Abstract
In recent years, the process of selective autophagy has received much attention with respect to the clearance of protein aggregates, damaged mitochondria, and bacteria. However, until recently, there have been virtually no studies on the selective autophagy of viruses, although they are perhaps one of the most ubiquitous unwanted constituents in human cells. Recently, we have shown that the ability of neuronal Atg5 to protect against lethal Sindbis virus central nervous system (CNS) infection in mice is associated with impaired viral capsid clearance, increased p62 accumulation, and increased neuronal cell death. In vitro, we showed that p62 interacts with the Sindbis capsid protein and targets it for degradation in autophagosomes. Herein, we review these findings and broadly speculate about potential roles of selective viral autophagy in the regulation of host immunity and viral pathogenesis.
By definition, all viruses—regardless of their genetic structure, taxonomic classification or intracellular site of genomic replication—are obligate intracellular parasites and must pass through the cytoplasm of the infected host cell at some point in their life cycle. As a corollary, the prediction is that eukaryotic cells should possess mechanisms to target intracytoplasmic viral components, such as assembled virus particles, viral proteins or viral nucleic acids, for autophagic destruction. It is not yet known whether this prediction is true, and if so, what are the underlying molecular mechanisms that govern the distinct stages of selective viral autophagy. Although much remains to be learned, we speculate that viruses are previously unrecognized important targets of selective autophagy, and that selective viral autophagy plays a crucial role in antiviral host defense.
Sindbis Virus Capsid: A Target of p62-Mediated Selective Autophagy
Our laboratory sought to examine the role of the endogenous autophagy machinery in host defense against a mammalian virus infection. In earlier reports, autophagy genes limited the spread of cell death during the hypersensitive response in plants infected with tobacco mosaic virus (TMV)Citation1 and protected flies against lethal infection with vesicular stomatitis virus (VSV).Citation2 However, no studies had established a direct role for autophagy in host defense against viral infection in a vertebrate model system. To examine this question, we studied the effect of neuronal inactivation of the Atg5 autophagy gene on the pathogenesis of neonatal CNS infection with Sindbis virus (SIN), an enveloped, positive-strand RNA virus in the alphavirus genus that serves as an animal model for human arthropod-borne encephalitides.Citation3
First, we performed in vitro studies to determine whether SIN induces autophagy, whether SIN is targeted to autophagosomes, and whether autophagy controls SIN replication.Citation3 We found that SIN infection induces autophagy in vitro, as evidenced by the induction of GFP-LC3 puncta, and the degradation of p62 in SIN-infected murine embryonic fibroblasts (MEFs) and mouse neuronal cells. In contrast to the findings of Shelly et al.,Citation2 SIN-induced autophagy requires viral replication, as autophagy is not observed in cells infected with UV-inactivated SIN. We also found that the SIN capsid protein colocalizes with GFP-LC3 in wild-type but not in Atg5-deficient MEFs. This colocalization represents the targeting of SIN capsid protein for autophagic degradation, as demonstrated by electron microscopy (EM) and live cell imaging. Although Atg5 is required for SIN capsid autophagic degradation, it is not required for the control of viral replication, since no differences in viral growth curves are observed in Atg5-deficient versus wild-type MEFs or embryonic stem cells. Thus, in vitro, SIN replication induces an autophagic response that results in the degradation of viral capsid, but not in the control of viral replication.
Next, we used three complementary approaches to inactivate the essential autophagy gene Atg5 in SIN-infected neurons in vivo. In the first model, a double subgenomic SIN vectorCitation4 was used to express a dominant-negative mutant version of Atg5 (Atg5K130R) in infections of wild-type mice. In the second model, the SIN vector was used to express Cre recombinase, resulting in the deletion of floxed alleles of Atg5 only in infected neurons of Atg5flox/flox mice. In the third model, we infected animals with neuronal specific deletion of Atg5 (Atg5flox/flox;nestin-Cre) with wild-type SIN. In each model, we observed a significant increase in mortality in animals in which Atg5 function was disrupted or Atg5 was deleted, demonstrating that the endogenous autophagy machinery functions to protect against viral infection in vertebrates.
