Abstract
Under normal growth conditions the mammalian target of rapamycin complex 1 (mTORC1) negatively regulates the central autophagy regulator complex consisting of Unc-51-like kinases 1/2 (Ulk1/2), focal adhesion kinase family-interacting protein of 200 kDa (FIP200) and Atg13. Upon starvation, mTORC1-mediated repression of this complex is released, which then leads to Ulk1/2 activation. In this scenario, Atg13 has been proposed as an adaptor mediating the interaction between Ulk1/2 and FIP200 and enhancing Ulk1/2 kinase activity. Using Atg13-deficient cells, we demonstrate that Atg13 is indispensable for autophagy induction. We further show that Atg13 function strictly depends on FIP200 binding. In contrast, the simultaneous knockout of Ulk1 and Ulk2 did not have a similar effect on autophagy induction. Accordingly, the Ulk1-dependent phosphorylation sites we identified in Atg13 are expendable for this process. This suggests that Atg13 has an additional function independent of Ulk1/2 and that Atg13 and FIP200 act in concert during autophagy induction.
Introduction
Macroautophagy (hereafter referred to as autophagy) is an essential and highly conserved lysosomal degradation process, helping to maintain cellular homeostasis by constitutive turnover of cytoplasmic material. Cellular components, such as long-lived proteins and entire organelles, are sequestered by a double membrane known as the isolation membrane or phagophore. After closure, the resulting autophagosome subsequently fuses with lysosomes, leading to the degradation of its content. The provided amino acids can be reused for protein synthesis as well as an energy source for ATP production. By this means, autophagy represents a cellular adaptation mechanism to starvation, and is hence strongly enhanced after nutrient deprivation. Despite its essential role for cell growth, survival and development, the molecular details of autophagy induction and its underlying signaling cascade are still poorly understood.
In yeast, two serine/threonine protein kinases could be identified as essential prerequisites for the regulation of autophagy induction. The nutrient-sensing kinase target of rapamycin (TOR) inhibits autophagy induction under nutrient-rich conditions through negative regulation of another serine/threonine kinase, Atg1. In yeast, Atg1 differentially interacts with the regulatory proteins Atg13 and Atg17, which are both important for Atg1 kinase activity.Citation1,Citation2 Under normal growth conditions Atg13 is hyperphosphorylated in a TOR-dependent manner. Upon starvation or rapamycin treatment, it is immediately dephosphorylated, leading to Atg1-Atg13-Atg17 complex formation and autophagy induction.
Vertebrates have at least two Atg1 homologs, Unc-51-like kinase 1 (Ulk1) and Ulk2,Citation3-Citation6 that seem to fulfill partially redundant functions.Citation7-Citation9 Although there is no Atg17 homolog, the focal adhesion kinase family-interacting protein of 200 kDa (FIP200) has been proposed as the functional counterpart of Atg17.Citation8 Furthermore, a vertebrate homolog of Atg13 has been predicted by sequence homologyCitation10 and has already been proven to be relevant for autophagy induction.Citation7 Ulk1/2, Atg13 and FIP200 form a large, stable complex of >3 MDa that, in contrast to the yeast Atg1-Atg13-Atg17 complex, is insensitive to the cellular nutrient status. Recently, several groups have simultaneously characterized the molecular details of this Ulk-Atg13-FIP200 complex and even more important, its direct regulation by mTOR complex 1 (mTORC1).Citation11-Citation15 Under nutrient-rich conditions mTORC1 joins the Ulk-Atg13-FIP200 complex, via interaction between Raptor and Ulk1/2,Citation14 and mTOR directly phosphorylates Atg13 and Ulk1/2Citation13-Citation15 presumably at inhibitory sites, to suppress the kinase activity of Ulk1/2. Under starvation conditions or when mTORC1 activity is repressed, Ulk1/2 phosphorylates FIP200 and Atg13 at yet unknown sites, and the entire complex translocates to phagophore assembly sites.Citation11,Citation14,Citation15 Thus, it is reasonable to assume that the phosphorylation of either FIP200 or Atg13 by Ulk1/2 is a prerequisite for this translocation and in turn for autophagy induction.
Using a newly generated Atg13-deficient cell line, we show that Atg13 is indispensable for starvation-induced autophagy as well as basal autophagy under normal growth conditions and that direct interaction with FIP200 is essential for this function. Interestingly, double knockout of Ulk1 and Ulk2 had no significant effect on LC3 lipidation and autophagosome formation. Accordingly, the Ulk1-dependent phosphorylation sites we identified in human Atg13 are not essential for Atg13 function. Since mTOR inhibition does not fully resemble autophagy induction by starvation, we conclude that Atg13 and FIP200 act in concert during induction of autophagy, but this function does not necessarily depend on the mTOR-mediated regulation of Ulk1 or Ulk2 kinase activity.
Results
Generation of Atg13 knockout cell line
The essential role of Atg13 for TOR-mediated autophagy induction has initially been shown in a genetic screening of Saccharomyces cerevisiae for autophagy defective mutants.Citation2,Citation16 The necessity of Atg13 seems to be conserved in higher metazoans, due to the complete autophagy-defective phenotype of the respective Drosophila melanogaster knockout mutant.Citation12 Until now, the importance of Atg13 for autophagy in higher vertebrates has been confirmed by siRNA-mediated knockdown. However, the observed effects vary depending on the respective cell line, the Atg13 knockdown efficiency and the readout used.Citation11,Citation13-Citation15
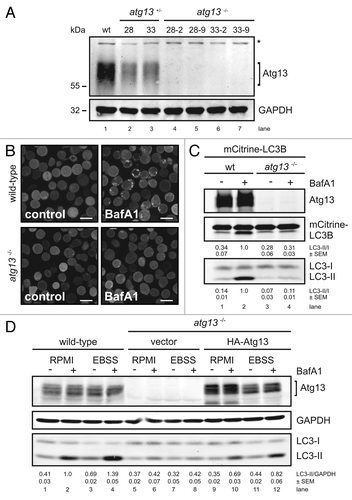
To analyze the role of Atg13 and its regulation by Ulk1 and Ulk2 in a genetically defined background, we first generated Atg13-deficient DT40 knockout cells by disrupting exon 1 of the atg13 gene (Fig. S1A and S1B). The DT40 cell system has already successfully been used to analyze autophagic processes.Citation17-Citation19 Notably, the primary structure of Atg13 is highly conserved between Homo sapiens and Gallus gallus, showing 92% amino acid identity (Fig. S1C). Four independent homozygous atg13−/− clones could be identified by genomic PCR, which have lost both wild-type alleles (Fig. S1B). The complete lack of protein expression was confirmed by immunoblotting for endogenous Atg13 protein ().
