Abstract
Macroautophagy is a highly conserved catabolic process that is crucial for organ homeostasis in mammals. However, methods to directly measure macroautophagic activity (or flux) in vivo are limited. In this study we developed a quantitative macroautophagic flux assay based on measuring LC3b protein turnover in vivo after administering the protease inhibitor leupeptin. Using this assay we then characterized basal macroautophagic flux in different mouse organs. We found that the rate of LC3b accumulation after leupeptin treatment was greatest in the liver and lowest in spleen. Interestingly we found that LC3a, an ATG8/LC3b homologue and the LC3b-interacting protein p62 were degraded with similar kinetics to LC3b. However, the LC3b-related proteins GABARAP and GATE-16 were not rapidly turned over in mouse liver, implying that different LC3b homologues may contribute to macroautophagy via distinct mechanisms. Nutrient starvation augmented macroautophagic flux as measured by our assay, while refeeding the animals after a period of starvation significantly suppressed flux. We also confirmed that beclin 1 heterozygous mice had reduced basal macroautophagic flux compared to wild-type littermates. These results illustrate the usefulness of our leupeptin-based assay for studying the dynamics of macroautophagy in mice.
Introduction
Macroautophagy is a highly conserved catabolic process by which cells transport macromolecules and organelles to lysosomes for degradation.Citation1 In this process, a double-membrane vesicle called an autophagosome is constructed around cytoplasmic components that are targeted for breakdown including, but not limited to, proteins and damaged organelles. After formation, the autophagosome undergoes fusion events with lysosomes, progressively acquiring hydrolytic enzymes and losing its distinctive double-membrane structure.Citation2 The products of digestion are recycled to the cytoplasm via lysosomal permeases.Citation3 After digestion is completed, primary lysosomes are then reconstituted via the budding of LAMP-1 positive structures from autolysosomes.Citation4 Under basal conditions, macroautophagy accounts for roughly one-third of total cellular proteolysis but it can be further stimulated by metabolic or environmental cellular stressors such as nutrient deprivation or hypoxia.Citation5,Citation6
At the molecular level, macroautophagy is a complex process that involves the participation of at least 30 proteins.Citation7 These proteins can be organized into three functional groups:Citation8 (1) a group of serine/threonine kinases, which include ATG1 and mTOR, that sense the external nutrient environment and initiate the process;Citation9 (2) a multiprotein complex containing Beclin 1 and PI3K activity that modifies the nascent autophagosomal membrane;Citation10–Citation12 and (3) a ubiquitin-like system that serves to insert a protein called ATG8/LC3b into both sides of the autophagosomal membrane by conjugating it to phosphatidylethanolamine.Citation13,Citation14 The lipid-conjugated form of LC3b (called LC3b-II) is critical for autophagosome maturation and closure,Citation15 and serves as a convenient marker for this structure.Citation16,Citation17 LC3b-II inserted into the luminal-facing surface of the autophagosome membrane is degraded by lysosomal cathepsins,Citation18 while LC3b-II on the cytoplasmic-facing side is recovered by delipidation.Citation19 The nonlipidated form of LC3b, called LC3b-I, can then be recruited for the construction of new autophagosomes.Citation14,Citation19
Traditionally, macroautophagic activity was inferred through a variety of techniques, including detection of autophagosomes via electron microscopy (EM),Citation20 static levels LC3b-II on western blot analysis,Citation21 or the localization of various macroautophagy proteins to punctate cytosolic structures.Citation16 However, such static measurements of the macroautophagy machinery do not reliably correlate with the rate of proteolytic activity generated by the system.Citation22–Citation24 Therefore there has been an increasing interest in methods to directly measure macroautophagic activity (or flux). The classical method of measuring macroautophagic flux in tissue culture is the bulk protein degradation assay,Citation16 which measures the liberation of radio-isotope tagged amino acids into the tissue culture medium in the presence or absence of 3-methyladenine. Recently, multiple groups have developed alternative techniques based on measuring the turnover of specific macroautophagy substrates, including LC3b,Citation25–Citation29 p62,Citation27,Citation30 NBR1,Citation27 BHMT,Citation31 and polyglutamine-protein aggregates.Citation32 However, existing methods to measure macroautophagic flux in vivo are less well developed and tend to employ semiquantitativeCitation32–Citation34 or qualitative readouts.Citation35
In this study, we developed a simple quantitative methodology to measure macroautophagic flux in mice via examining endogenous LC3b protein turnover. Our method involves the administration of leupeptin, a membrane-permeable thiolprotease inhibitor that inhibits Cathepsin B, H and L,Citation36,Citation37 and also impairs amphisome-lysosome fusion.Citation38 We demonstrated the utility of this assay by quantitatively measuring macroautophagic flux in multiple organs. We also tested the sensitivity of the leupeptin assay for detecting changes in macroautophagic flux as a consequence of nutritional manipulation and genetic deletion of the macroautophagy gene beclin 1. Our results indicate that the leupeptin assay represents a useful approach for analyzing macroautophagy dynamics in vivo.
Results
Comparison of lysosomal inhibitors for measuring macroautophagic flux in vivo.
To develop an LC3b turnover assay for in vivo use, we first generated dose-response curves for chloroquine, bafilomycin B1 and leupeptin, three major compounds that have been used to detect LC3b turnover in tissue culture cells ().Citation16 The inhibitors were administered to mice via intraperitoneal (i.p.) injection and the animals were sacrificed 4 h thereafter. We defined macroautophagic flux as the increase in the 14 KDa lipidated LC3b form (LC3b-II) on western blot analysis that was induced by the addition of inhibitor.Citation22 While most LC3b-II protein is autophagosome membrane-associated and most LC3b-I is located in the cytosol, this distinction is not always absolute.Citation39,Citation40 To ensure that the LC3b-II signal being detected was membrane-associated, we fractionated tissue extracts via centrifugation at 20,000 g to obtain a lysosome and autophagosome-enriched pellet fraction (LE fraction).Citation41 The LE fraction was enriched for lysosomal membrane proteins and LC3b-II and was depleted of cytosolic proteins such as LDH (Fig. S1). Cytoskeletal proteins such as β-actin distributed in both the cytosolic and the LE fractions (Fig. S1). We quantified LC3 band intensities on western blots by extrapolating from a standard curve of purified GST-LC3 protein that was run alongside in each gel.
All three lysosomal inhibitors promoted the accumulation of LC3b-II in mouse liver (). However, chloroquine was less potent than previously reported in reference Citation33 and doses greater than 60 mg/kg were needed to obtain a significant increase in LC3b levels ( and B). While bafilomycin B1 was effective at increasing LC3b-II levels (), the animals receiving this compound developed signs of toxicity within two h of administration including piloerection and lethargy. In contrast leupeptin was well tolerated by the animals and produced a strong, dose-dependent increase in LC3b-II in both the total tissue extracts and the LE fraction ( and B). To further confirm that leupeptin stabilized LC3b-II protein in lysosomes, we fractionated lung extracts on a Nycodenz density gradient (). While leupeptin had a minimal effect on the levels of LC3b in low density cytoplasmic fractions, it increased LC3b-II content in dense vesicular fractions consistent with lysosome and/or autophagosome localization (). Leupeptin treatment also led to accumulation of LC3a, a homologue of LC3b that is 82% identical to LC3b at the amino acid level, in dense fractions ().