Somewhat surprisingly, but nonetheless consistent with our in vitro findings in Atg5-deficient cells, the increased mortality in mice disrupted of Atg5 is not associated with increased viral titers in the brain. Also, there were no differences observed in the levels of type I interferon, a critical mediator of host defense against SIN infection, in the brains of mice with intact versus those with disrupted neuronal Atg5 function. Yet, there were striking changes noted at the histopathological level. Mice with disrupted neuronal Atg5 have a marked delay in the clearance of SIN antigens, increased accumulation of p62 aggregates, and increased apoptosis of infected neurons. While it is difficult to conclude cause and effect relationships from in vivo viral pathogenesis studies, these results are most consistent with a scenario in which defective cell-autonomous clearance of protein aggregates produced during viral infection results in neuronal death, ultimately leading to decreased survival of the infected animal.
Our in vivo studies led us to ask the question—how are SIN proteins targeted to the autophagosome? As discussed in detail in other articles in this issue of Autophagy, p62 is an adaptor protein, containing a ubiquitin-binding association (UBA) domain and an LC3-interaction region (LIR), that targets ubiquitinated substrates to the autophagosome. In recent years, the role of p62 in the selective autophagy of cellular proteins and bacteria has received much attention, whereas its potential role in targeting viral proteins had been unexplored. However, in retrospect, a potential link between selective autophagy adaptors and host defense against viruses was suggested decades ago; ref(2)P, the Drosophila melanogaster p62 ortholog, was found to be responsible for restriction of sigmavirus replicationCitation5,Citation6 and ref(2)P was found to bind sigmavirus capsid in coimmunoprecipitation studies.Citation7 In addition, mutations in the UBA domain of p62 are associated with Paget disease of the bone (reviewed in ref. Citation8), which has long been suspected to have an underlying viral etiology, as intracellular inclusions resembling paramyxovirus nucleocapsids have been detected in ultrastructural studies of Pagetic bone lesions.Citation9
These reports, coupled with our observations of p62 aggregates in the neurons of SIN-infected mice lacking intact Atg5 function, led us to hypothesize that p62 might target SIN antigens for autophagic clearance. Indeed, we identified SIN capsid as a p62-interacting protein in coimmunoprecipitation studies, and found that siRNA-mediated knockdown of p62 blocks SIN capsid colocalization with GFP-LC3, delays clearance of SIN capsid from infected cells, and accelerates apoptosis of infected cells. Thus, the SIN capsid protein is delivered to the autophagosome by a mechanism involving the p62 adaptor, which helps to protect against virus-induced cell death. Our findings provide the first example of a viral protein serving as a target for selective autophagy, and suggest a role for selective viral autophagy in cell survival during viral infection.
SIN Capsid Recognition by p62: Unanswered Questions
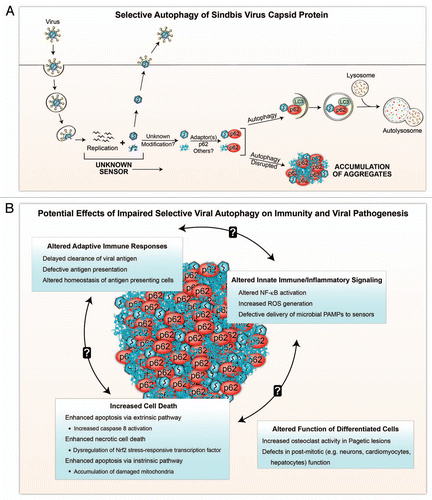
We demonstrated that the SIN capsid protein is delivered to the autophagosome in a p62-dependent manner;Citation3 however, much remains to be learned about this process (). Three crucial unanswered questions include: (1) How does the cell sense the presence of SIN infection to activate SIN capsid autophagy? (2) Is p62-mediated recognition of SIN capsid ubiquitin-dependent or ubiquitin-independent? (3) What form(s) of capsid (monomers, aggregates or assembled nucleocapsids) are recognized by p62?