Figure 1. Generation of Atg13-deficient DT40 cells. (A) Atg13-deficient DT40 B cell lines (atg13−/−) were generated by targeted disruption of both atg13 alleles. Successful targeting was confirmed by genomic PCR using primers specific for wild-type, hisD- or bsr-targeted alleles (see Fig. S1 for details). Equal amounts of protein from cleared cellular lysates of either wild-type (wt) cells (lane 1), atg13+/− clones (lanes 2–3) and atg13−/− clones (lanes 4–7) were analyzed for Atg13 and GAPDH by immunoblotting. The asterisk indicates an unspecific background band. (B) Wild-type and atg13−/− DT40 cells, retrovirally transfected with cDNA encoding mCitrine-LC3B, were treated with 10 nM bafilomycin A1 (BafA1) or DMSO (control) for 6 h and directly visualized by confocal laser scanning microscopy (bars: 10 µm). (C) Cleared cellular lysates of cells described in (B) were subjected to anti-Atg13 and anti-LC3 immunoblotting. LC3-II/LC3-I ratios are represented as mean values of three independent experiments ± SEM (D) atg13−/− cells reconstituted with HA-tagged full-length chicken Atg13 isoform A (lanes 9–12) were incubated in normal growth medium (RPMI) or starvation medium (EBSS) for 1 h in the presence or absence of 10 nM (BafA1). Equal protein amounts from cleared cellular lysates were analyzed for Atg13, GAPDH and LC3 by immunoblotting. As control, wild-type cells (lanes 1–4) and atg13−/− cells reconstituted with empty vector (lanes 5–8) were analyzed in parallel. LC3-II/GAPDH ratios are represented as mean values of three independent experiments ± SEM.
Atg13 deficiency blocks autophagy induction
In order to analyze their autophagic capacity, we first studied LC3 lipidation and autophagosome formation using fluorescently labeled chicken LC3B (mCitrine-LC3B) in wild-type and atg13−/− cells in the presence and absence of bafilomycin A1. LC3 is a cytosolic protein that is lipidated by the autophagic machinery during autophagosome formation (LC3-II) and is thus recruited to autophagosomes.Citation20 The vacuolar H+-ATPase inhibitor bafilomycin A1 prevents the lysosomal degradation of autophagosomesCitation21 and therefore is frequently used to analyze the autophagic flux.
Strikingly, while bafilomycin A1 significantly increased the number and size of autophagosomes in wild-type DT40 cells, no dot formation could be observed in atg13−/− cells (). Next, we investigated the effect of Atg13 knockout on LC3 lipidation by immunoblotting. In wild-type cells, incubation with bafilomycin A1 led to a prominent accumulation of endogenous and fluorescently labeled LC3-II under normal growth conditions (, lanes 1 and 2), while atg13−/− cells showed almost no detectable accumulation of lipidated LC3 (, lanes 3 and 4). Strikingly, in wild-type cells endogenous LC3 lipidation could be further enhanced by starvation in EBSS (, lanes 1–4). In contrast, atg13−/− cells did not show any prominent accumulation of lipidated LC3 after starvation (, lanes 5–8). Notably, the reconstitution of atg13−/− cells with HA-tagged full-length chicken Atg13 was partially able to rescue the autophagy-defective phenotype (, lanes 9–12). In order to verify that LC3 lipidation correlates with autophagosome formation, we further investigated the presence of autophagosomes by transmission electron microscopy. While wild-type DT40 cells displayed numerous double-membrane autophagosomes per cell after starvation (), almost no autophagosomes could be detected in atg13−/− cells (). Noteworthy, Atg13-deficient cells in contrast displayed enlarged and swollen mitochondria under starvation conditions (). In addition, we investigated the formation of autolysosomes using a tandem mRFP-EGFP-rLC3 construct.Citation22 Again, while wild-type DT40 cells showed numerous autophagosomes and autolysosomes after 2 h starvation in EBSS (), mRFP-EGFP-rLC3 was mainly distributed throughout the cytoplasm in atg13−/− cells and almost no autolysosomes could be detected in these cells ().
Figure 2. Atg13 is essential for autophagosome generation (A) DT40 wild-type and (B) atg13−/− cells were incubated in normal growth medium (control) or EBSS for 2 h. Cells were fixed and analyzed by transmission electron microscopy. A representative cell from each condition is shown in two different magnifications. Autophagosomes are indicated by black arrow heads in the image with higher magnification, swollen mitochondria are indicated by asterisks (bars: 500 nm). (C) Wild-type and (D) atg13−/− cells, retrovirally transfected with cDNA encoding mRFP-EGFP-rLC3 were incubated in EBSS for 2 h and analyzed by confocal laser scanning microscopy. The mRFP signal is shown in red and the EGFP signal in green in the merged image. Autolysosomes are indicated by white arrow heads (bars: 10 µm). The percentage of autolysosome containing cells (>200 cells/experiment) is represented as mean ± range of two independent experiments.