At the EM level, leupeptin induced the accumulation of electron-dense vesicular structures that, in hepatocytes, were visible by 60 min after treatment (). These structures tended to congregate around bile canaliculi, which is the subcellular localization expected for lysosomes in hepatocytes ().Citation42 The structures most resembled engorged lysosomes, late autophagosomes (AVd) and amphisomesCitation38 in that they were delimited by single unit membranes and contained heterogeneous, decomposed material as well as the occasional glycogen granule (). The abundance of early autophagosomes (AVi) was not increased by leupeptin (data not shown), consistent with prior observations.Citation36 On average, leupeptin increased the cross-sectional area of the AVd/lysosomal compartment from 0.36% ± 0.27% (±1 SD) of total cytoplasmic area, to 2.50% ± 0.33%, a 6.88-fold increase (p = 0.013, 2-tailed Student's t-test, n = 4 animals per group; ). Importantly, the magnitude of leupeptin-induced increases in AVd/lysosomal area correlated with the degree of induction in LC3b-II levels in the LE fraction (). Collectively, these data suggest that leupeptin enhanced LC3b-II levels in vivo by protecting this protein from being degraded inside lysosomes. It therefore represents a valid “chemical clamp” for determining the rate of LC3b turnover, which is a marker of macroautophagic flux.
Kinetics of LC3 protein turnover in vivo.
To determine the rate of LC3b-II accumulation after leupeptin injection we performed western blot analysis of the LE fraction from mouse solid organs harvested 0–240 min after leupeptin treatment (). Leupeptin treatment led to a time-dependent increase in LC3b-II that was evident by 30–60 min after administration in vivo (). Of the organs tested, liver demonstrated the largest and most durable accumulation of LC3b-II, while spleen demonstrated the least ( and B, and ). In liver, LC3b-II accumulated almost linearly with time after leupeptin injection, whereas in lung, kidney and spleen LC3b-II levels reached a plateau by 60 min post-treatment. In heart there was a strong initial increase in LC3b-II signal that then declined after 60 min, perhaps reflecting inactivation of the drug in heart tissue or redistribution to other organs ( and B). In addition to LC3b, leupeptin treatment also led to accumulation of LC3a-II (), and p62, an LC3b-binding protein that targets ubiquitinated protein aggregates to the autophagosome (Fig. S2).Citation43–Citation45 Leupeptin treatment led to the accumulation of p62 monomer, as well as SDS-stable, slower migrating p62-cross reactive complexes (Fig. S2A). The effect of leupeptin on p62 was readily detectable in the LE fraction (Fig. S2A), but was obscured when analyzing total liver extract (Fig. S2B), implying only a minority of p62 is targeted for degradation in lysosomes under basal conditions. Interestingly, levels of GATE-16 and GABARAP, both LC3b-related proteins,Citation14 did not increase in liver after leupeptin treatment even up to several h post-injection ( and data not shown).
While GABARAP and GATE-16 are highly similar to LC3b at the tertiary-structure level,Citation46 they are only roughly 30% homologous to LC3b at the amino-acid level. As such the apparent differences in turnover between the LC3 protein homologues might be explained by a lower vulnerability of GATE-16 and GABARAP to leupeptin-sensitive proteases, rather than a true difference in turnover rate. To examine this possibility, we directly measured the half-lives of the LC3-family proteins in vivo. We reasoned that if LC3b was indeed degraded more rapidly than GATE-16 and GABARAP, then LC3b should have a shorter half-life. Moreover the decay of LC3b should be strongly attenuated by leupeptin, consistent with lysosomes being the dominant site of LC3b clearance. To investigate these hypotheses, we injected mice with cycloheximide (CHX), a classic inhibitor of translation used to determine protein half-life in vitro and in vivo.Citation47,Citation48 CHX was previously shown to rapidly inhibit protein translation in mouse liver when administered via i.p. injection.Citation47,Citation48 Importantly, while prolonged exposure to cycloheximide can inhibit macroautophagy, short term treatment with CHX (on the order of 1 h) was shown not to impair macroautophagic flux.Citation49,Citation50 We found that CHX treatment led to a dose-dependent decrease in LC3b and LC3a levels in mouse liver (). We observed that the optimal dose of CHX was 40 mg/kg, a dose previously shown to inhibit translation by over 95% in mouse liver.Citation47 At this dose, LC3b-II levels were reduced by approximately 80% one h after CHX administration (). In contrast, CHX treatment had a minimal effect on the levels of GABARAP and GATE-16 ().
To determine the kinetics of LC3 protein decay, mice were sacrificed 0–90 min after CHX injection. We found that, in liver, LC3b-II and LC3a-II decayed with a half-life of roughly 10 min in the presence of CHX ( and B). Pretreatment of the animals with leupeptin 60 min prior to CHX administration stabilized levels of both LC3b-II and LC3a-II ( and B). Specifically, leupeptin pre-treatment led to retention of LC3b-II and LC3a-II signal in the LE fraction but not in the cytosolic fraction ( and B). Levels of GABARAP and GATE-16 were not significantly affected by CHX treatment (). Levels of p62 in total liver extracts were also not reduced by CHX in the 90 min timeframe we analyzed (Fig. S2C), suggesting only a minority of p62 proteins are targeted for rapid degradation in lysosomes.
To investigate whether the organ-specific differences that we observed in LC3b turnover using the leupeptin assay coincided with measurable differences in half-life, we compared the half-life of LC3b in the lungs versus the liver in CHX-treated mice. We found that LC3b-II decay in lung was more gradual compared to liver, with an approximate half-life of 30 min as opposed to 10 min (). The three-fold difference in LC3b-II half-life between lung and liver was similar to the approximately 2.9-fold difference in LC3b-II accumulation between these organs after leupeptin treatment (Table 1).
Finally, to confirm that the rapid decay of LC3b seen in presence of CHX was a product of translational inhibition rather than an off-target effect, we measured LC3b half-life in the presence of puromycin, a chemically distinct inhibitor of translation (Fig. S4).Citation51 At a puromycin dose of 300 mg/kg i.p. (a dose sufficient to inhibit liver translation by 90%Citation51), LC3b-II levels rapidly declined similar to CHX treatment, although the half-life was somewhat longer (approximately 38 min as opposed to 10 min). In summary we found that leupeptin treatment can be used to measure LC3b protein turnover in multiple organs, and that LC3a and p62 can serve as surrogate markers for flux measurements. However, while the LC3b homologues GABARAP and GATE-16 can be recovered from the LE fraction, their rate of degradation within lysosomes was slower despite their structural similarity to LC3b.