Several sensors of pathogen-associated molecular patterns (PAMPs) have been shown to induce autophagy (reviewed in ref. Citation10 and Citation11). Most attention has focused on bacterial recognition and autophagy induction via Toll-like receptors (TLRs) and Nod-like receptors (NLRs). Endosomal TLRs that sense viral nucleic acids (TLR3, 7, 8, 9) have also been reported to activate autophagy. In the case of SIN infection, we postulate that autophagy induction is mediated by a cytoplasmic RNA sensor and/or by danger-associated molecular patterns (DAMPs) such as ER stress and reactive oxygen species (ROS) production that increase during infection and induce autophagy. One candidate sensor, the double-stranded RNA-activated kinase, PKR, is required for herpes simplex virus type I (HSV-1)-induced autophagy.Citation12,Citation13 In unpublished studies from our laboratory, we observed a lack of autophagosomes (at the EM level) in SIN-infected pkr-/- primary sympathetic neurons as well as increased mortality in SIN-infected pkr-/- mice. Thus, PKR may represent a sensor that initiates the autophagy pathway in response to viral dsRNA replication intermediates during SIN infection. Other candidates include members of the RIG-I-like helicase (RLH) family, such as RIG-I and MDA5, which sense cytoplasmic viral RNA, although it is not yet known whether these sensors activate autophagy during viral infection. The NLR family member NOD2, which activates autophagy in response to bacterial PAMPs,Citation11 also recognizes single-stranded viral RNA and activates innate antiviral immunity,Citation14 suggesting that it might also be a candidate sensor for autophagy induction during viral infection. A further possibility is that autophagy activation during SIN replication proceeds via a novel pathway that involves as-of-yet unidentified nucleic acid sensors or alternatively, other signals generated during viral replication (such as newly synthesized viral proteins or cellular stress signals elicited in response to viral replication).
An even bigger “black box” than the identity of the sensor is the nature of the cellular events that link the presumed viral RNA-sensing signal with the orchestration of selective viral protein substrate recognition and targeting to the autophagosome. Along these lines, our laboratory has completed a high-content genome-wide siRNA screen to identify novel host factors that are required for targeting SIN capsid to LC3-positive autophagosomes (unpublished data). The further analyses of these factors, which include molecules involved in substrate recognition, mRNA processing, and cytoskeletal trafficking, may begin to shed light on the intermediate events in selective viral autophagy.
Although the general paradigm is that p62 recognizes ubiquitinated substrates through its UBA domain (reviewed in ref. Citation15–Citation18), it is possible that p62-mediated targeting of SIN capsid occurs in a ubiquitin-independent manner. In contrast to the aggregates that accumulate in the brains of older mice with neuronal-specific deletion of Atg5 or Atg7, which stain positive for both ubiquitin and p62,Citation19,Citation20 we did not find any ubiquitin-staining in association with the p62-positive, SIN capsid-positive aggregates that accumulate in the brains of SIN-infected mice with disrupted neuronal Atg5 function.Citation3 Moreover, we were unable to detect capsid ubiquitination during SIN infection in vitro, even after treatment with the proteasome inhibitor MG132, or overexpression of HA-tagged ubiquitin. Thus, although we cannot formally exclude a role for capsid ubiquitination in p62-mediated autophagy, the preponderance of our data suggest that p62 recognizes capsid in a ubiquitin-independent manner. However, we have only examined the SIN capsid-p62 interaction in coimmunoprecipitation studies and do not yet know whether the interaction is direct or indirect. Therefore, it is possible that an as-of-yet unidentified capsid-binding protein that is ubiquitinated may serve as the direct target for p62, although this would not explain the lack of detection of ubiquitin aggregates in the brains of SIN-infected mice with disrupted neuronal Atg5 function and p62 aggregates. Future studies are needed to determine whether the UBA domain of p62 is required for SIN capsid binding and for SIN capsid autophagy; to determine whether p62 and SIN capsid bind directly in vitro; and to identify other components of the SIN capsid-p62 complex. Such studies may reveal novel mechanisms by which p62 recognizes autophagy substrates.