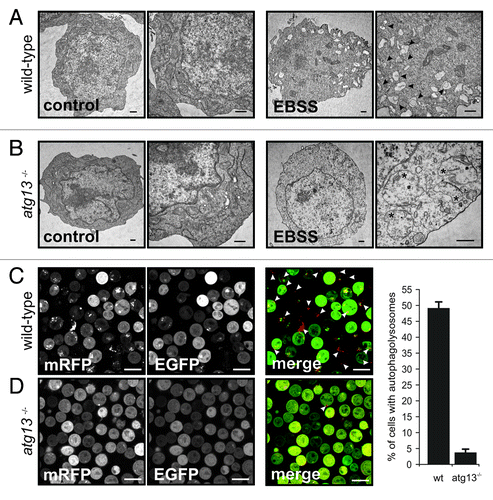
Collectively, these results indicate that in DT40 cells Atg13 is essential for autophagy induction and autophagosome generation under starvation conditions as well as for basal autophagy.
Deficiency for Ulk1 and Ulk2 does not resemble the phenotype of Atg13 knockout
Based on previous observations,Citation12-Citation15 the following attractive working model has been proposed for starvation-induced autophagy:Citation11 Under nutrient-rich growth conditions, mTORC1 associates with the Ulk-Atg13-FIP200 complex, phosphorylates Ulk1 and Atg13, and is thereby suppressing Ulk1 kinase activity. Following starvation, Ulk1 phosphorylates both Atg13 and FIP200, finally leading to autophagy induction.Citation11
Since Ulk2 was reported to compensate for the lack of Ulk1,Citation9,Citation23 we generated DT40 cells deficient for both Ulk1 and Ulk2 (for targeting strategies see Fig. S2A and S2B). Starting from ulk1−/− cells (clone 16–10), two homozygous ulk1−/−ulk2−/− cell lines (2–6 and 17–16) could be identified by genomic PCR (Fig. S2C). The complete absence of transcripts was verified by RT-PCR () and quantitative real-time PCR (Fig. S6B).
Figure 3. Ulk1 and Ulk2 are dispensable for autophagy induction in DT40 cells. (A) DT40 cells deficient for Ulk1 (ulk1−/−), Ulk2 (ulk2−/−) or Ulk1 and Ulk2 (ulk1/2−/−) were generated by gene targeting and loss of wild-type alleles was confirmed by genomic PCR (for details see Figure S2). The absence of ulk1 and ulk2 transcripts was verified by RT-PCR. (B) Wild-type cells and two independent double deficient ulk1/2−/− clones (2–6 and 17–16) were incubated in full medium (RPMI) or EBSS in the presence or absence of 10 nM BafA1 for 1 h. Equal amounts of protein from cleared cellular lysates were analyzed for Atg13, GAPDH and LC3 by immunoblotting. LC3-II/GAPDH ratios are represented as mean values of three independent experiments ± SEM (C) ulk1/2−/− cells (clone 2–6) were incubated in normal growth medium (control) or EBSS for 2 h, cells were fixed and analyzed by transmission electron microscopy. For starvation condition, a representative cell is shown in two different magnifications. Autophagosomes are indicated by black arrow heads in the image with higher magnification (bars: 500 nm). (D) ulk1/2−/− cells (clone 2–6) retrovirally transfected with cDNA encoding mRFP-EGFP-rLC3 were incubated in EBSS for 2 h and analyzed by confocal laser scanning microscopy. The mRFP signal is shown in red and the EGFP signal in green in the merged image. Autolysosomes are indicated by white arrow heads (bars: 10 µm). The percentage of cells with autolysosomes (> 100 cells/experiment) is represented as mean value ± SD from three independent experiments. n.s. indicates a non-significant difference between wild-type and ulk1/2−/− cells (Student’s t-test).
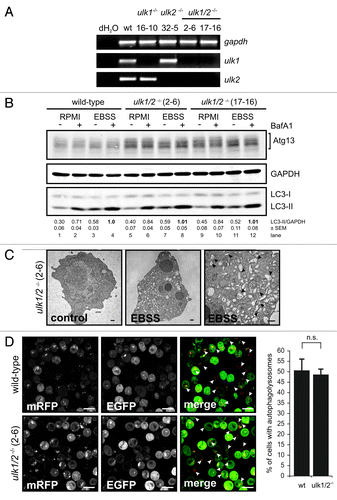
Interestingly, neither the single-deficient ulk1−/− and ulk2−/− cells (Fig. S3A) nor both double-deficient ulk1−/−ulk2−/− cell lines resembled the phenotype of atg13−/− cells since the absence of Ulk1 and Ulk2 had obviously no effect on starvation-induced autophagy. Autophagy induction was assessed by endogenous LC3 lipidation in the presence and absence of bafilomycin A1 (), autophagosome generation using electron microscopy (, S3B and S3C) and autolysosome formation using the mRFP-EGFP-rLC3 tandem construct ().
We additionally found that the five Ulk1-dependent in vitro phosphorylation sites we could identify in human Atg13 were dispensable for Atg13 function in DT40 cells (Fig. S4 and S5). Although it is commonly accepted that only the highly related Ulk1 and Ulk2 have the capability to interact with Atg13 via their unique C-terminal domain (Fig. S6A) and thus can complement each other, it is nevertheless conceivable that one of the other Ulk homologs (Ulk3, Ulk4 and STK36/Fused) may regulate starvation-induced autophagy in the absence of Ulk1 and Ulk2. To address this question, we analyzed the abundance of all known Ulk homologs (Ulk1, Ulk2, Ulk3, Ulk4 and STK36/Fused) by quantitative real-time PCR in wild-type DT40 cells, ulk1−/−, ulk2−/− and both double-deficient ulk1−/−ulk2−/− cell lines (Fig. S6B). Ulk2 seems to be the most highly abundant Ulk homolog in wild-type DT40 cells. Notably, none of the other Ulk homologs is significantly upregulated in the ulk1−/−ulk2−/− cells. Since Ulk3 has been implicated in autophagy induction after overexpressionCitation24 and ulk3 mRNA is found at moderate levels in DT40 cells (Fig. S6B), we analyzed autophagy induction after transient siRNA mediated knockdown of Ulk3 in the ulk1−/−ulk2−/− cells. However, starvation-induced autophagy was not affected by additional Ulk3 downregulation (Fig. S6C).
