Abstract
The dawn of next generation sequencing technologies has opened up exciting possibilities for whole genome sequencing of a plethora of organisms. The 2nd and 3rd generation sequencing technologies, based on cloning-free, massively parallel sequencing, have enabled the generation of a deluge of genomic sequences of both prokaryotic and eukaryotic origin in the last seven years. However, whole genome sequencing of bacterial viruses has not kept pace with this revolution, despite the fact that their genomes are orders of magnitude smaller in size compared with bacteria and other organisms. Sequencing phage genomes poses several challenges; (1) obtaining pure phage genomic material, (2) PCR amplification biases and (3) complex nature of their genetic material due to features such as methylated bases and repeats that are inherently difficult to sequence and assemble. Here we describe conclusions drawn from our efforts in sequencing hundreds of bacteriophage genomes from a variety of Gram-positive and Gram-negative bacteria using Sanger, 454, Illumina and PacBio technologies. Based on our experience we propose several general considerations regarding sample quality, the choice of technology and a “blended approach” for generating reliable whole genome sequences of phages.
Introduction
Bacteriophages (phages) are natural viral predators of bacteria. They are in a constant evolutionary arms race with host bacteria; the survival of phages over millions of years is a testament to their ability to overcome bacterial resistance mechanisms by constantly evolving in parallel with their hosts. Isolation of new phages is rapid, facile and inexpensive, and there is an abundant supply of phages in nature, making them ideal weapons to combat bacterial infections. Despite the fact that they are non-toxic to animals and plants,Citation1 phages are not as widely used for biocontrol and therapeutics as one would imagine. Since the introduction of antibiotics in the 1940s to treat bacterial infections in humans and livestock, the widespread use, and in many instances misuse, has resulted in the current crisis with multi-drug resistant bacteria. This activity, combined with a decline in the discovery of new classes of antibiotics that are effective against these resistant bacteria in the past several decades, has brought about a renewed interest in alternatives to antibiotics, such as phages or phage-encoded lytic enzymes.Citation2-Citation5
Since their discovery around 1915–1917, phages have served as excellent research tools,Citation6 although the promise of their antibacterial potential has not been fully realized.Citation7 Despite the apparent attractiveness of phages as antimicrobials, history is replete with false starts that have suppressed the field for decades at a time.Citation1-Citation3,Citation7
Besides human therapy approaches, whole-phage preparations have also been widely evaluated as biocontrol agents for food production. Numerous studies attest to the efficacy of selected phages or phage cocktails against foodborne pathogens, such as Listeria, Salmonella or E. coli.Citation8-Citation13 First phage preparations, such as ListexTM (Micreos) or ListShieldTM (Intralytix), have received approval from regulatory agencies and are being used in food production. Phage lytic enzyme application in food production has received intensive research interest, as they present highly effective and practical means of decontamination (reviewed in refs. Citation14, Citation15). Phage particles or their components have also been used successfully as detection agents for pathogens. Phage-based detection methods confer a faster and more sensitive detection. Newer developments include phage-amplification assays coupled with MALDI-MS,Citation16,Citation17 detection by lysis products (reviewed in ref. Citation18), reporter bacteriophages (reviewed in ref. Citation19) or detection by receptor binding, to list just a few.
Today, technologies exist that allow cost-effective sequencing of hundreds of viral or bacterial genomes per year, and we can anticipate in the not-too-distant future further advances that might allow routine whole-genome screening of every pathogen encountered in a clinicCitation20 or on contaminated foodstuff. The majority of the sequenced bacterial genomes reveal the presence of one or more partial or complete prophage genomes. Even closely related genomes appear to possess different sets of prophages.Citation21 Thus, the phage gene pool is larger and more diverse than the rest of the chromosome. In our experience, some prophage regions are recalcitrant to cloning, most likely due to toxicity of the gene products to the bacterium. Genes revealed by whole genome sequencing and screening of phage collections will potentially yield new generations of antimicrobials. Whole-genome sequencing has become mandatory for regulatory approval of any healthcare or food-industry application of phage or phage products,Citation22-Citation24 but today’s researchers are faced with an array of sequencing platforms and assembly options and the massive amounts of data they produce.Citation25
The current wave of high throughput sequencing efforts began in 2005 with the introduction of the Roche/454 sequencer followed by other platforms such as SOLiD, Solexa (Illumina), Helicos, Ion Torrent and PacBio and another wave of platforms yet to be released such as a nanopore-based platform (MinIon and GridIon of Oxford Nanopore).Citation26 Furthermore, the relatively small footprint, both in terms of laboratory space and personnel, required by these technologies brought about the democratization of genome sequencing in the sense that whole-genome sequencing can be done in any laboratory with limited resources and is therefore no longer just a prerogative of large Genome Centers. Each of the 2nd and 3rd generation sequencing platforms has its own unique features and distinct advantages over other platforms. However, all these platforms produce very high sequence outputs compared with the throughput of conventional Sanger sequencing platforms. Conservative estimates by the Genomic Standards Consortium in 2009 placed the prokaryotic and eukaryotic genomes completed by 2012 at over 10,000 and ~2000 respectively.Citation27 According to the GOLD genomes database, currently there are a total of approximately 15,000 prokaryotic and 3,000 eukaryotic genomes listed, of which only about 20% are finished genomes.
