3′ and 5′-TCA AGG CTG TGC CCA CAG CA-3′ for Atg16L1-C (amino acid residues 266–623); 5′-GGA TCC ATG GCA GGA CCT GGC GCC-3′ and 5′-CAA TTG GTC AGG GCC TGT-3′ for Atg16L2-N (amino acid residues 1–84); 5′-CAA TTG TCA TCA CCA GCC-3′ and 5′-AAG CTT TGA GAA CTC AGG CAG-3′ for Atg16L2-M (amino acid residues 85–280); and 5′-AAG CTT GAG ACT TGT GAA AAA-3′ and 5′-TCA GTG CCA GAG CAC CAC CT-3′ for Atg16L2-C (amino acid residues 281–623). Two Atg16L swapping mutants, designated as Atg16L-121 and Atg16L-212, were produced by combining three fragments, L1-N, L2-M and L1-C, and L2-N, L1-M and L2-C, respectively, using MunI and HindIII sites. Atg16Ls (111, 121, 212 and 222) were subcloned into the pMRX-IRES-puro-T7 vector.Citation33 Mutants of Atg16L1-N and Atg16L2-N fused to the CAAX motif sequence (17 amino acids) of human K-ras protein at the C terminus were constructed as described previously.Citation10 Six Atg16L1 deletion mutants, i.e., Atg16L1-1–200, -1–214, -1–228, -1–242, -1–256 and -ΔC (amino acid residues 1–265), were similarly constructed by conventional PCR techniques and subcloned into the pMRX-IRES-puro-T7 vector as described above. The human ULK1 cDNACitation34 was subcloned into the pMRX-IRES-puro-HA vector.Citation33 Other expression plasmids, i.e., pEF-FLAG-Atg5, pGEX-4T-3-Atg5 and pGEX-4T-3-Rab33B, were prepared as described previously.Citation16
RT-PCR analysis
Mouse first-strand cDNAs prepared from adult mouse heart, brain, spleen, lung, liver, skeletal muscle, kidney and testis and from 7-, 11-, 15- and 17-d mouse embryos were obtained from BD Biosciences Clontech (MTC Multiple Tissue cDNA Panels, 636745). RT-PCR analysis was performed as described previouslyCitation35 using the following sets of oligonucleotides designed at exons 6 and 10 (for Atg16L1) or 7 and 10 (for Atg16L2) (see also ): 5′-GCA GCA AAG GAA CCT CTA CCT-3′ and 5′-AGT TGG GAC TCT CAC ATC TTT ACC-3′ for Atg16L1; and 5′-AAG CTT GAG ACT TGT GAA AAA-3′ and 5′-AGC ATT GAC CTC AGA GAG AT-3′ for Atg16L2.
Cell culture, transfection and infection
COS-7 cells and MEF cells were maintained in Dulbecco′s modified Eagle′s medium (DMEM; Sigma-Aldrich Corp., D5796) containing 10% fetal bovine serum and antibiotics under 5% CO2 at 37°C. Atg16L1-KO MEF cells, which express Atg16L1α and β lacking the CC domain, were prepared as described previously.Citation8 Starvation was achieved by washing the cells once with HBSS (Sigma-Aldrich Corp., H9269) and transferring them to HBSS for 1–1.5 h. Transfection of plasmids into COS-7 cells (for immunoprecipitation) and MEF cells (for immunofluorescence) was performed by using Lipofectamine Plus and Lipofectamine 2000, (Invitrogen Corp., 18324-020 and 11668-019), respectively, each according to the manufacturer’s instructions. Transfection of siRNAs into MEF cells was also performed by using Lipofectamine RNAiMAX (Invitrogen Corp., 13778-150) according to the manufacturer’s instructions. Retrovirus infection was performed essentially as described previously.Citation33
Immunoprecipitation
Co-immunoprecipitation assays in COS-7 cells were performed as described previously.Citation36,Citation37 Direct binding experiments between T7-Atg16L1 (or T7-Atg16L2) and GST-Atg5 (or GST-Rab33B) were also performed as described previously.Citation36,Citation38 In brief, T7-agarose beads coupled with T7-Atg16L1/L2 were incubated for 1 h with various concentrations of purified GST-Atg5 or GST-Rab33B. After washing the beads three times, proteins bound to the beads were analyzed by 10% SDS-PAGE followed by immunoblotting with HRP-conjugated anti-T7 tag antibody and HRP-conjugated anti-GST antibody as described previously.Citation36 Immunoreactive bands were visualized by enhanced chemiluminescence (ECL). The intensity of the bands on the X-ray film was captured and quantified with MetaMorph software (Molecular Devices). The statistical analyses and curve fitting were performed with the GraphPad PRISM computer program (version 4.0). Immunoprecipitation of endogenous Atg16L1 (or Atrg16L2) molecules and immunoblotting with specific antibodies were essentially performed as described previously.Citation36 The blots shown in this paper are representative of at least three independent experiments.