These results strongly suggest that in DT40 cells, the crucial function of Atg13 in autophagy induction is autonomous from Ulk1 and Ulk2. This surprising finding might be further explained by the fact that in our cells mTOR appears to play a minor role in autophagy induction. While starvation in EBSS leads to a prominent LC3-II accumulation already after 1 h, direct mTOR inhibition by rapamycin or Torin1 does not show a similar effect, although mTOR activity is repressed almost immediately (Fig. S7A–C). Interestingly, Atg13 did not display an altered migrational behavior in SDS-PAGE after starvation or mTOR inhibition (Fig. S7D). Thus, we conclude that Atg13 has an additional function in starvation-induced autophagy independent of mTORC1-regulated Ulk1/2 kinase activity.
Atg13 function depends on FIP200 binding
Since Ulk1 and Ulk2 seem to be dispensable for autophagy induction in this cell line, we investigated next whether FIP200 binding is essential for Atg13 function, or if Atg13 has additional functions completely independent of the Ulk1/2-Atg13-FIP200 complex.
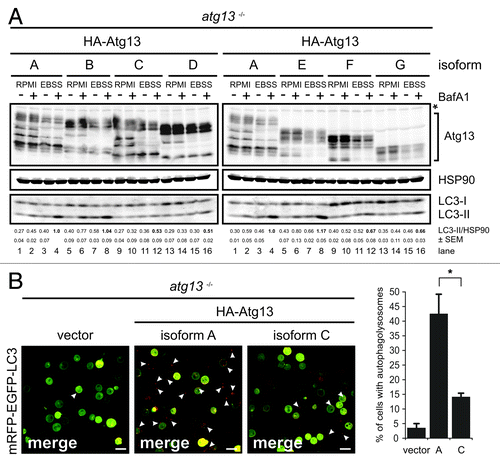
Interestingly, during cloning of chicken Atg13 we found that atg13 mRNA is alternatively spliced in DT40 cells (), which is consistent with previous findings in other species.Citation15 We determined the absolute abundance of these isoforms (named A-G) by quantitative real-time PCR using splice variant-specific primer combinations (Fig. S8A) and found that the full-length splice variant A, corresponding to human isoform 215, is most highly expressed (). Although the additional exon found in human isoform 1 is present on genomic level, it is not part of mature mRNA in DT40 cells (data not shown). Notably, this high variety of splice variants implicates a rather complex regulatory mechanism and hence might explain the incomplete reconstitution of atg13−/− cells with a single isoform ().
Figure 4. FIP200 binding site in Atg13 is encoded by exon 12. (A) Schematic representation of Atg13 splice variants (named A-G) amplified from DT40 cells. (B) The relative abundance of these splice variants was analyzed by qRT-PCR using splice variant-specific primer combinations (see Supplementary Material and Methods and Fig. S8A). (C) atg13−/− cells were reconstituted with HA-tagged versions of splice variants A-G and lysates were subjected to anti-HA immunoprecipitation and analyzed for Atg13 and FIP200 by immunoblotting. Asterisk indicates an unspecific background band.
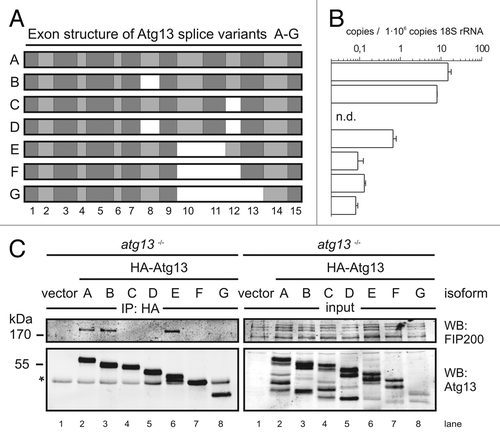
In order to analyze their capacity to interact with FIP200 we transfected atg13−/− cells and ulk1−/−ulk2−/− cells with cDNAs encoding HA-tagged versions of these splice variants and immunopurified Atg13 using anti-HA agarose. The cDNA of isoform C was generated by molecular cloning and is not expressed at detectable level (). Interestingly, FIP200 could be successfully co-immunopurified only with isoforms containing exon 12 both in the presence () and absence of Ulk1 and Ulk2 (Fig. S8B). This suggests that the FIP200 binding site within Atg13, previously narrowed down to the C-terminal region by Jung et al.,Citation15 is exclusively encoded by exon 12 and that the interaction between Atg13 and FIP200 does not depend on Ulk1 and Ulk2. Accordingly, only isoforms capable of FIP200 interaction (B and E) were able to reconstitute the autophagy-defective phenotype in atg13−/− cells to the same extent as full-length isoform A (). Furthermore, only the full-length isoform A but not isoform C (Δexon12) was able to efficiently restore autophagolysosome generation in atg13−/− cells (). These results clearly demonstrate that in DT40 cells, Atg13 has an essential function for basal as well as starvation-induced autophagy. It appears that this function necessarily requires the FIP200 binding capacity of Atg13 but seems to be completely independent of Ulk1 and Ulk2 and its regulation by mTOR.
Figure 5. FIP200 binding site is essential for Atg13 function. (A) atg13−/− cells were reconstituted with HA-tagged versions of splice variants A-G and incubated in full medium (RPMI) or EBSS for 1 h in the presence or absence of 10 nM BafA1. Equal protein amounts from cleared cellular lysates were analyzed for Atg13, HSP90 and LC3 by immunoblotting. LC3-II/HSP90 ratios from three independent experiments are represented as mean values ± SEM (B) atg13−/− retrovirally transfected with cDNAs encoding either HA-Atg13 isoform A (full-length) or HA-Atg13 isoform C (?exon12) or the empty vector were stably transfected with pmRFP-EGFP-rLC3. Cells were incubated in EBSS for 2 h and analyzed by confocal laser scanning microscopy. The mRFP signal is displayed in red and the EGFP signal in green in the merged image. The percentage of autolysosome containing cells (>100 cells/experiment) from three independent experiments is represented as mean value ± SD *p < 0.05, Student’s t-test.