Although phage genomes are orders of magnitude smaller in size, whole-genome sequencing of phage has not kept pace with the current trend in high throughput sequencing of bacteria and other organisms. There has only been a slow increase in the number of complete bacteriophage genomes published.Citation28 The NCBI genome database contains around 600 Caudovirales genomes to date as well as some unclassified phage genomes.
The lack of parity in phage genome sequencing can be attributed to several problems unique to sequencing and assembly of phage genomes. (1) Phages are not self-replicating and rely on their host macromolecular machinery for their replication and growth and hence isolation of phage genomic material completely devoid of host genetic material involves extensive purification steps. Although one can, in many cases, separate the reads pertaining to the host bioinformatically post-sequencing, the presence of prophage sequences in the host chromosomes may pose a problem for such filtration. (2) Sometimes phage preparations are associated with host debris and cellular membrane fractions that contaminate the genomic material and interfere with subsequent steps of DNA sequencing. (3) Phages, especially the exclusively lytic phages, have notoriously highly methylated genomes because bacteria possess restriction-modification systems to safeguard the integrity of their genomes from invading DNAs. In order to overcome such restriction systems, phages have evolved mechanisms such as genome methylation so that they are able to infect and grow in their host bacteria. From a practical standpoint, such highly methylated sequences are recalcitrant to many of the routine genetic manipulations including shearing, cloning and DNA sequencing. In conventional cloning-based shotgun Sanger sequencing, many of the phage fragments are underrepresented and/or unclonable due to toxicity of the genes for the cloning host, usually an E. coli strain. This problem is avoided in the next generation sequencing platforms, by virtue of cloning free PCR amplification of fragments in oil and water microreactors, or emulsion PCR. However, in many instances, highly methylated DNA is a poor template for PCR and sometimes even for fragmentation of the DNA by usual procedures, such as nebulization by compressed Nitrogen gas. (4) Some phage genomes are notoriously rich in extreme GC content that is different from that of their host. Such extremes may pose a problem for PCR and sequencing. (5) Phage genomes are also known to contain complex genomic structures such as extremely long direct or inverted repeats and terminal redundancies that are problematic for assembly of the whole-genome sequence from the reads. Many assembly algorithms break the contigs at these repeats, requiring further evaluation by the human eye and confirmatory sequencing by other methods, such as PCR, restriction analysis or Sanger sequencing.Citation29,Citation30 (6) Regions of uneven sequence depth along the length of the genome, when amplifying or generating libraries using random-priming methods, may cause problems for many of the common assembly algorithms because the programs assume that this uneven coverage is due to repeats or contamination, resulting in artificially poor assemblies. (7) Almost 80% of the genome sequences in the genome online database (GOLD) are unfinished draft sequences. For bacteria and other organisms, complete genome finishing may not be a requisite for many applications, but for small genomes such as bacteriophages, finishing the genome sequencing is essential to obtain a more complete understanding of their biology, i.e., obtain confirmation of their lifestyle by identification/exclusion of genes encoding lysogeny control functions; or to identify their potential for generalized transduction of host DNA by assessing the physical genome structure.Citation29 Hence, phage researchers are faced with an increased demand in resources in order to finish and polish phage genomes before publication is possible. (8) Whereas in bacterial and human genomics, mapping of reads to a finished reference genome can be a powerful analytical tool not just for genome assembly, but for discovery of genetic variations such as insertions/deletions (indels) and single nucleotide polymorphisms (SNPs), in phage genomics this is very seldom feasible due to the absence of a reference genome for any given phage. Phage genomes are extremely mosaic in natureCitation31 and even closely related phages are highly divergent, rendering reference mapping a futile effort. (9) In general, a lack of resources for phage genome sequencing within the reach of individual phage researchers coupled with a general lack of interest and support for phage genomics by the journals and the funding agencies have resulted in too few complete phage reference genomes.
Despite all the challenges outlined above, 2nd and 3rd generation sequencing platforms offer the best opportunity for whole-genome sequencing of phages. In this report, we describe our efforts to sequence a large number of bacteriophages using both conventional and 2nd/3rd generation sequencing approaches. We also present general guidelines for obtaining a complete genome sequence of phages using a blended approach.