Validation of the leupeptin assay for measuring macroautophagy regulation in vivo.
To test the sensitivity of our leupeptin assay for detecting regulation of macroautophagy in vivo we measured LC3b turnover in the setting of calorie starvation, which is a known stimulator of macroautophagic activity (). As judged by the leupeptin assay, starvation increased mean LC3b-II turnover in liver from 37.90 to 53.94 ng LC3b-II per mg total protein (a 45% increase), compared to control fed mice ( and ). These differences were statistically significant (p = 0.04, Student's 2-tailed t-test, ). Re-feeding the mice after the starvation period, a maneuver which stimulates mTOR activity,Citation52 significantly inhibited macroautophagic flux ( and ).
Previously, beclin 1 heterozygous mice were shown to have impaired macroautophagic activity as evidenced by reduced GFP-LC3b clustering in muscle in response to starvation.Citation53 This phenotype was seen in conjunction with a 50% reduction in Beclin 1 protein in heterozygous animals.Citation53 However many generations of inbreeding since the initial description of the beclin 1 depleted strain may have led to a reduction in the gene-dosage effect due to negative selection pressure. Indeed one later report found near normalization of basal levels of Beclin 1 protein in the heterozygous animals.Citation54 Therefore we wondered whether more contemporary generations of beclin 1 heterozygous mice retained the original defect in macroautophagy. To investigate this, we compared basal macroautophagic flux in the livers of beclin 1+/+ and beclin 1+/− animals using the leupeptin assay (). We found a mild decrease in LC3b turnover in beclin 1+/− mice compared beclin 1+/+ litter mates that did not reach statistical significance via the leupeptin assay (). We also noted a mild prolongation in LC3b-II half-life in beclin 1+/− mice compared to wild-type litter mates (Fig. S5). However, there was a more significant reduction in p62 turnover in beclin 1+−/− animals compared to wild-type litter mates (). Surprisingly, these differences were evident despite the fact that our contemporary colony of beclin 1 heterozygotes no longer demonstrated a reduction of Beclin 1 at either the protein or mRNA level, at least in the livers of 6–8-week-old male animals (Fig. S6). Taken together we found that the leupeptin assay is sufficiently sensitive to detect regulation in macroautophagic activity caused by nutritional manipulation or by macroautophagy gene deletion.
Discussion
In this study we established a new in vivo assay for measuring LC3b turnover, which is a popular approach for estimating macroautophagic flux.Citation16,Citation22 In our assay, leupeptin is administered to mice via i.p. injection, producing an increase in LC3b-II signal in autolysosomes that can be quantitatively measured. Previously leupeptin has been used to measure LC3b turnover in tissue culture cellsCitation16,Citation55 and also to detect the sequestration of cytosolic enzymes in lysosomes in vivo.Citation56,Citation57 Our protocol combines aspects of these earlier reports to enable the quantitative measurement of LC3b turnover via macroautophagy in vivo.
Currently there is a need for new methods to measure macroautophagic flux that are technically simple, and which can complement or refine previously described approaches. For example, the bulk protein degradation assay has been used in vivo,Citation50 but it is cumbersome and has not been extensively employed in animal models. Static levels of p62 have been advanced as a marker for macroautophagic flux in vivo, with reduced levels of this protein indicating increased catabolic activity.Citation35 However static levels of p62 represent a balance between new protein synthesis and clearance, and so this readout requires validation to ensure that changes in p62 quantity are not a function of altered transcription or translation rather than degradation. It may be more straightforward to directly measure p62 turnover using the leupeptin assay rather than trying to infer its rate of turnover based on steady-state levels. Ju et al.Citation32 developed an assay to measure macroautophagic flux in skeletal muscle based on the decay of transfected polyglutamine-luciferase fusion proteins. This assay directly measures macroautophagic flux but relies on the clearance of exogenous substrates that are overexpressed, and has not yet been extended to other organs beyond muscle. The lysosomal inhibitor chloroquine has been used to measure endogenous LC3b turnover in heart muscle.Citation33 While straightforward, the use of chloroquine has not been validated for measuring macroautophagic flux in other organs. Chloroquine also has anti-inflammatory effects,Citation58,Citation59 which make it less desirable for measuring macroautophagic flux in the setting of animal models of inflammation or infection. Finally, Ju et al.Citation34 recently developed an LC3b turnover assay to measure macroautophagic flux in skeletal muscle using colchicine treatment. As with previous efforts their assay has so far been validated for only a single organ (skeletal muscle).Citation34 Moreover their assay requires a two-day treatment with colchicine,Citation34 and so provides a two-day average of LC3b turnover that may miss dynamic changes in macroautophagic flux that occur over shorter time-frames.
The leupeptin assay we describe here offers some advantages over these previously described methods to measure macroautophagic flux. First, our assay detects the turnover of endogenous LC3b, LC3a and p62, as opposed to exogenous polyglutamine repeat-luciferase fusion proteinsCitation32 or GFP-LC3b whose rate of decay might differ from endogenous LC3b.Citation27 Second, we validated the leupeptin-based assay in multiple solid organs including liver, heart, lung, kidney and spleen. Third, by utilizing a lysosome and autophagosome enriched fraction for measuring flux, our method adds a level of specificity that is not present when analyzing unfractionated lysates. Fourth, we employed a straightforward means of quantifying macroautophagic flux by extrapolating the intensity of LC3b-II against a standard curve of purified GST-LC3b. Finally, our method offers an alternative to chloroquine and colchicine for the in vivo measurement of LC3 protein turnover. While both of these compounds are effective at increasing LC3b-II levels they are potent anti-inflammatory agents that interfere with TNFα activity.Citation59,Citation60 As such, the leupeptin assay may be advantageous when measuring macroautophagic flux in mouse models of infection or inflammation.
In validating the leupeptin assay we made several novel observations of basal macroautophagic activity. First, we found that the amount LC3b-II accumulation after i.p. injection of leupeptin differed considerably between organs, and was highest and most sustained in the liver ( and ). Potentially, this could reflect a differential potency or bioavailability for leupeptin in different organ types.Citation61,Citation62 An alternate explanation for our data is that liver harbors higher levels of basal macroautophagic activity. Such a conclusion might be supported by the shorter half life of LC3b-II we observed in liver compared to lung after cycloheximide treatment, a compound which is chemically dissimilar to leupeptin (). Irrespective, a direct comparison of flux between organs is potentially misleading, since macroautophagy likely contributes to cellular homeostasis somewhat differently in different specialized tissue types. Nevertheless, the fact that LC3b-II turnover is very robust in the liver may suggest that macroautophagy plays a significant role in the physiologic contributions of this organ even under nutrient-rich conditions. More research is needed to clarify the role of macroautophagy in metabolism.
Second, we found that LC3b turnover is very rapid, with a half-life of approximately 10 to 40 min, depending on the organ studied and the translation inhibitor used. This data is consistent with the results of Schworer et al.Citation50 who determined the half life of autophagosomes in hepatocytes to be roughly 8 min, based on an EM analysis. Our results are also in line with those of Fass et al. who estimated the average life span of GFP-LC3 puncta to be 35–40 min.