Discussion
The mTORC1-regulated Ulk-Atg13-FIP200 complex plays a crucial role in autophagy induction. In particular, the mTORC1 substrates Ulk1 and Ulk2 have been suggested as major players in the initiation of autophagosome formation. In this report we investigated the role of the Ulk-Atg13-FIP200 complex in higher vertebrates by targeted disruption of the atg13, ulk1 and ulk2 loci in the same cellular system. Using this approach, we would like to propose the existence of an additional function of Atg13 that is completely independent of Ulk1/2 for the following reasons: First, the knockout of Atg13 clearly confirms its necessity for autophagy induction and hence the suitability of our system. Second, the function of Atg13 strongly depends on its interaction with FIP200, underlining their mutual dependence in autophagy regulation. Third, the simultaneous knockout of Ulk1 and Ulk2 does not resemble the phenotype of Atg13 knockout cells. Thus, we conclude that the joint action of Atg13 and FIP200 is a prerequisite for autophagy induction, whereas the mTOR-dependent regulation of Ulk1 and Ulk2 kinase activity seems to be expendable for starvation-induced autophagy in this model system.
The identification of autophagy-related genes in yeast and the direct regulation of their products by TOR once ushered in the molecular era of autophagy research. Meanwhile orthologs of most of these genes have been identified in higher eukaryotes.Citation25 However, it becomes more and more apparent that the regulation of the molecular machinery is more heterogenous than initially thought. While yeast, C. elegans and Drosophila possess only one Atg1 gene, higher vertebrates have at least five different kinases highly related to Atg1 (Ulk1-Ulk4 and STK36). Ulk1 and Ulk2 were the first identified homologs of C. elegans uncoordinated-51 (Unc-51). Although the knockdown of Ulk1 is sufficient to inhibit autophagy in some cell linesCitation3,Citation26 the respective knockout mouse has only a mild phenotype,Citation9 indicating that the most closely related Ulk1 and Ulk2 have at least partially redundant functions and can compensate for each other. This is further supported by the observation that Ulk1 and Ulk2 both can interact with Atg13 and FIP200, though with different affinities.Citation15 Notably, the C-terminal domain in Ulk1 and Ulk2 mediating this interaction is not conserved in Ulk3, Ulk4 and STK36.Citation27 Accordingly, only Ulk1 and Ulk2 have been found in a complex with Atg13, FIP200 and Atg101.Citation28
Based on previous findings it has been proposed that Atg13 primarily serves as an adaptor molecule mediating the interaction between Ulk1/2 and its substrate FIP200 as well as enhancing Ulk1/2 kinase activity.Citation29 Surprisingly, Ulk1 and Ulk2 appear to be dispensable for autophagy induction in DT40 cells. In contrast, atg13−/− cells do resemble the phenotype of FIP200−/− MEFsCitation8 and show a complete blockage of the autophagic flux. Furthermore, Atg13 function necessarily depends on FIP200 binding, since only those isoforms interacting with FIP200 were able to reconstitute the autophagy-defective phenotype of atg13−/− cells. The FIP200 binding site in Atg13 was found to be exclusively encoded by exon 12. Thus, it seems that Atg13 has an additional role, besides the mediation of Ulk1/2-dependent FIP200 phosphorylation.
Nutrient deprivation is one of the most potent and rapid inducers of autophagy. In yeast, the dephosphorylation of TOR-dependent phosphorylation sites in Atg13 is the initial step and sufficient for autophagy induction under starvation conditions.Citation30 In this regard, mTOR has been proposed as the major junction for nutrient status dependent signaling in higher vertebrates. Indeed, mTOR activity is immediately and dramatically reduced after nutrient deprivation (Fig. S7D). However, the mTOR inhibitors rapamycin and Torin1 fail to induce autophagy to a similar extent as starvation, although mTOR activity is repressed immediately after treatment with inhibitors (Fig. S7A–C). Furthermore, Atg13 protein did not display any prominent downshift in SDS-PAGE upon starvation or mTOR inhibition (Fig. S7D). If the release of mTOR-mediated repression on the Ulk-Atg13-FIP200 complex would fully account for the immediate autophagic response after starvation, one would expect the same kinetic response of autophagy induction following mTOR inhibition by rapamycin or Torin1. However, the recently identified direct regulation of Ulk1 kinase activity by AMPK might provide an explanation for this discrepancy.Citation31,Citation32
The dissimilar phenotypes of atg13−/− and ulk1/2−/− DT40 cells let us assume that Atg13 has a more basal function in these cells that is closely related to FIP200, but not necessarily regulated by Ulk1 or Ulk2. Interestingly, since FIP200−/− MEFs are resistant to autophagy induction by LiClCitation8 it has been suggested that FIP200 is additionally involved in mTOR-independent pathways. Although DT40 wild-type cells failed to respond to LiCl (data not shown), this might provide an explanation for the observed independence of autophagy induction from mTOR, Ulk1 and Ulk2 in DT40 cells. Furthermore, during revision of this manuscript Cheong et al. provided further evidence for an Ulk1/2-independent autophagy induction pathway in mammalian cells.Citation33 Though ulk1/2−/− MEFs were resistant to amino acid starvation, they did respond to bioenergetic stress from glucose withdrawal.Citation33 Thus, although we cannot exclude a cell type-specific feature, it is conceivable that DT40 cells preferentially use such an unconventional autophagy induction pathway upon starvation (for schematic model see ).