DNA Preparation and Quality
A common prerequisite to all DNA sequencing technologies is the necessity for high-quality nucleic acid preparations, free from contaminating RNA, proteins or solvents. Traditionally, the organic extraction method for DNA purification originally developed for bacteriophage LambdaCitation32 is used to produce highly pure DNA samples, which are sufficient even for the rigorous sample quality demands of the newest sequencing technologies. Superior results have been obtained from high-titer phage stocks purified by CsCl, sucrose or Optiprep® stepped density gradient ultracentrifugation and subsequently dialyzed against buffer of choice. Genomic DNA is then extracted by cracking the phage capsid with heat and proteinase K and purified using organic extraction as described elsewhere.Citation32 The DNA usually features A280/260 values of ~1.8 and forms a clear, sharp band on agarose gel electrophoresis runs. Co-purification of host RNA or proteins is virtually impossible using these ultracentrifugation techniques.
Alternatively to ultracentrifugation, pure, phage plate lysates or lysates from liquid cultures can be filtered through membranes with pore diameters of 200 nm and concentrated by high-speed centrifugation for several hours. Phage particles collected in the centrifugation pellet are resuspended in buffer and the DNA prepared by organic solvent extraction as described elsewhere.Citation32 A DNase and RNase-treatment before centrifugation and the inclusion of a second phenol-extraction step is recommended to increase DNA purity. DEAE anion exchange chromatography can also be used to remove contaminating free-floating nucleic acids.Citation33
In contrast, we found that DNA purified with commercial phage DNA preparation kits was often not completely free of contaminants, and additional purification steps were necessary. The overall yield is quite low (which may be attributable to smaller starting sample sizes) compared with the methods outlined above, although the kits are far less labor-intensive and do not require a preparative ultracentrifuge.
If it is difficult to obtain sufficient quantity of DNA for library construction, a variety of random-priming DNA amplification methods exist for generating large quantities of pure DNA from a few nanograms of starting material, including, multiple displacement amplification (MDA) using the phi-29 DNA polymeraseCitation34 or sequence-independent single primer amplification (SISPA).Citation35,Citation36
Sanger Chain-Termination Sequencing
Most bacteriophage DNA sequences have been obtained using a shotgun library approach followed by Sanger sequencing on capillary sequencers (e.g., ABI 3730XL). In this procedure, a phage genome is fragmented enzymatically, by sonication or by other methods, to a convenient fragment size and these fragments are randomly cloned into a high copy-number plasmid, such as pBluescript (Stratagene) or pHOS2.Citation37 Using primers located on the vector sequence, the unknown insert can be sequenced. Sanger reads usually exceed 1500 bp in length on a capillary sequencer and yield a Phred20 quality-correctedCitation38-Citation40 length of approximately 950–1100 basepairs of high-quality sequence with high accuracy. Although labor-intensive, the Shotgun-sequencing approach features a considerable advantage: Dependent on the choice of fragment size, reads from both flanking regions result in a mate-pair of sequences, which is either overlapping (fragment size below 2 kb) or has a known distance (fragment size above 2 kb) between the reads from both sides of the insert. This additional information on the physical distance between two reads can be computed into the sequence assembly in state-of-the-art software suites, such as products from CLC Bio, Geneious, DNAstar and many more, and aids scaffolding (called linkage) and correct placement of individual sequence reads within the contigs.
Usually, a shotgun sequencing approach leaves the bacteriophage researcher with a handful of high-quality contigs. Gap-closure between contigs can be done by Sanger sequencing on PCR amplified regions between contigs, generated using combinations of primers facing outward of the contigs, or by primer walking directly on phage DNA. Such direct genome primer walking, if done by an expert technician on a well-calibrated capillary sequencer, has advantages over PCR-based gap closure; it is equally fast and omits the error-prone amplification step. Primer walking can also be used to check regions of low coverage or low confidence contig-join regions in the sequence assembly.
Additionally, restriction maps of the phage genome should be generated and if possible pulsed-field gel electrophoresis performed, in order to verify computed genome size and contig alignment with experimentally obtained data. This step is especially critical in the discovery of multiple phage variants, such as in the case of Gamma/Cherry phages of Bacillus anthracis.Citation41
Some potential drawbacks of shotgun sequencing have been mentioned. One concern is cloning bias, which can occur when the target DNA exhibits extensive secondary structures or stretches of non-clonable DNA (e.g., due to encoded proteins/enzymes toxic to the cloning host, usually an E. coli strain). A second shotgun library with smaller insert sizes (i.e., < 500 bp) might be able to help circumvent the cloning problem, because the toxic ORF might be incomplete in the clone, and therefore no functional protein would be made in E. coli.