We also found that the LC3b-related proteins LC3a, GABARAP and GATE-16 were turned over with different kinetics in liver. LC3a-II levels were enhanced by leupeptin treatment similar to LC3b and also had a similar half-life after CHX treatment, while GABARAP and GATE-16 were not as rapidly turned over in our assays. This is striking because all three of these proteins have conformational structures that are similar to LC3b, and all have been shown to be targeted to autophagosome membranes.Citation13 Moreover, a recent proteomic screen for ATG8/LC3 interacting proteins found that the known mammalian LC3 homologues largely share the same interacting proteins.Citation64 One explanation for our findings is that LC3b and LC3a might be more abundantly recruited to the interior of developing autophagosomal membranes compared to GABARAP and GATE-16, and so the steady-state levels of LC3a and LC3b are more greatly influenced by macroautophagic turnover. In fact, a recent functional analysis of the LC3 and GABARAP subfamilies in HeLa cells suggested that LC3b is recruited to autophagosome membranes during the elongation phase of their biogenesis.Citation65 In contrast GATE-16 is required for sealing of the nascent autophagosome membrane and dissociation of the ATG5-12-16L complex. This might imply that GATE-16 and its close homologue GABARAP are enveloped by autophagosomes in lower quantity than LC3b and LC3a, thereby explaining the different turnover rates of these proteins in our assays.
Finally, our assay confirmed that beclin 1 heterozygous mice have mildly decreased macroautophagic flux under basal conditions. While this result was intuitively expected, a quantitative measurement of flux in these mice in vivo had not previously been reported. Flux measurements are important in mutant mice, such as beclin 1 heterozygotes, where the defect in macroautophagy is partial and selection pressure over many generations of inbreeding can lead to phenotypic drift. Given the growing availability of genetically altered mice containing complex mutations or deletions in macroautophagy genes it will be important going forward to perform in vivo measurements of macroautophagic flux when using these mice, in order to better understand the contribution of macroautophagy to the observed phenotypes. Moreover, the assays described here provide a way of analyzing potential synergisms between macroautophagy gene mutations and external factors such as diet,Citation66,Citation67 gender,Citation68 age,Citation69 and circadian rhythm.Citation70
Currently, LC3b turnover is one of the most commonly utilized means for inferring macroautophagic flux.Citation22 While our experience confirms the value of LC3b turnover assays for in vivo measurement of flux, it is worth noting some caveats to this approach. LC3b is specifically recruited to autophagosome membranes and as such the quantity of LC3b engulfed by a given autophagosome is a function of its surface area. In contrast, the rate of bulk degradation generated by the macroautophagy system is a function of the volume of cytoplasm being sequestered by autophagosomes. This distinction is important because autophagosomes produced under starvation conditions are roughly 50% larger than those produced under basal conditions,Citation50 and therefore have a lower surface area to volume ratio. As such the quantitative relationship between LC3b turnover and bulk protein turnover can vary to some degree. Utilizing assays based on p62 or NBR-1 turnoverCitation27 will not circumvent this issue because the engulfment of these proteins by autophagosomes is dependent on binding to LC3b-II,Citation45,Citation71,Citation72 and so these proteins are themselves indirectly membrane-associated. One additional caveat of LC3b turnover assays is the assumption that LC3b is recruited to autophagosome membranes at similar densities irrespective of the stimuli used to elicit autophagosome production. To our knowledge there is no literature directly addressing this point but, interestingly, autophagosomes produced in response to starvation have higher fusogenic activity in vitro than autophagosomes produced in the basal state.Citation67 Since yeast ATG8 can promote the vesicle tethering and hemifusion in vitro,Citation73 it may be important to investigate the degree to which LC3b density on autophagosome membranes might vary under differing stimuli. Finally, LC3b turnover assays will not detect degradation occurring through the recently described non-canonical macroautophagy pathway because autophagosomes produced by this process do not incorporate LC3b.Citation74 Although these caveats should be kept in mind when interpreting results, we found that the approach of measuring LC3b turnover is clearly capable of detecting physiologically relevant differences in macroautophagic flux between organs and between animals of different genetic backgrounds. As such it represents both a powerful and feasible approach for gauging macroautophagic flux in vivo.
In summary, we demonstrate a useful leupeptin-based assay for estimating macroautophagic flux in vivo. This method should prove valuable for investigating the regulation of macroautophagy in mammalian models of complex diseases.
Materials and Methods
Reagents.
LC3b antibodies were obtained from Novus Biologicals (NB 100-2220 and NB600-1384), LC3a antibodies were obtained from Abcam (ab52628). We verified that our LC3b and LC3a antibodies were selective for their specific homologues by western blot analysis against purified GST-LC3b and LC3a-His fusion proteins (data not shown). Antibodies to GATE-16 were obtained from Abcam (ab52470). Antibodies to GABARAP (SC-28938), LAMP1 (SC-19992), LAMP-2 (SC-71492), β-actin (SC-69879), Cathpesin B (SC-13985), Cytochrome C (SC-7159) and α-tubulin (SC-12462), were obtained from Santa Cruz Biotechnologies. Leupeptin hemisulfate (L-2884 and L-8511), chloroquine (C6628), cycloheximide (01810), bafilomycin B1 (11707), p62-antibody (P0067) and puromycin dihydrochloride (P7255) were obtained from Sigma. GST-LC3b fusion protein was generated by transformation of pGST-LC3 (a generous gift of Tamotsu Yoshimori), into B21DE3 cells (Strategene 200131) as described in reference Citation14. Recombinant p62-His6 protein (NBP1-44490) was obtained from Novus Biologicals. Recombinant His6-LC3a protein (UL-430) was obtained from BostonBiochem.
Mice.
C57BL/6NCrl male mice (6–8 weeks old, 20–25 g) were obtained from Charles River Laboratories. Beclin 1+/+ and beclin 1+/− mice were a generous gift from Beth Levine.Citation53 Mice were housed in shoe box-type cages under a 12 h day/light cycle and fed a standard 20% protein rodent diet, according to the regulations of the American Association for the Accreditation of Laboratory Animal Care. For experiments involving calorie starvation, food was withdrawn for 17–18 h but water was provided ad libitum (ad lib.). For all experiments mice were euthanized with phenobarbital (120 mg/kg i.p.) and exsanguinated via direct cardiac puncture. All animal experimental protocols were approved by the Harvard Medical School Institutional Review Board.
Leupeptin assay for measuring macroautophagic flux.