It will also be of significant interest to determine whether similar p62-dependent targeting mechanisms underlie the recognition of viral capsids from other virus families. As noted above, the Drosophila p62 ortholog, ref(2)P, coimmunoprecipitates with sigmavirus capsid protein,Citation7 indicating the biochemical interactions between viral capsid proteins and p62 are not restricted to the alphavirus genus. Although it remains controversial whether infection with paramyxoviruses (such as measles virus, respiratory syncytial virus and canine distemper virus) are etiologically linked to the pathogenesis of Paget disease bone lesions in patients with germline p62 mutations,Citation8,Citation21 our findings with p62-mediated clearance of SIN capsid provide a potential explanation for the clinical association that has been observed between patients with p62 mutations, Paget disease and the accumulation of paramyxovirus nucleocapsid in Pagetic bone lesions. Mutations in p62 occur in 25–30% of patients with familial Paget disease, and about 10% of patients with sporadic Paget disease. In the 1970s, nuclear and cytoplasmic inclusions that were similar to nucleocapsids from paramyxoviruses were observed by ultrastructural analysis in osteoclasts from Paget disease patients,Citation22 and measles virus and/or respiratory syncytial virus nucleocapsid antigens were detected in osteoclasts from Paget disease patients.Citation9,Citation23 More recent studies include the identification of canine distemper virus nucleocapsid protein gene transcripts in osteoclasts from Paget disease patients (11/25 using in situ hybridization;Citation24 12/12 using in situ PCRCitation25) and the detection of measles virus nucleocapsid protein gene transcripts in approximately 70% of patients harboring the most common p62 mutation associated with Paget disease (p62P392L).Citation21 We speculate that the accumulation of paramyxovirus nucleocapsids in patients with mutations in p62 may result from deficient p62-mediated selective autophagy of paramyxovirus nucleocapsids.
An interesting question vis-à-vis our findings with SIN capsid protein, as well as other viral capsid proteins that may potentially interact with p62, is whether p62 interacts with the nascent protein and/or the assembled viral nucleocapsid. The SIN capsid assembles with SIN viral RNA to form a nucleocapsid in the cytoplasm (rapidly after viral genomic replication) and buds from the plasma membrane (where it acquires its two envelope glycoproteins). From a teleological point of view, it makes sense for the host cell to target the assembled nucleocapsid to the autophagy pathway, so that the viral genetic material can also be degraded, thereby restricting viral replication. Indeed, our EM analyses of SIN-infected cells reveal the presence of SIN nucleocapsids inside autophagosomesCitation3 although EM analyses are insufficient to rule out the possibility that other forms of capsid protein may also be targeted for degradation. However, we did not observe an increase in SIN replication in vivo in neurons with deficient Atg5 function or in vitro in cells with Atg7 or p62 knockdown. This suggests that autophagy-independent pathways are sufficient to control cellular production of SIN infectious virus, but not to clear excess capsid protein and/or nucleocapsids that may accumulate in virally-infected cells. This is perhaps not surprising in view of observations that only a small percentage of the total amount of SIN capsid that is produced correctly assembles with SIN RNA to form intact viral nucleocapsids; the majority is thought to form defective cytoplasmic particles that may contain other single-stranded RNA (personal communication, Richard Kuhn, Purdue University). Further studies are required to determine which form(s) of SIN capsid, and potentially other viral capsid proteins, are substrates of p62 during selective viral autophagy. The strong prediction is that autophagy may be particularly important in degrading assembled SIN capsid particles since these are too large to be degraded by the ubiquitin-proteasome system.