Figure 6. Model of differential Atg13-dependent autophagy induction pathways. Based on previous findings it has been proposed that mTORC1 associates with the Ulk-Atg13-FIP200 complex under nutrient-rich conditions, phosphorylates Ulk1/2 and Atg13 at inhibitory sites and suppresses Ulk1/2 kinase activity. Following starvation or direct mTORC1 inhibition, this negative regulation is released, Ulk1/2 autophosphorylates itself at activating sites and subsequently phosphorylates both Atg13 and FIP200. This in turn leads to autophagy induction (mTORC1-Ulk1/2-axis). In cells that do respond to mTORC1 inhibition by autophagy induction and do depend on Ulk1 and Ulk2, this pathway is most likely favored. However, the incomparable phenotypes of atg13−/− and ulk1/2−/− DT40 cells let us assume that Atg13 has a more basal function that is not necessarily regulated by Ulk1 or Ulk2, but necessarily requires FIP200 binding capacity. Thus, Atg13 surprisingly has an additional role, besides its proposed function as an adaptor molecule that bridges Ulk1/2 and its substrate FIP200. Several modes of action are conceivable: (A) Atg13 acts in a kinase-independent way, e.g. by stabilizing or recruiting FIP200, (B) the Atg13-FIP200 complex is regulated in a kinase-independent manner, or (C) Atg13-FIP200 is regulated by other kinases than Ulk1/2.
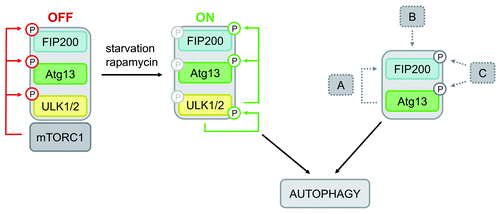
Since Atg13-FIP200 can obviously be triggered by upstream signaling events other than the already known major mTOR-Ulk1/2 axis, it is conceivable that Atg13 constitutes a central signaling node that integrates different upstream pathways. In yeast, PKA is known to regulate autophagy induction, in addition to TOR, by direct phosphorylation of Atg13.Citation34 However, none of these PKA phosphorylation sites is conserved in Atg13 orthologs of higher vertebrates. Hence, it would be reasonable to assume an additional regulation of Atg13 and FIP200 by other kinases in response to starvation. However, in a recent publication the authors failed to identify nutrient-regulated phosphorylation sites in Atg13.Citation35 Thus, future studies have to reveal the relevance of those alternative induction pathways in more detail.
Supplementary information is available at Autophagy’s website.
Material and Methods
Cell culture
Chicken DT40 cells were cultured in RPMI 1640 (BE12-702F, Lonza) supplemented with 10% FCS, 1% chicken serum, 3 mM L-glutamine, 50 µM β-mercaptoethanol, 50 U/ml penicillin and 50 µg/ml streptomycin (full medium). For starvation, DT40 cells were washed twice and incubated in EBSS (14155-048, Gibco) for the indicated time. Flp-InTM T-RExTM 293 cells (R780–07, Invitrogen) stably expressing FLAG-Ulk1 wt or FLAG-Ulk1 kinase dead (D165A) and Plat-E cells (kindly provided by Toshio Kitamura, Tokyo, Japan)Citation36 were cultured in DMEM (4.5 g/l D-Glucose, E15-810, PAA) supplemented with 10% FCS, 50 U/ml penicillin and 50 µg/ml streptomycin. All cell lines were maintained at 37°C in a 5% CO2 incubator.
Antibodies and reagents
Anti-p70S6K (9202), anti-phospho-p70S6K (p-Thr389) (9206) and anti-LC3B antibodies (2775) were purchased from Cell Signaling Technology, anti-GAPDH (6C5, ab8245) from Abcam, anti-HSP90 from BD Transduction laboratories (610418) and anti-GFP from Boehringer Mannheim (1814460). Monoclonal anti-HA agarose (A2095) was purchased from Sigma. Anti-Atg13 antibodies were kindly provided by Do-Hyung Kim (Department of Biochemistry and Biophysics, University of Minnesota, Minneapolis, USA).Citation15 Anti-FIP200 antibodies were kindly provided by Noboru Mizushima (Department of Physiology and Cell Biology, Tokyo Medical and Dental University, Japan).Citation8 IRDye®800 and IRDye®680 conjugated secondary antibodies were purchased from LI-COR Biosciences (926-32210/11 and 926-68020/21). Torin1 was kindly provided by Nathanael Gray (Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts, USA) and David Sabatini (Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, USA),Citation37 rapamycin (553210) and Bleocin® (203408) were purchased from Calbiochem; G418 from Biochrom (A2912), bafilomycin A1 (B1793), histidinol (H6647) and mycophenolic acid (M3536) from Sigma. Blasticidin (ant-bl-1) and puromycin (ant-pr-1) were purchased from InvivoGen.
Expression constructs and retroviral infection
Chicken atg13 cDNAs for isoforms A, B, D, E, F and G were amplified by RT-PCR from DT40 RNA and cloned into pCR®-II TOPO® vector (Invitrogen, 10351-021). Coding sequence for HA tag was introduced at the 5′-end by PCR. Isoform C was generated from isoforms A and D by molecular cloning, using a unique BbsI site in exon 9. Amino acid substitutions in Atg13 were generated by site-directed mutagenesis of atg13 cDNA and verified by sequencing. For transfection, Atg13 cDNA was subcloned into retroviral expression vector pMSCVpuro (Clontech, PT3303-5) via EcoRI. pMSCV-mRFP-EGFP-rLC3 expression vector was generated by cloning of mRFP-EGFP-rLC3 cDNA from pmRFP-EGFP-rLC3 (kindly provided by Tamotsu Yoshimori, Department of Genetics, Osaka, Japan) via NheI/EcoRI into the HpaI site of pMSCVpuro. Alternatively, cells were transfected with DraIII linearized pmRFP-EGFP-rLC3 by electroporation and selected in medium containing 2 mg/ml G418. The pMSCV-mCitrine-chLC3B expression vector has been previously described.Citation17 For the production of recombinant retroviruses, Plat-E cells were transfected with pMSCV-based vector using FuGENE transfection reagent (Roche, 11988387001). The MMLV was pseudotyped with VSV-G. 1 × 106 DT40 cells were incubated with retroviral supernatant containing 3 µg/ml Polybrene (Sigma, H9268) and selected in medium containing 0.5 µg/ml puromycin.