Mice received i.p. injections of 0.5 ml sterile Phosphate Buffered Saline (PBS, GIBCO 10010) or 0.5 ml PBS containing 9–40 mg/kg leupeptin hemisulfate. In other experiments (), mice alternatively received 28–112 mg/kg chloroquine in PBS or 0.1–0.3 mg/kg Bafilomycin B1 in PBS. After injection, the mice were returned to their cages and provided free access to food and water unless they were being subjected to calorie-starvation for experimental purposes. At specified time points after injection, the mice were euthanized and their solid organs were manually dissected and flash frozen in liquid nitrogen. In experiments in which macroautophagic flux was compared between treatments (for example starvation versus fed; ) or genotypes (beclin 1+/+ versus beclin 1−/−, ), care was taken to dissect the different experimental groups in parallel to ensure they were exposed to leupeptin for equal amounts of time. Since ischemic conditions can potentially affect macroautophagic flux, we sought to minimize the time between euthanizing the animals and flash-freezing their organs (preferably less than 5 min per animal). Organ extracts were prepared using a 15 ml capacity dounce homogenizer (Belco Glass Inc., NC9791614). For liver extracts the left lobe was placed in 7 ml of detergent-free, cold Homogenization Buffer (10 mM Tris pH 8.0, 5 mM EDTA, 250 mM Sucrose, 1 protease inhibitor tablet per 50 ml (Sigma, S8830)), and subjected to 10 strokes with the “loose” pestle and 15 strokes with the “tight” pestle. The crude homogenate was then centrifuged at 700 g for 10 min at 4°C to pellet nuclei and debris and the supernatant was saved (post-nuclear S1). To obtain a lysosome enriched (LE) fraction, 2 mg of post-nuclear S1 in 1 ml homogenization buffer was transferred to 1.5 ml microcentrifuge tubes and centrifuged at 20,000 g for 30 min at 4°C.Citation41 The supernatant was kept and labeled cytoplasmic fraction. The pellet was washed twice with 1 ml cold Homogenization Buffer and re-suspended in 200 µl of 1x LDS SDS-PAGE Sample Buffer (Invitrogen, NP0007) containing 1 mM DTT (labeled LE fraction). The LE fraction was directly solubilized in SDS-PAGE sample buffer to ensure maximum recovery of LC3b in line with prior suggestions.Citation16 To quantify LC3b levels we performed western blot analysis, with a standard curve consisting of purified GST-LC3b protein run alongside in the same gel (range 10 ng–0.1 ng protein per lane). While it is preferable to perform SDS-PAGE on the LE fraction directly after it is prepared, we found this fraction was stable for up to two freeze-thaw cycles in SDS-PAGE sample buffer when the samples are stored in liquid nitrogen (data not shown). The blots were analyzed by densitometry and LC3b or p62 band intensity was converted to protein quantity by extrapolation from the standard curve using regression analysis tools in Microsoft Excel 2003 (Microsoft Corporation). GST-LC3b content was converted to endogenous LC3b content by multiplying by a factor of 2.5, and this value was then normalized to mg of total protein. To standardize our quantification methods, we defined macroautophagic flux as the difference in LC3b-II quantity on western blots obtained in the presence versus the absence of inhibitor. For experiments where p62 turnover was quantified (), we defined flux as the difference in the sum of all p62-cross reactive bands greater or equal to 62 kDa in order to include consideration of the high-molecular weight species. Since the recombinant protein we used for p62 quantification was a 40 kDa partial fragment we adjusted the extrapolated values by diving by a factor of 1.55 and this value was then normalized to mg of total protein.
Density gradient centrifugation.
30% Nycodenz density gradients were prepared in 5 ml Polyallomar tubes (Beckman, 326819) by freeze-thawing as described in reference Citation75. Lung homogenate (3 mg pooled from n = 3 animals) in 0.5 ml homogenization buffer was layered on top, and centrifuged at 35,000 rpm (116,000 RCFavg) in a Beckman sw55Ti rotor at 4°C for 1 h. 400 µl fractions were collected from the top and 10 µl from each fraction was analyzed by western blot for LC3 proteins or protein markers of cytosol (α-tubulin), mitochondria (Cytochrome C) or lysosomes (LAMP-1, Cathepsin B). Fraction 1 represented the lowest density fraction and fraction 8 the highest density. Alternatively, 10 µl of each fraction was assayed for acid phosphatase activity, a lysosome membrane marker, using a commercial kit (Sigma, CS0740) according to the manufacturer's instructions.
EM morphometric analysis.
Approximately 2 mm thick liver fragments were cut from the tip of the left lobe and then fixed by immersion in 2.5% gluteraldehyde overnight. This was followed by osmication in 1% osmium tetroxide/1.5% potassium ferrocyanide and staining with 1% uranyl acetate. The samples were then dehydrated and embedded in Taab 812 Resin (Marivac Ltd., Nova Scotia, Canada). 95 nm sections were cut with the Leica ultracut microtome, picked up on 100 µm formvar coated Cu grids, stained with 0.2% lead citrate and imaged under the Philips Technai BioTwin Spirit Electron Microscope. For each tissue section, 20 random pictures of periportal hepatocytes were digitally captured at 1,900x (which allowed capture of an entire hepatocyte in the photographic field), and at 5,800x. The microscopist generating the pictures was blinded to the sample identity. Morphometric analysis of the 1,900x samples was conducted by a different investigator, who was also blinded to sample identity, using ImageJ software (available at http://rsbweb.nih.gov/ij/) essentially as described in reference Citation20. We defined early autophagosomes (Avi) as vesicles fully enclosed by two-unit membranes that contained material of similar density and consistency to the nearby cytoplasmic ground substance. Structures meeting these criteria for AVi but which were located close to a hepatocyte free-margin were excluded. Late autophagosomes (AVd) or lysosomes were defined as vesicles enclosed by a single unit membrane that contained dense, heterogeneous or partially digested-looking material when compared to the nearby cytoplasmic ground substance or nearby mitochondria.
Quantitative real time PCR.
Total RNA was isolated from beclin 1+/+ and beclin 1+/− mouse liver tissues using the RNeasy Mini Kit (Qiagen, 74104) as per the manufacturer's protocol. On-column DNase I treatment was performed at room temperature for 15 min using the Rnase free DNase (Qiagen, 79254) to remove the traces of genomic DNA, and the isolated RNA was eluted with 30 µl of RNase-free water. RNA concentrations were measured with a Nanodrop spectrophotometer (Nanodrop Technologies). RNA quality was assessed in each sample using the RNA 6000 Nano chip Kit (Agilent Technologies, 5065-4776) on the Agilent 2100 Bioanalyzer (Agilent Technologies). The RNA Integrity Number (RIN) of the samples analyzed was 9.2 ± 0.6 (mean ± SD).
Reverse transcription was carried out using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813). One microgram total RNA from each sample was reverse transcribed into cDNA using random hexamer primers in a final volume of 20 µl as per the manufacturer's protocol.
