Genetic inactivation of autophagy in liver or brain leads to the appearance of ubiquitin- and p62-positive inclusions coincident with liver dysfunction and neurodegeneration, respectively. In our recent study we measured the abundance of polyubiquitin species in autophagy-deficient tissues and demonstrated that a specific polyubiquitin chain linkage is not the decisive autophagic substrate-targeting signal. Instead our data suggest that aggregation or oligomerization of a misfolded protein, in the absence of detectable polyubiquitin modification, is an important signal for autophagic degradation. We determined that the ubiquitin accumulation observed upon autophagy inhibition is caused by p62-mediated activation of Nrf2 resulting in global transcriptional changes to ubiquitin-associated genes. Thus, substrate polyubiquitination does not appear to be the major autophagy substrate-targeting signal and the primary role of p62 appears to be Nrf2 activation, not ubiquitin-dependent substrate degradation.
Selective targeting of proteins to distinct subcellular machineries is fundamental to the regulation of cellular decisions between catabolism and anabolism. In particular, ubiquitin (Ub)-mediated targeting of proteins is essential in maintaining cellular homeostasis, and has been predominantly associated with nonlysosomal-mediated degradation. However, liver- and brain-specific autophagy knockout mice exhibit accumulation of Ub- and p62-positive inclusions suggesting that Ub-modification targets cargo for selective autophagic degradation. Subsequent reports have established the requirement of p62 oligomerization for the appearance of Ub-positive inclusions and suggest that p62 is a selective ‘autophagy adaptor’ for recognition and delivery of Ub-modified cargo to autophagosomes. Autophagy contributes to the detoxification of misfolded, aggregated proteins commonly associated with neurodegenerative disorders (Alzheimer disease, Parkinson disease, Huntington disease, and Lou Gehrig's disease [amyotrophic lateral sclerosis] and prion encephalophathies) and the presence of these misfolded proteins also correlates with the appearance of Ub- and p62-positive inclusions. Revealing how proteins are ‘marked’ for selective recognition by the autophagy machinery is essential.
To determine whether specific polyubiquitin linkages target substrates for selective autophagic degradation we used Ub absolute quantification (AQUA) mass spectrometry to measure the amount of Ub that accumulates in both liver- and brain-specific autophagy knockout mouse models. We observed a global increase in all Ub isopeptide and non-isopeptide species and the results were similar between two different autophagy-deficient models in two separate tissues. Moreover the increased levels of Ub conjugates observed in samples of Atg7-null tissues were suppressed when this mutation was combined with deletions of p62 or Nrf2. These results are significant for two reasons. First, Nrf2 controls expression levels of detoxification enzymes by regulating genes that contain an antioxidant response element (ARE). If substrate polyubiquitination was the major autophagy targeting signal then we would not expect Ub accumulation in autophagy-deficient tissue to depend upon the presence of this transcription factor. Second, whereas p62 has been hypothesized to be an adaptor facilitating the autophagic degradation of ubiquitinated substrates, it has been previously shown that loss of p62 protects tissues from autophagy deficiency by preventing Nrf2 activation. Our observations are consistent with a role for p62 in controlling Nrf2—and not with a role as a simple Ub-dependent autophagy adaptor. Our findings suggest that accumulation of Ub during autophagy deficiency is a result of Nrf2 stress signaling downstream of the multifunctional scaffolding protein p62 rather than polyubiquitin functioning as the substrate targeting signal for selective autophagy.
To identify Ub-related Nrf2 target genes that might explain the Ub dysregulation observed in autophagy-deficient tissues, we used bioinformatics, functional genomics and RT-PCR. Nrf2 (and its heterodimeric binding partner Maf) transcription factor-binding sites were found to be enriched among Ub-associated genes and the autophagy network. Within the autophagy network, there are 35 Ub-associated Nrf2 targets, and 22 are differentially affected in Atg7-/- mice compared to wild-type mice. Importantly, these changes are reversed in the Atg7-/Nrf2-double knockout mice confirming Nrf2-dependent regulation. Our overall interpretation of these data is that autophagy deficiency results in a stress response that activates Nrf2, globally affecting numerous Ub-related proteins. It is important to note that upregulation of ARE-containing genes has been reported for numerous neurodegenerative disorders. Understanding the mechanistic details of how Nrf2 binds ARE elements within Ub-associated genes, with what binding partner it binds, and Nrf2 nuclear/cytoplasmic trafficking in response to autophagy inhibition/p62 accumulation will reveal the physiological relevance of Nrf2-p62 Ub-associated signaling in neurodegenerative disorders.