p62-Mediated Selective Viral Autophagy: Broad Implications for Immunity and Viral Pathogenesis
We speculate that selective viral autophagy may play a crucial role in life and death decisions of the infected cell, and may also help govern the “quality of life” of virally-infected cells and surrounding tissue. Based on the synthesis of our findings (in which the defective clearance of SIN capsid-p62 aggregates is associated with increased cell death),Citation3 and the growing body of literature on the diverse functions of p62 (reviewed in ref. Citation26), we propose that the abnormal accumulation of viral capsid-p62 aggregates may potentially alter innate and adaptive immunity, cell death and homeostatic and specialized functions of nondividing cells ().
p62 not only functions as an adaptor protein that targets substrates to the autophagosome, but also as a scaffold protein that recruits and oligmerizes signaling proteins to regulate inflammatory and apoptotic signaling pathways. In addition to the UBA domain and LIR region, p62 has several other functionally important domains, including a PB1 domain, which is a protein-protein interaction module present in many signaling molecules, and a TRAF6-interacting domain. The oligomerization/aggregation of p62 results in the formation of “signal-organizing centers” where p62 interacts with TRAF6 and caspase 8.Citation26,Citation27 The interaction of p62 with TRAF6 promotes TRAF6 oligomerization and subsequent activation, which leads to K63-linked polyubiquitination of TRAF6 and activation of NFκB. Accordingly, most studies have shown that increased p62 accumulation results in increased activation of NFκB signaling.Citation26,Citation27 Therefore, by extrapolation, the accumulation of viral capsid-p62 aggregates may enhance pro-inflammatory NFκB signaling during viral infection.
There is a potential precedent in the literature for synergy between increased p62 function and viral capsid accumulation in disease pathogenesis. Knock-in mutant mice that express the most common mutation found in p62 in association with Paget disease display increased NFκB signaling and enhanced osteoclastogenesis.Citation26 Among osteoclast precursors derived from Paget disease patients with this mutation (p62P392L), only those that also have detectable measles virus nucleocapsid transcripts form osteclasts with a Pagetic phenotype in vitro.Citation21
We speculate that the accumulation of viral capsid-p62 aggregates may alter the specialized functions of other differentiated cells besides osteoclasts, in particular those that do not routinely turn over, such as neurons, cardiomyocytes and hepatocytes. One mechanism which has been proposed by which p62 aggregates may alter cellular function is by sequestering ubiquitinated proteins and preventing their access to the proteasome, thereby increasing the levels of short-lived proteins.Citation28 This was demonstrated for two proteasome substrates, p53 and β-catenin; a similar mechanism involving these and other proteasome substrates could underlie abnormal cellular function during virus infection. In our model of SIN CNS infection, this seems unlikely, as no ubiquitin accumulation is detected in association with SIN capsid and p62 neuronal aggregates; however, this concept remains a possibility for SIN infection in other cell types and for other viral infections. Additional, as-of-yet undefined effects of viral capsid-p62 aggregates could also interfere with the normal function of differentiated cells.