Generation of DT40 knockout cell lines
For Atg13 targeting vectors, a 6 kb fragment was amplified from DT40 genomic DNA and cloned into pBluescript II SK(-) vector. The start codon in exon 1 was mutated and a histidinol (hisD) or blasticidin (bsr) resistance cassette was inserted into this newly generated BamHI site, respectively. DT40 cells were transfected with linearized targeting vectors by electroporation at 250 V and 975 µF and selected in 1 mg/ml histidinol and 50 µg/ml blasticidin. Atg13-deficient clones were screened by genomic PCR and immunoblotting. For further details and generation of Ulk1 and Ulk2 targeting vectors see Supplementary Material and Methods.
Immunoblotting and immunopurification
DT40 cells were incubated in full medium or EBSS, containing 10 nM bafilomycin A1 or DMSO as control, at a density of 1 × 106 cells/ml for the indicated time. Cells were lysed in lysis buffer [10 mM TRIS-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1% Triton X-100, 10 mM NaF, 2.5 mM NaPP, 10 µM Na2MoO4, 1 mM Na3VO4, and protease inhibitors (P2714, Sigma)] and clarified by centrifugation at 16,000 g for 20 min. Equal total protein amounts, as determined by Bradford, were separated on an 8–15% gradient SDS-polyacrylamide gel and transferred to PVDF membrane (Millipore). Immunoblot analysis was performed using the indicated primary antibodies and appropriate IRDye®800 or IRDye®680 conjugated secondary antibodies (LI-COR Biosciences). Signals were detected with an Odyssey® Infrared Imaging system (LI-COR Biosciences) and quantified using ImageJ 1.41 (http://rsb.info.nih.gov/ij/). For immuno-purifications, cells were lysed in IP buffer [40 mM HEPES pH 7.5, 2 mM EGTA, 0.3% CHAPS, 10 mM NaPP, 1 mM Na3VO4 and protease inhibitors (P2714, Sigma)] and equal total protein amounts were incubated with monoclonal anti-HA-agarose (A2095, Sigma) over night. Beads were washed four times with lysis buffer (without protease inhibitors), boiled in sample buffer and supernatant was subjected to SDS-PAGE.
Quantitative real-time PCR
Quantitative real-time RT-PCR analysis was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems) and SYBR Green-containing qPCR Mastermix Plus (Fermentas, K0222). Total RNA from 1 × 106 cells was isolated using the NucleoSpinTM RNA II-Kit (Macherey & Nagel, 740955.50). cDNA was generated from 1 µg of total RNA with 200 U RevertAid H MinusTM reverse transcriptase (Fermentas, EP0452), 50 µM random hexamers (Fermentas, SO142), 400 µM dNTPs (Fermentas, R0192), and 1.6 U/µl RiboLockTM RNase inhibitor (Fermentas, EO0381) according to manufacturer’s recommendations. 80/8/0.8 ng of the resulting cDNA (in duplicates) were applied to qRT-PCR analyses and amplified in the presence of 300 nM primers (for sequences see Supplementary Materials) with the standard temperature profile (2 min 50°C, 10 min 95°C, 40 cycles 15 sec 95°C, 1 min 60°C). For absolute quantification of splice variants, cDNA standards for each variant were generated by EcoRI digestion of the corresponding plasmid, isolated from agarose gels and quantified by spectrophotometry. The copy number of standard DNA molecules was calculated using the following formula: X (g/µl) DNA / [DNA fragment length (bp) × 660 (g/mole)] × 6.022 × 1023 = Y (copies/µl). Standard curves (plot of CT value/crossing point against log of copy number) were generated from serial 10-fold dilutions, ranging from 1 × 1010 to 1 × 100 copies per point. The detection limits were 10 copies per reaction for Ulk1, Ulk2, Ulk3, Ulk4 and 100 copies per reaction for STK36 and 18S rRNA. Results of absolute copy numbers detected in the samples were normalized to the absolute copy numbers of 18S rRNA to account for RT-PCR efficiency and are displayed as copies per 1 × 109 copies 18S rRNA. Mean values ± SD over three dilutions (80/8/0.8 ng cDNA per reaction) are given.
Confocal laser scanning microscopy
For starvation conditions, cells were washed once with EBSS, resuspended in EBSS and seeded onto chambered coverglasses (Nunc). After 2 h cells were analyzed on a Leica TCS SP II confocal laser scanning microscope. As control, cells were resuspended in Krebs Ringer solution [10 mM HEPES (pH 7.0), 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 10 mM glucose] directly prior to analysis. EGFP and mCitrine were excited at 488 nm, and mRFP at 543 nm wavelength.
Transmission electron microscopy
Transmission electron microscopy was performed as previously decribed.Citation38,Citation39 Briefly, DT40 cells were incubated in normal growth medium or EBSS at a density of 1 × 106 cells/ml for 2 h at 37°C. Cells were harvested, washed with prewarmed PBS, fixed with warm Karnovsky’s solution for 10 min at 37°C and stored at 4°C. For electron microscopic analysis, cells were embedded in 3.5% agarose at 37°C, coagulated at room temperature and fixed again in Karnovsky’s solution. Post-fixation was based on 1.0% osmium tetroxide containing 1.5% K-ferrocyanide in 0.1 M cacodylate buffer for 2 h. After following standard methods, blocks were embedded in glycide ether and cut using an ultra microtome (Ultracut). Ultra-thin sections (30 nm) were mounted on copper grids and analyzed using a Zeiss LIBRA 120 transmission electron microscope (Carl Zeiss) operating at 120 kV.