Real-time PCR was performed on the ABI PRISM 7300 Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Master Mix (Applied Biosystems, 4369016) and Pre-Designed TaqMan Assays specific to mouse beclin 1 (Mm01265461_m1), LC3b (Mm00782868_sH) and the housekeeping genes β-actin (Mm00607939_s1), GAPDH (Mm99999915_g1) and TATA box-binding protein, TBP (Mm00446973_m1). Among the three house keeping genes tested, β-actin had the least expression variation across all samples, including across beclin 1 genotypes. PCR was performed in a final reaction volume of 25 µl and the amplification was carried out for 40 cycles. Relative quantities of mRNA were calculated using the 2−ΔΔCt method with β-actin as the housekeeping gene. Similar results were obtained when TBP was utilized as the housekeeping gene in lieu of β-actin.
PCR genotyping of beclin 1 heterozygous animals.
PCR genotyping of genomic DNA obtained from livers of beclin 1 strain mice was performed essentially as described in reference Citation53. For detecting the wild-type beclin 1 allele, the sense primer 5′-CTG GAC ACG AGT TTC AAG ATC CTG-3′ and antisense oligo 5′-GGG CAT GGT AGC ACA CAG ACC TC-3′ was used. For detecting the knockout allele, the sense primer 5′-TGC GGG CCA GAG GCC ACT TGT GTA GC-3′ and antisense primer 5′-GGG CAT GGT AGC ACA CAG ACC TC-3′ was used.Citation53
Abbreviations
| ad lib. | = | ad libitum |
| Avi | = | early autophagosome |
| AVd | = | late autophagosome |
| CTSB | = | cathepsin B |
| CHX | = | cycloheximide |
| CytC | = | cytochrome C |
| EM | = | electron microscopy |
| I.P. | = | intraperitoneal |
| LDH | = | lactate dehydrogenase |
| LE | = | lysosome enriched |
Figures and Tables
Figure 1 Comparison of lysosomal inhibitors for measuring macroautophagic flux in vivo. Mice received intraperitoneal (i.p.) injections of leupeptin, chloroquine or bafilomycin B1 at the indicated doses and were sacrificed 4 h later. (A) Western blot analysis of LC3b in liver total extracts (20 µg per lane, n = 2 animals per group). (B) Western blot analysis of LC3b in the liver LE fractions (10 µl per lane equivalent to 5% of the fractions by volume, n = 2 animals per group). LAMP-1 western blots are shown as loading controls.
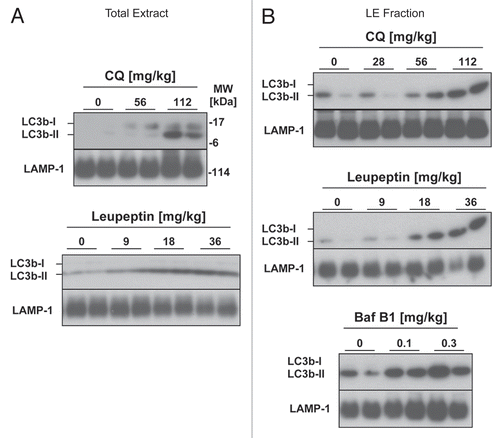
Figure 2 Subcellular fractionation and localization of LC3b following leupeptin treatment. Mice were treated with PBS or leupeptin (20 mg/kg i.p.) and sacrificed 1 later. Lung homogenates (3 mg total protein pooled from n = 3 animals per group), were separated by centrifugation through a 30% Nycodenz density gradient. Fraction 1 represents the lowest density fraction and fraction 8 the highest density fraction. (A) Acid phosphatase activity, a marker of lysosomal membranes, was measured for each fraction (10 µl per fraction). (B) Western blot analysis for the indicated proteins (10 µl per lane). Peak fractions for acid phosphatase activity, indicating the presence of lysosomes, are underlined. Data are representative of three independent experiments.
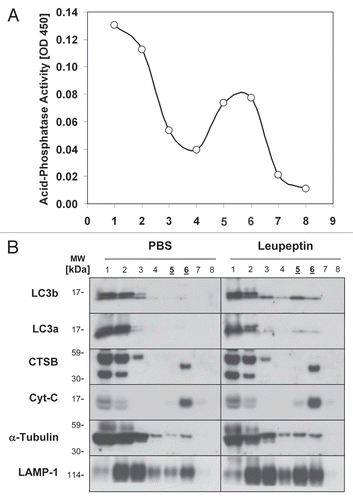
Figure 3 Ultrastructural changes in autophagosomes and lysosomes induced by leupeptin treatment. (A) Representative electron micrograph at 1,900x of a hepatocyte from a PBS treated animal. Scale bar = 2 µm. (B) Representative micrograph at 1,900x of a hepatocyte 60 min after injection of 40 mg/kg leupeptin. A bile canaliculus (BC) is denoted by a yellow arrow. Scale bar = 2 µm. (C) 5,800x magnification of the framed area in (B) demonstrating engorged lysosomes and late autophagosomes (yellow arrowheads). The nucleus (N) and a representative mitochondria (M) are labeled in blue. Scale bar = 500 nm. (D) Morphometric analysis of autophagosome (AV) and lysosome cross-sectional area in hepatocytes as a fraction of total cytoplasmic area. Filled bar represents the mean ± SD for leupeptin treated livers and the open bar represents the mean ± SD for PBS treated livers (n = 4 animals per treatment pooled from two independent experiments; n = 20 randomly selected hepatocytes per specimen). LC3b western blots for all eight liver specimens that were included in the morphometric analysis are depicted above the bar graph. *p = 0.01, Students 2-tailed t-test. (E) Scatter plot depicting the induction of AV-lysosome area versus the induction in LC3b-II content in the LE fraction of the four leupeptin-treated livers depicted in (D). A linear regression line is also depicted.
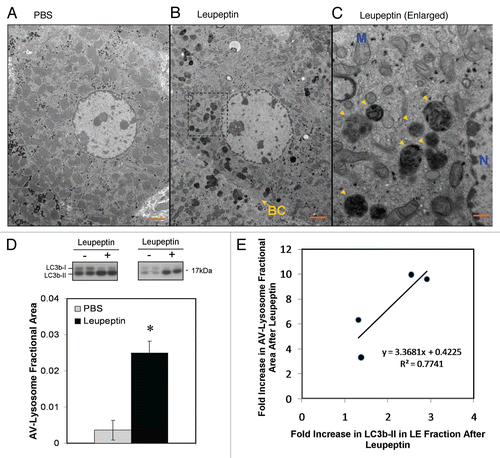
Figure 4 Kinetics of LC3 accumulation in different organs after injection of leupeptin. Mice were injected with 40 mg/kg leupeptin and sacrificed at the indicated time points. (A) Western blot analysis of LC3b in the LE fraction of each indicated organ (10 µl per lane, n = 4–5 animals per time point). LC3b-II is indicated by an arrowhead. LAMP-1 in liver is shown as a loading control (see Fig. S3 for additional controls from other organs). (B) Quantification of LC3b-II content in the LE fraction normalized to total protein (see also ). Each point represents the mean ± 1 SD. Data are representative of two independent time-course experiments. (C) Western blot analysis of LC3a, GABRAP and GATE-16 content in the liver LE faction 90 min after treatment with PBS or 40 mg/kg leupeptin. LAMP-1 is shown as a loading control. Data are representative of three independent experiments.