Several scenarios might contribute to the increased death of virally-infected cells with p62 aggregates. The interaction of p62 with caspase 8 serves as a scaffold for the aggregation of ubiquitinated caspase 8, leading to its autoproteolytic processing and activation, and thereby potentiating the extrinsic apoptotic signaling pathway that is triggered by death ligands.Citation29 Another possibility is suggested by the discovery that p62 controls the levels of Nrf2-dependent gene transcription by binding to the E3 ubiquitin ligase Keap1, which normally targets Nrf2 for degradation.Citation30 While the target genes of Nrf2 are normally cytoprotective, their constitutive activation in the context of deficient autophagy somewhat paradoxically results in necrotic cell death of hepatocytes. Also, defects in other forms of p62-dependent selective autophagy, such as mitophagy, in virally-infected cells could potentially contribute to cellular damage (through ROS generation) and/or activation of the intrinsic apoptotic pathway. It is not yet known which, if any, of these mechanisms contribute to the accelerated cell death that is observed in autophagy-deficient cells infected with SIN,Citation3 a virus that induces death via a classical apoptotic pathway.Citation31
In addition to effects on NFκB signaling, we speculate that the accumulation of viral capsid-p62 aggregates may exert a wide range of other effects on immunity. One potential mechanism is via increased ROS generation. Enforced p62 expression in autophagy-deficient cells results in increased ROS accumulation,Citation32 and increased ROS generation in the setting of deficient autophagy enhances type I interferon responsesCitation33 and contributes to increased inflammasome activation.Citation34 Although not yet directly tested, it seems likely that the autophagy-dependent delivery of endogenously synthesized viral nucleic acids to endosomal TLRs for activation of type I interferon responsesCitation35 may involve the selective autophagy machinery. If so, the formation of viral capsid-p62 aggregates could potentially interfere with this normal function of p62 in the delivery of microbial PAMPs to pattern recognition receptors. In terms of adaptive immunity, the delayed clearance of viral antigen in the setting of defective selective viral autophagy could alter the kinetics and nature of antigen presentation. Moreover, similar to the effects of deficient selective autophagy in maintaining homeostasis in other cell types,Citation36 the accumulation of viral capsid-p62 aggregates in antigen presentation cells could disrupt their homeostasis and function.
At present, it is not known precisely how viral capsid-p62 aggregates may contribute to altered immunity, cellular function and cell survival. Nonetheless, several emerging observations suggest that further investigations into this question are warranted. In our recent study, we observed increased cell death in mice with deficient autophagy and SIN capsid-p62 aggregates.Citation3 There is increasing evidence for a link between Paget disease of the bone, p62 mutations that affect NFκB signaling, and the accumulation of paramyoxoviruses.Citation8,Citation21 Beyond these observations in viral systems, p62-dependent aggregate formation contributes to liver disease in Atg7-deficient mice,Citation37 and increased p62 levels alter inflammatory and death signaling pathways.Citation26 Thus, it is possible that defective selective viral autophagy, through the accumulation of viral protein-p62 aggregates may have a panoply of adverse effects on the host. As a corollary, the successful clearance of viral proteins through selective autophagy may represent an integral component of the normal host antiviral defense response.
Concluding Remarks
Viral infections are the most common form of illness that inflict mankind. It has been estimated that each of the approximately 6.75 billion people in the world harbors between eight and twelve chronic viral infections,Citation38 and the most common acute viral syndrome—the common cold—is estimated to occur between two and four times per year in adults and six and twelve times per year in children. During the productive replication phases of chronic viral infection and during acute viral infection, the cell is overloaded with a large quantity of viral proteins that are unwelcome intruders with the potential to kill the cell and/or derail its functions. Accordingly, we propose that the removal of these proteins by selective autophagy represents a critical cellular function that helps us stay healthy.
Figures and Tables
Figure 1 Schematic model of selective viral autophagy (A) and the potential biological consequences of viral protein-p62 aggregate accumulation during impaired selective autophagy (B). (A) After entry into the host cell and uncoating, Sindbis virus (SIN) replicates in the cytosol, generating newly synthesized viral nucleic acids and proteins. Autophagy is triggered by an unknown sensor(s) during viral replication, and SIN capsid protein is targeted to the autophagic machinery in a process that requires the selective autophagy adaptor protein, p62. It is unknown whether p62 recognizes free capsid (monomeric or aggregated) or assembled capsid (containing viral or other nucleic acids), whether the targeted capsid proteins undergo modification prior to recognition by p62, or whether interactions with additional adaptor proteins are required for selective autophagy of SIN capsid. Disruption of autophagy results in the accumulation of aggregates of viral proteins and p62 within the host cell. (B) In the absence of selective autophagy, aggregates containing viral capsid and p62 may perturb innate and adaptive immune response, and result in increased cell death and disruption of cellular functions. The processes listed are speculations based on extrapolations from the literature on p62 functions (see text for more detailed explanations). Further studies are required to test these speculations in models of viral infection.