Immunofluorescence analysis
Immunostaining was performed as described previously,Citation39 and the stained cells were examined for fluorescence with a confocal fluorescence microscope (Fluoview 1000, Olympus). To quantitatively measure phagophore/autophagosome formation, the cells were incubated in HBSS for 1 h before fixation. Phagophores and autophagosomes were separately visualized by staining with anti-Atg12 antibody and anti-LC3 antibody, respectively. The images of the cells were captured at random with the confocal microscope, and the number of fluorescent dots was counted with the MetaMorph software.
Gel filtration analysis
Gel filtration analysis was performed as described previously.Citation6,Citation16 In brief, MEF cells were homogenized in homogenization buffer (50 mM TRIS-HCl, pH 7.5, 150 mM NaCl, and Protease inhibitor cocktail; Roche Molecular Biochemicals, 1873580) by repeated passage (15 times) through a 1-ml syringe with a 23-gauge needle. The homogenate was centrifuged at 10,000 × g for 10 min, and the supernatant was further centrifuged at 100,000 × g for 60 min. The resulting supernatants (i.e., cytosolic fraction) were separated by size exclusion chromatography on a Superose 6 column (GE Healthcare, 17-5172-01) followed by immunoblotting with anti-Atg5 antibody, anti-Atg16L1 antibody, anti-Atg16L2 antibody and HRP-conjugated anti-T7 tag antibody as described above.
Sequence analysis
Sequence alignment and depiction of the phylogenetic tree of the Atg16L proteins were performed by using the ClustalW program (available at http://clustalw.ddbj.nig.ac.jp/top-e.html) and CLUSTAL X (1.8) program set at the default parameters.Citation40
Yeast two-hybrid assay
Yeast two-hybrid assay using a panel of putatively GTP-locked Rab1~43 was performed as described previously.Citation24,Citation41
| Abbreviations: | ||
| Atg | = | autophagy-related |
| CC | = | coiled-coil |
| EGFP | = | enhanced green fluorescent protein |
| GST | = | glutathione S-transferase |
| HBSS | = | Hanks’ Balanced Salt Solutions |
| HRP | = | horseradish peroxidase |
| KD | = | knockdown |
| KO | = | knockout |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MEF | = | mouse embryonic fibroblast |
| RT | = | reverse transcriptase |
| siRNA | = | small interfering RNA |
| ULK1 | = | Unc-51-like kinase 1 |
Additional material
Download Zip (1.6 MB)Acknowledgments
We thank Dr. Tatsuya Saitoh and Dr. Shizuo Akira (Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) for kindly donating Atg16L1-KO MEF cells, Dr. Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for kindly donating Atg5-KO MEF cells, Dr. Toshio Kitamura (The University of Tokyo, Tokyo, Japan) for kindly donating Plat-E cells and retroviral vectors, Megumi Aizawa for technical assistance, and members of the Fukuda Laboratory for valuable discussions. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, and Technology (MEXT) of Japan (to M.F. and T.I.), by a grant from the Global COE Program (Basic & Translational Research Center for Global Brain Science) of the MEXT of Japan (to M.F.), and by the Uehara Memorial Foundation (to T.I.). K.I. was supported by the Japan Society for the Promotion of Science.
Note
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AB476646-8.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
References
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211 - 6; http://dx.doi.org/10.1038/35056522; PMID: 11265251
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Yoshimori T, Noda T. Toward unraveling membrane biogenesis in mammalian autophagy. Curr Opin Cell Biol 2008; 20:401 - 7; http://dx.doi.org/10.1016/j.ceb.2008.03.010; PMID: 18472412
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 2007; 581:2156 - 61; http://dx.doi.org/10.1016/j.febslet.2007.01.096; PMID: 17382324
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679 - 88; http://dx.doi.org/10.1242/jcs.00381; PMID: 12665549
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032 - 6; http://dx.doi.org/10.1038/nature03029; PMID: 15525940
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008; 456:264 - 8; http://dx.doi.org/10.1038/nature07383; PMID: 18849965
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 302; http://dx.doi.org/10.1074/jbc.C700195200; PMID: 17986448
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092 - 100; http://dx.doi.org/10.1091/mbc.E07-12-1257; PMID: 18321988
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165 - 78; http://dx.doi.org/10.1016/j.cell.2007.05.021; PMID: 17632063
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39:207 - 11; http://dx.doi.org/10.1038/ng1954; PMID: 17200669
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39:596 - 604; http://dx.doi.org/10.1038/ng2032; PMID: 17435756