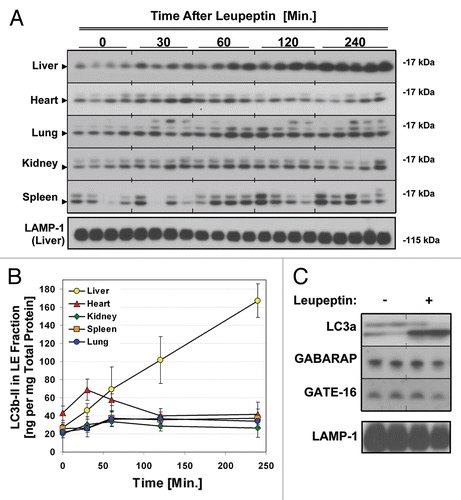
Figure 5 Cycloheximide decreases LC3b and LC3a protein levels in a dose-dependent manner. Mice were injected with 0–40 mg/kg cycloheximide and sacrificed 1 hlater. (A) Western blot analysis of indicated proteins in liver total extract (20 µg per lane). LC3b-II and LC3a-II are indicated by arrows. Western blot analysis of Lactate Dehydrogenase (LDH), a known long-lived protein, is shown as a negative control. (B) Quantification of LC3b-II signal, normalized to LC3b-II content in the control-treated (PBS) animals. Bars represent the mean ± 1 SD. *p < 0.05 (Student's two-tailed t-test).
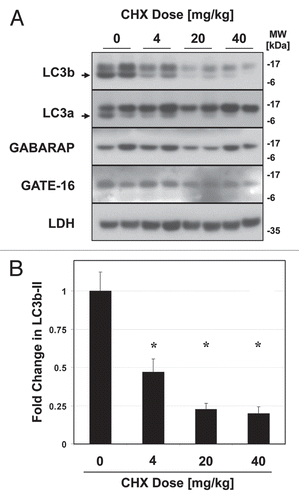
Figure 6 Kinetics of LC3b decay after injection of cycloheximide. Mice (n = 2–3 per group) were injected with 40 mg/kg cycloheximide and sacrificed at 0–90 min later. Additionally, mice were pretreated with 40 mg/kg leupeptin 1 h prior to administering cycloheximide (“Leup.”), and then sacrificed 60 min after the cycloheximide dose. (A) Western blot analyses of LC3b in liver total extracts (“Total”, 20 µg per lane), cytoplasmic fractions (“Cyto”, 10 µl per lane) and LE fractions (“LE”, 10 µl per lane). LC3B-II is indicated by an arrowhead. Analysis of µ-actin in liver total extract is shown as a loading control. Data is representative of two independent time-course experiments. (B) Western blot analysis of LC3a, GABARAP and GATE-16 levels in liver LE fractions (10 µl per lane). LC3a-II is indicated by an arrowhead. Analysis of β-actin is shown as a loading control. (C) Analysis of LC3b decay in lung total extracts after injection of cycloheximide (10 µl per lane). Quantification of LC3b-II decay in liver and lung total extracts is graphically depicted below. Open circles represent liver and closed squares represent lung. Each data point represents the mean ± 1 SD. *p < 0.05 lung versus liver by Student's 2-tailed t-test.
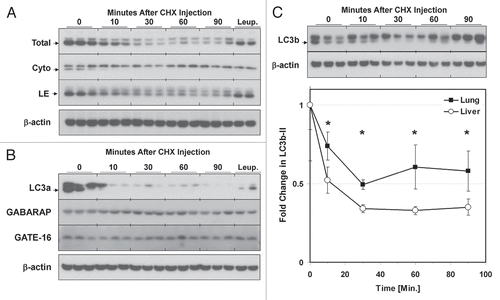
Figure 7 Comparison of LC3b turnover in starved and refed animals. Mice were fed ad lib., calorie-starved for 18 h or starved for 17 h and then given access to food for 1 h (“Refed”). The mice were then injected with leupeptin (40 mg/kg) or PBS and sacrificed 1 and 3 h thereafter. (A) Western blot analysis of LC3b levels in liver LE fractions (10 µl per lane). (B–D) Quantification of LC3b-II in the LE fractions of mice fed ad lib. (B), calorie starved (C) and refed (D). Closed squares represent the mean ± 1 SD of PBS treated mice (n = 3) and open squares represent the mean ±1 SD of leupeptin treated mice (n = 3). (E) LC3b-II turnover (flux), defined as the LC3b-II content in leupeptin treated animals minus the mean LC3b-II content in the PBS-treated animals. Each symbol represents the mean ± 1 SD (n = 3). Red squares represent results for calorie-starved animals, black squares for ad lib.-fed animals and blue triangles for refed animals. *p < 0.05 1-tailed Student's t-test. #p < 0.05 2-tailed Student's t-test. Data is representative of three independent experiments. See also for additional results.
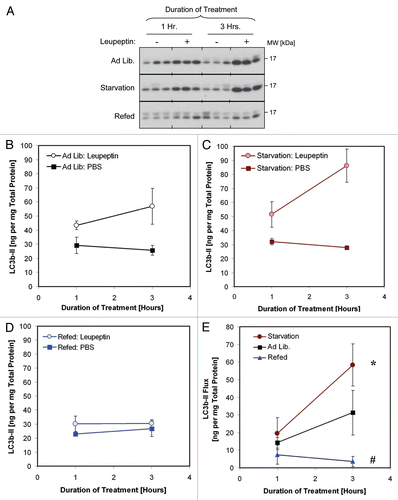
Figure 8 Analysis of basal macroautophagic flux in beclin 1+/+ and beclin 1+/− mice using the leupeptin assay. Mice were injected with PBS or 40 mg/kg leupeptin and sacrificed 0–180 min later (n = 3–4 mice per time point). (A) Western blot analysis for LC3b (top part), p62 (middle part) and β-actin (loading control; lower part) are shown. Each lane represents the liver LE fraction derived from an individual animal (10 µl per lane). The positions of LC3b-II, p62 monomer and β-actin are depicted by arrows. (B) Quantification of LC3b-II content (left graph) and p62 content (right graph) derived from the western blots depicted in (A). Symbols represent the mean ± 1 SD (n = 3–4). Solid lines represent values for beclin 1+/+ animals and dashed lines represent values for beclin 1+/− litter mates. *p < 0.05 wild-type versus beclin 1+/− mice (Student's 2-tailed t-test). (C) Bar graphs depicting LC3b-II and p62 flux in liver 1 and 3 h after leupeptin treatment. Solid bars represent mean ± 1 SD (n = 3–4) for beclin 1+/+ mice and grey bars represent mean results for beclin 1+/− litter mates. *p < 0.05 beclin 1+/− versus beclin 1+/+ mice (Student's 2-tailed t-test).