Since our results from mouse indicate that polyubiquitination is not the major autophagy substrate targeting signal, we used quantitative mass spectrometry and flow cytometry to measure Ub-modification of a selective autophagy substrate in an autophagy-regulatable stable cell line. Ub has been implicated to function as a signal in several forms of selective autophagy such as pexophagy, mitophagy and xenophagy. However, in these studies, the dependence of p62 and Ub on autophagic clearance was shown by immunofluorescence studies in which the absence of p62 results in the loss of substrate puncta formation or Ub colocalization with the substrate. Colocalization, though indicative of a signaling role, does not demonstrate covalent Ub modification of substrates. Our work addresses directly whether substrate modification by polyubiquitin chains targets proteins for selective autophagy and enables us to identify additional features of selective substrates. We developed a flow cytometry cell-based assay to measure selective autophagy using an autophagy-regulatable cell line stably expressing a bicistronic reporter construct containing both the misfolded, aggregation-prone protein huntingtin with an expanded polyglutamine tract (htt(Q47)) fused to GFP (green fluorescent protein), and the non-aggregation-prone protein cherry chFP (cherry fluorescent protein). Following autophagy shut-off, the reporter cell lines provide the ability to quantify and compare the relative accumulation of the two different fluorescent reporters, where autophagic selectivity is indicated by a ratio of greater than one. Using our flow cytometry assay we measured the selective accumulation of the aggregation-prone protein htt(Q47) compared to the non-aggregation prone protein chFP and although there is global accumulation of polyubiquitin chains following autophagy shut-off there is no increase in polyubiquitin-modified htt(Q47). Overall, these data demonstrate that aggregation or oligomerization of a misfolded protein, in the absence of detectable Ub-modification, results in selective accumulation following genetic ablation of autophagy.
Based on these observations the major conclusion of our manuscript is that oligomerization targets proteins for selective autophagy in mammalian cells, and that polyubiquitination does not appear to be the major autophagy targeting signal; the primary role of p62 in autophagy deficiency appears to be Nrf2 activation, not Ub-dependent substrate degradation. Moreover, our data also demonstrate the broad importance of Nrf2-driven Ub signaling as an important cellular detoxification mechanism acting in addition to the arsenal of Nrf2-oxidative stress genes, and suggest that sustained activation could be detrimental to the cell ().
Abbreviations
| Ub | = | ubiquitin |
| AQUA | = | absolute quantification |
| ARE | = | antioxidant response element |
| Nrf2 | = | nuclear factor E2-related factor 2 |
| KEAP1 | = | kelch-like ECH-associated protein |
| Maf | = | musculoaponeurotic fibrosarcoma oncogene |
| GFP | = | green fluorescent protein |
| chFP | = | cherry fluorescent protein |
| htt | = | huntingtin |
Figures and Tables
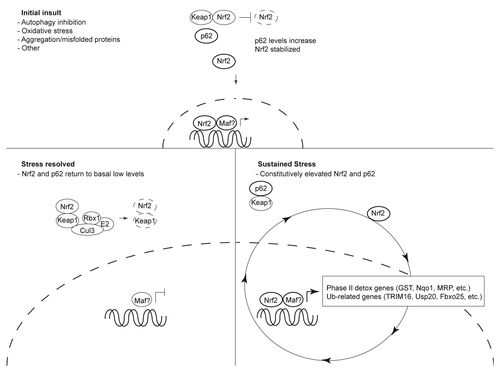
Figure 1 Schematic of Ub-associated signaling regulated by the Nrf2-p62 axis. The Keap1-Cul3-Rbx1 E3 Ub ligase maintains low levels of Nrf2 in the cell. In response to oxidative insult, accumulation of misfolded, aggregation-prone proteins or additional unknown stressors, p62 levels increase. Interaction of p62 with the Keap1:Nrf2 binding pocket results in Nrf2 stabilization and nuclear translocation. Once in the nucleus, Nrf2—and its heterodimeric binding partner Maf (or additional undefined binding partners)—bind antioxidant response elements (ARE) to activate phase II antioxidant genes and Ub-associated genes. Resolution of the stress resets the Nrf2-p62 axis by returning Nrf2 and p62 back to low levels. In the presence of continual cellular stress, Nrf2 and p62 levels are constitutively elevated contributing to a feedback loop resulting in sustained activation of phase II antioxidant and Ub-related genes.
Punctum to: Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 2010; 191:537 - 552; PMID: 21041446; http://dx.doi.org/10.1083/jcb.201005012