Acknowledgements
The work in the authors' laboratory was supported by the Ellison Medical Foundation Senior Scholars Award in Infectious Diseases (B.L.) and NIH RO1 AI151367 (B.L.). We thank Angela Diehl for expert scientific illustration.
References
- Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005; 121:567 - 577
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009; 30:588 - 598
- Orvedahl A, MacPherson S, Sumpter RJ, Tallóczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host & Microbe 2010; 7:115 - 127
- Hardwick JM, Levine B. Sindbis virus vector system for functional analysis of apoptosis regulators. Methods Enzymol 2000; 322:492 - 508
- Gay P. [Drosophila genes which intervene in multiplication of sigma virus (author's transl)]. Mol Gen Genet 1978; 159:269 - 283
- Dru P, Bras F, Dezelee S, Gay P, Petitjean AM, Pierre-Deneubourg A, et al. Unusual variability of the Drosophila melanogaster ref(2)P protein which controls the multiplication of sigma rhabdovirus. Genetics 1993; 133:943 - 954
- Wyers F, Dru P, Simonet B, Contamine D. Immunological cross-reactions and interactions between the Drosophila melanogaster ref(2)P protein and sigma rhabdovirus proteins. J Virol 1993; 67:3208 - 3216
- Goode A, Layfield R. Recent advances in understanding the molecular basis of Paget disease of bone. J Clin Pathol 2010; 63:199 - 203
- Mills BG, Singer FR. Nuclear inclusions in Paget's disease of bone. Science 1976; 194:201 - 202
- Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ 2009; 16:976 - 983
- Sumpter R Jr, Levine B. Autophagy and innate immunity: Triggering, targeting and tuning. Semin Cell Dev Biol 2010; 21:699 - 711
- Tallóczy Z, Jiang W, Virgin HW IV, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad of Sci USA 2002; 99:190 - 195
- Tallóczy Z, Virgin HW IV, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006; 2:24 - 29
- Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol 2009; 10:1073 - 1080
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836 - 841
- Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells 2010; 15:923 - 933
- Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol 2010; In press
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431 - 3434
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 884
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 889
- Roodman GD. Insights into the pathogenesis of Paget's disease. Annals NY Acad Sci 2010; 1192:176 - 180
- Rebel A, Malkani K, Basle M, Bregeon C. Is Paget's disease of bone a viral infection?. Calcified Tissue Res 1977; 22:283 - 286
- Mills BG, Singer FR, Weiner LP, Suffin SC, Stabile E, Holst P. Evidence for both respiratory syncytial virus and measles virus antigens in the osteoclasts of patients with Paget's disease of bone. Clin Orthopaedics Related Res 1984; 303 - 311
- Gordon MT, Mee AP, Sharpe PT. Paramyxoviruses in Paget's disease. Semin Arthritis Rheum 1994; 23:232 - 234
- Mee AP, Dixon JA, Hoyland JA, Davies M, Selby PL, Mawer EB. Detection of canine distemper virus in 100% of Paget's disease samples by in situ-reverse transcriptase-polymerase chain reaction. Bone 1998; 23:171 - 175
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis and cancer. Cell 2009; 137:1001 - 1004
- Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374 - 1378
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517 - 527
- Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009; 137:721 - 735
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213 - 223
- Levine B. Apoptosis in viral infections of neurons: a protective or pathologic host response?. Curr Top Microbiol Immunol 2002; 265:95 - 118
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 1075
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 2009; 106:2770 - 2775
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008; 456:264 - 268
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007; 315:1398 - 1401
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 830
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 1163
- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell 2009; 138:30 - 50