Table 1 Comparison of basal LC3b turnover in different mouse organs
Table 2 LC3b turnover is stimulated by starvation and suppressed by refeeding
Additional material
Download Zip (2.3 MB)Acknowledgements
We thank Beth Levine for the generous gift of beclin 1 heterozygous mice and Tamotsu Yoshimori for his GST-LC3b expression construct. We also thank Louise Trakimas for her technical assistance with electron microscopy and Stefan Ryter and Robyn Haspel for their critique of this manuscript. This work was funded by NIH T32 HL007633, NIH R03HL097005 and a FAMRI Clinical Innovator Award.
References
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 1721
- Ericsson JL. Studies on induced cellular autophagy. I. Electron microscopy of cells with in vivo labelled lysosomes. Exp Cell Res 1969; 55:95 - 106
- Yang Z, Klionsky DJ. Permeases recycle amino acids resulting from autophagy. Autophagy 2007; 3:149 - 150
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942 - 946
- David H, Ellermann J, Uerlings I. Primary phase of hepatocytic autophagocytosis under ischaemic conditions. Exp Toxicol Pathol 1992; 44:74 - 80
- Schworer CM, Cox JR, Mortimore GE. Alteration of lysosomal density by sequestered glycogen during deprivation-induced autophagy in rat liver. Biochem Biophys Res Commun 1979; 87:163 - 170
- Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007; 12:209 - 218
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839 - 849
- Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 2009; 5:534 - 536
- Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol-3-phosphate and exerts an essential function. J Biol Chem 2008; 283:23972 - 23980
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805 - 2812
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 5728
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651 - 4659
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 175
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 1111
- Ueno T, Takahashi K. A cathepsin L-specific inhibitor preferentially inhibits degradation of autophagosomal LC3 and GABARAP in HeLa and Huh-7 cells. Autophagy 2009; 5:878 - 879
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503 - 2518
- Swanlund JM, Kregel KC, Oberley TD. Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy 2010; 6:270 - 277
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 545
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 326
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy 2009; 5:585 - 589
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84 - 91
- Shvets E, Elazar Z. Flow cytometric analysis of autophagy in living mammalian cells. Methods Enzymol 2009; 452:131 - 141
- Farkas T, Hoyer-Hansen M, Jaattela M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 2009; 5:1018 - 1025
- Larsen KB, Lamark T, Overvatn A, Harneshaug I, Johansen T, Bjorkoy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010; 6:784 - 793
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 460
- Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy 2010; 6:634 - 641
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181 - 197
- Dennis PB, Mercer CA. The GST-BHMT assay and related assays for autophagy. Methods Enzymol 2009; 452:97 - 118
- Ju JS, Miller SE, Jackson E, Cadwell K, Piwnica-Worms D, Weihl CC. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy 2009; 5:511 - 519
- Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, et al. A method to measure cardiac autophagic flux in vivo. Autophagy 2008; 4:322 - 329
- Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 2010; 6:929 - 935
- Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F, et al. Autophagy is essential for mouse sense of balance. J Clin Invest 2010; 120:2331 - 2344
- Furuno K, Ishikawa T, Kato K. Appearance of autolysosomes in rat liver after leupeptin treatment. J Biochem 1982; 91:1485 - 1494
- Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition and degradation capacity—formation of crinosomes. Exp Mol Pathol 1987; 47:346 - 362
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 1998; 273:21883 - 21892
- Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy 2007; 3:553 - 560
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009; 458:1131 - 1135
- Graham JM. Isolation of lysosomes from tissues and cells by differential and density gradient centrifugation. Curr Protoc Cell Biol 2001; 3:3 - 6
- Sewell RB, Dillon C, Grinpukel S, Yeomans ND, Smallwood RA. Pericanalicular location of hepatocyte lysosomes and effects of fasting: a morphometric analysis. Hepatology 1986; 6:305 - 311
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847 - 22857
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 1163
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 24145
- Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells 2004; 9:611 - 618
- Trakatellis AC, Montjar M, Axelrod AE. Effect of Cycloheximide on Polysomes and Protein Synthesis in the Mouse Liver. Biochemistry 1965; 4:2065 - 2071
- Zhou P. Determining protein half-lives. Methods Mol Biol 2004; 284:67 - 77
- Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol 1990; 111:941 - 953
- Schworer CM, Shiffer KA, Mortimore GE. Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J Biol Chem 1981; 256:7652 - 7658
- Hofert JF, Boutwell RK. Puromycin-Induced Glycogenolysis as an Even Independent from Inhibited Protein Synthesis in Mouse Liver; Effects of Puromycin Analogs. Arch Biochem Biophys 1963; 103:338 - 344
- Anand P, Gruppuso PA. Rapamycin inhibits liver growth during refeeding in rats via control of ribosomal protein translation but not cap-dependent translation initiation. J Nutr 2006; 136:27 - 33
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 1820
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 2007; 117:1782 - 1793
- Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 2008; 105:20567 - 20574
- Katunuma N, Kominami E. Lysosomal sequestration of cytosolic enzymes and lysosomal thiol cathepsins. Adv Enzyme Regul 1985; 23:159 - 168
- Kominami E, Hashida S, Khairallah EA, Katunuma N. Sequestration of cytoplasmic enzymes in an autophagic vacuole-lysosomal system induced by injection of leupeptin. J Biol Chem 1983; 258:6093 - 6100
- Jang CH, Choi JH, Byun MS, Jue DM. Chloroquine inhibits production of TNFalpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology (Oxford) 2006; 45:703 - 710
- Weber SM, Levitz SM. Chloroquine interferes with lipopolysaccharide-induced TNFalpha gene expression by a nonlysosomotropic mechanism. J Immunol 2000; 165:1534 - 1540
- Ding AH, Porteu F, Sanchez E, Nathan CF. Downregulation of tumor necrosis factor receptors on macrophages and endothelial cells by microtubule depolymerizing agents. J Exp Med 1990; 171:715 - 727
- Salminen A, Kihlstrom M, Vihko V. Acid proteolytic activities in mouse liver and muscle tissues after treatment with protease inhibitor leupeptin. Comp Biochem Physiol C 1984; 79:93 - 95
- Sutherland JH, Greenbaum LM. Paradoxical effect of leupeptin in vivo on cathepsin B activity. Biochem Biophys Res Commun 1983; 110:332 - 338
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 2006; 281:36303 - 36316
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792 - 1802
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008; 8:325 - 332
- Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J 2010; 24:3052 - 3065
- Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, et al. Starving neurons show sex difference in autophagy. J Biol Chem 2009; 284:2383 - 2396
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, et al. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol 2003; 38:519 - 527
- Sachdeva UM, Thompson CB. Diurnal rhythms of autophagy: implications for cell biology and human disease. Autophagy 2008; 4:581 - 589
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 516
- Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 2008; 4:1063 - 1066
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165 - 178
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 658
- Murayama K, Fujimura T, Morita M, Shindo N. One-step subcellular fractionation of rat liver tissue using a Nycodenz density gradient prepared by freezing-thawing and two-dimensional sodium dodecyl sulfate electrophoresis profiles of the main fraction of organelles. Electrophoresis 2001; 22:2872 - 2880