Additional material
Download Zip (592.7 KB)Acknowledgment
We thank Toshio Kitamura for providing Plat-E cells, Do-Hyung Kim for Atg13 antibodies, Nathanael Gray and David Sabatini for Torin1, Tamotsu Yoshimori for the pmRFP-EGFP-rLC3 construct, and Noboru Mizushima for FIP200 antibodies and helpful discussions. We thank Maria Deak (MRC Protein and Phosphorylation Unit, University of Dundee, UK) and protein purification teams of the Division of Signal Transduction Therapy (University of Dundee) for GST-Atg13. We are grateful to Dario Alessi for carefully reading the manuscript and for helpful suggestions. We also thank Tassula Proikas-Cezanne for helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft SFB 773 (to S.W., B.S. and M.S.), GRK 1302 (to S.W. and B.S.), from the Interdisciplinary Center of Clinical Research, Faculty of Medicine, Tübingen (Nachwuchsgruppe 1866-0-0, to B.S.) and the UK Medical Research Council (to D.G.C.). The Division of Signal Transduction Therapy (University of Dundee) is supported by the following pharmaceutical companies: AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck-Serono and Pfizer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 2005; 16:2544 - 53; http://dx.doi.org/10.1091/mbc.E04-08-0669; PMID: 15743910
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507 - 13; http://dx.doi.org/10.1083/jcb.150.6.1507; PMID: 10995454
- Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 2007; 282:25464 - 74; http://dx.doi.org/10.1074/jbc.M703663200; PMID: 17595159
- Kuroyanagi H, Yan J, Seki N, Yamanouchi Y, Suzuki Y, Takano T, et al. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics 1998; 51:76 - 85; http://dx.doi.org/10.1006/geno.1998.5340; PMID: 9693035
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 1999; 24:833 - 46; http://dx.doi.org/10.1016/S0896-6273(00)81031-4; PMID: 10624947
- Yan J, Kuroyanagi H, Tomemori T, Okazaki N, Asato K, Matsuda Y, et al. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene 1999; 18:5850 - 9; http://dx.doi.org/10.1038/sj.onc.1202988; PMID: 10557072
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29:157 - 71; http://dx.doi.org/10.1128/MCB.01082-08; PMID: 18936157
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497 - 510; http://dx.doi.org/10.1083/jcb.200712064; PMID: 18443221
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493 - 502; http://dx.doi.org/10.1182/blood-2008-02-137398; PMID: 18539900
- Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007; 3:106 - 16; PMID: 17204848
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal 2009; 2:pe51; http://dx.doi.org/10.1126/scisignal.284pe51; PMID: 19690328
- Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell 2009; 20:2004 - 14; http://dx.doi.org/10.1091/mbc.E08-12-1250; PMID: 19225150
- Ganley IG. Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297 - 305; http://dx.doi.org/10.1074/jbc.M900573200; PMID: 19258318
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 91; http://dx.doi.org/10.1091/mbc.E08-12-1248; PMID: 19211835
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003; http://dx.doi.org/10.1091/mbc.E08-12-1249; PMID: 19225151
- Funakoshi T, Matsuura A, Noda T, Ohsumi Y. Analyses of APG13 gene involved in autophagy in yeast, Saccharomyces cerevisiae. Gene 1997; 192:207 - 13; http://dx.doi.org/10.1016/S0378-1119(97)00031-0; PMID: 9224892
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914 - 25; http://dx.doi.org/10.1016/j.cellsig.2010.01.015; PMID: 20114074
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem 2010; 285:16912 - 20; http://dx.doi.org/10.1074/jbc.M110.114207; PMID: 20308071
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006 - 17; http://dx.doi.org/10.1038/cdd.2009.34; PMID: 19325567
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 2006; 281:36303 - 16; http://dx.doi.org/10.1074/jbc.M607031200; PMID: 16963441
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139
- Lee EJ, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 2011; 7:689 - 95; http://dx.doi.org/10.4161/auto.7.7.15450; PMID: 21460635
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798 - 803; http://dx.doi.org/10.1101/gad.519709; PMID: 19279323
- Meléndez A, Neufeld TP. The cell biology of autophagy in metazoans: a developing story. Development 2008; 135:2347 - 60; http://dx.doi.org/10.1242/dev.016105; PMID: 18567846
- Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006; 119:3888 - 900; http://dx.doi.org/10.1242/jcs.03172; PMID: 16940348
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132 - 9; http://dx.doi.org/10.1016/j.ceb.2009.12.004; PMID: 20056399
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 95; http://dx.doi.org/10.1016/j.febslet.2010.01.017; PMID: 20083114
- Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol 2010; 30:1049 - 58; http://dx.doi.org/10.1128/MCB.01344-09; PMID: 19995911
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456 - 61; http://dx.doi.org/10.1126/science.1196371; PMID: 21205641
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA 2011; 108:11121 - 6; http://dx.doi.org/10.1073/pnas.1107969108; PMID: 21690395
- Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci USA 2009; 106:17049 - 54; http://dx.doi.org/10.1073/pnas.0903316106; PMID: 19805182
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA 2011; 108:4788 - 93; http://dx.doi.org/10.1073/pnas.1100844108; PMID: 21383122
- Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 2000; 7:1063 - 6; http://dx.doi.org/10.1038/sj.gt.3301206; PMID: 10871756
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284:8023 - 32; http://dx.doi.org/10.1074/jbc.M900301200; PMID: 19150980
- Gröbner S, Fritz E, Schoch F, Schaller M, Berger AC, Bitzer M, et al. Lysozyme activates Enterococcus faecium to induce necrotic cell death in macrophages. Cell Mol Life Sci 2010; 67:3331 - 44; http://dx.doi.org/10.1007/s00018-010-0384-9; PMID: 20458518
- Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, et al. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 2011; 469:419 - 23; http://dx.doi.org/10.1038/nature09674; PMID: 21248850