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259 - 63; http://dx.doi.org/10.1038/nature07416; PMID: 18849966
- Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell 2008; 19:2916 - 25; http://dx.doi.org/10.1091/mbc.E07-12-1231; PMID: 18448665
- Pfeffer SR. Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol 2001; 11:487 - 91; http://dx.doi.org/10.1016/S0962-8924(01)02147-X; PMID: 11719054
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001; 2:107 - 17; http://dx.doi.org/10.1038/35052055; PMID: 11252952
- Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 2006; 103:11821 - 7; http://dx.doi.org/10.1073/pnas.0601617103; PMID: 16882731
- Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. Rab GTPases at a glance. J Cell Sci 2007; 120:3905 - 10; http://dx.doi.org/10.1242/jcs.015909; PMID: 17989088
- Fukuda M, Itoh T. Direct link between Atg protein and small GTPase Rab: Atg16L functions as a potential Rab33 effector in mammals. Autophagy 2008; 4:824 - 6; PMID: 18670194
- Fujita N, Saitoh T, Kageyama S, Akira S, Noda T, Yoshimori T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem 2009; 284:32602 - 9; http://dx.doi.org/10.1074/jbc.M109.037671; PMID: 19783656
- Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F. Dimeric coiled-coil structure of Saccharomyces cerevisiae ATG16 and its functional significance in autophagy. J Biol Chem 2010; 285:1508 - 15; http://dx.doi.org/10.1074/jbc.M109.053520; PMID: 19889643
- Fukuda M, Kanno E, Ishibashi K, Itoh T. Large scale screening for novel Rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics 2008; 7:1031 - 42; http://dx.doi.org/10.1074/mcp.M700569-MCP200; PMID: 18256213
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580:2623 - 9; http://dx.doi.org/10.1016/j.febslet.2006.04.008; PMID: 16647067
- Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041 - 52; http://dx.doi.org/10.1016/j.devcel.2010.05.005; PMID: 20627085
- Itoh T, Kanno E, Uemura T, Waguri S, Fukuda M. OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol 2011; 192:839 - 53; http://dx.doi.org/10.1083/jcb.201008107; PMID: 21383079
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Fukuda M, Mikoshiba K. A novel alternatively spliced variant of synaptotagmin VI lacking a transmembrane domain: implications for distinct functions of the two isoforms. J Biol Chem 1999; 274:31428 - 34; http://dx.doi.org/10.1074/jbc.274.44.31428; PMID: 10531344
- Fukuda M, Kanno E, Mikoshiba K. Conserved N-terminal cysteine motif is essential for homo- and heterodimer formation of synaptotagmins III, V, VI, and X. J Biol Chem 1999; 274:31421 - 7; http://dx.doi.org/10.1074/jbc.274.44.31421; PMID: 10531343
- Fukuda M, Kojima T, Aruga J, Niinobe M, Mikoshiba K. Functional diversity of C2 domains of synaptotagmin family: mutational analysis of inositol high polyphosphate binding domain. J Biol Chem 1995; 270:26523 - 7; PMID: 7592870
- Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 2000; 7:1063 - 6; http://dx.doi.org/10.1038/sj.gt.3301206; PMID: 10871756
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651 - 9; http://dx.doi.org/10.1091/mbc.E08-03-0312; PMID: 18768752
- Fukuda M, Saegusa C, Mikoshiba K. Novel splicing isoforms of synaptotagmin-like proteins 2 and 3: identification of the Slp homology domain. Biochem Biophys Res Commun 2001; 283:513 - 9; http://dx.doi.org/10.1006/bbrc.2001.4803; PMID: 11327731
- Fukuda M, Kanno E. Analysis of the role of Rab27 effector Slp4-a/granuphilin-a in dense-core vesicle exocytosis. Methods Enzymol 2005; 403:445 - 57; http://dx.doi.org/10.1016/S0076-6879(05)03039-9; PMID: 16473610
- Kuroda TS, Fukuda M. Identification and biochemical analysis of Slac2-c/MyRIP as a Rab27A-, myosin Va/VIIa-, and actin-binding protein. Methods Enzymol 2005; 403:431 - 44; http://dx.doi.org/10.1016/S0076-6879(05)03038-7; PMID: 16473609
- Tsuboi T, Fukuda M. The C2B domain of rabphilin directly interacts with SNAP-25 and regulates the docking step of dense core vesicle exocytosis in PC12 cells. J Biol Chem 2005; 280:39253 - 9; http://dx.doi.org/10.1074/jbc.M507173200; PMID: 16203731
- Fukuda M, Itoh T. Slac2-a/melanophilin contains multiple PEST-like sequences that are highly sensitive to proteolysis. J Biol Chem 2004; 279:22314 - 21; http://dx.doi.org/10.1074/jbc.401791200; PMID: 15145961
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 25:4876 - 82; http://dx.doi.org/10.1093/nar/25.24.4876; PMID: 9396791
- Itoh T, Satoh M, Kanno E, Fukuda M. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-containing proteins based on their Rab-binding activity. Genes Cells 2006; 11:1023 - 37; http://dx.doi.org/10.1111/j.1365-2443.2006.00997.x; PMID: 16923123