Small, temperate siphoviruses, such as A500 of ListeriaCitation42 or TP21-L of BacillusCitation43 usually feature a genome size of approximately 40 kb and are easy targets for sequencing. A shotgun library of roughly 250 clones carrying distinct inserts, sequenced from both sides is sufficient to assemble a high-quality draft genome, which needs generally no more than 10–15 runs of primer walking to finish and polish. Most small siphoviruses can be sequenced efficiently in a timeframe of 4–5 weeks, using the approach outlined above. Average genome coverage of 4–7-fold is obtained, which is sufficient for a reliable assembly. If necessary, primer walking can be used to ensure that each part of both DNA strands was sequenced completely at least once. Higher throughput Sanger-sequencing pipelines, such as the JCVI pipeline, sequence in a minimal unit of 384-well plates or blocks. By sequencing the ~40kb phage phiEf11 using 384 clones of 2–3 kb insert size in both directions, we were able to assemble the complete genome with no further finishing needed.Citation44 summarizes the sequencing data on a selected number of bacteriophages and the approximate time for sequencing and assembly in working days. Sequencing of larger viruses or complex myoviruses might require significantly more time and resources than that outlined for sequencing of small temperate siphoviruses.
Table 1. Summary of bacteriophage shotgun genome sequencing projects
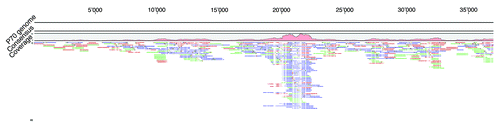
However, some viruses exhibit an exceptional cloning bias, which makes them unsuitable for shotgun cloning. depicts a read pile-up in one contig of Listeria phage P70 genome assembly from 589 Sanger reads using CLC Genomics Workbench 5.1. Although the reads equal an average overall coverage of 8.5-fold, no complete genome could be assembled due to the biased representation of clones from certain regions of the phage genome. No apparent difference between these regions and the rest of the genome could be found, besides the presence of putative promoter sequences and a slightly elevated GC content.
Figure 1. Sanger read pile-up in the assembly of a shotgun library sequencing approach of Listeria phage P70. Image captured from CLC Genomics Workbench 5.1. Upper scale shows sequence length in bp. Green are forward reads, red are reverse reads. Blue are mate-pair reads. Light green and light read color indicates trimmed sequence parts. The coverage plot shows the region of sequence and cloning bias, which features a significant higher coverage (up to 55-fold) than the rest of the contig sequence (2–21 fold).
Roche / 454 Sequencing
The 454 pyrosequencing technology, marketed by Roche, was the first of the second generation technologies available to researchers since 2005. Several thousand genomes and genomic fragments have been sequenced using the 454 technology and it has become somewhat the standard in genome sequencing. The newest FLX+ system together with Titanium XL+ reagents promises a read length of up to 1000 bp (mode read length 700 bp) and an average sequence output of 700 Mbp per sequencing plate in 23 h of runtime. Roche claims 99.997% accuracy at 15-fold coverage and provides the possibility to multiplex up to 132 samples on one plate or 16 samples when using gaskets. At a theoretical maximum output of 700 Mbp, an unbelievable number of 582 phage genomes of 40 kb could be sequenced in one run with 30-fold coverage, provided the plate would allow for more than 132 samples per run (http://my454.com/products/gs-flx-system) and each DNA species would be equally distributed on the sequencing plate. This can be done using the SISPA method (sequence-independent single-primer amplification) since up to 1,500 error-correcting barcodes can be designed.Citation36,Citation45
We have used the current 454 FLX Titanium sequencing for a number of Listeria and Bacillus phages with mixed outcomes. Generally, the large amount of sequence data poses problems with regard to IT requirements. Also, the choice of sequence assembly algorithm has to be made based on the phage genome in question and no universal solution is available. Due to the lack of reference genomes for most phages, de novo assembly is often the only option. For some phages, the GSAssembler softwareCitation46 that comes with the instrument is sufficient to quickly produce a single contig or only a handful of contigs, but in many cases, due to repeat issues discussed above, the software produces a hundred or more contigs even for a small phage genome. If mate-pair information is available, we have found the Celera AssemblerCitation47 version 7.0 (http://wgs-assembler.sourceforge.net) to give very good results.
An advantage of the Roche/454 technology is the option for sample multiplexing combined with average to long read lengths. We have run up to 16 standard phage DNA libraries on a single 454 plate, each individually barcoded for later read separation. The results of one such run on a FLX machine using XLR70 chemistry are depicted in . The total read number of 85461924 bases is quite equally distributed between most of the phages, with exception of Bacillus CP-51 phage, ( sample 7, ) which did not sequence well in all 454 approaches, most likely due to problems with library preparation or unequal pooling of multiple MID libraries (). In addition, we have run up to 25 SISPA individually bar-coded libraries on a half 454 plate with mixed results (e.g., only one phage assembling completely) ().
Table 2. Results of 454 sequencing of bacteriophage genomes A) 16 bacteriophage genomes on one sequencing plate
Illumina
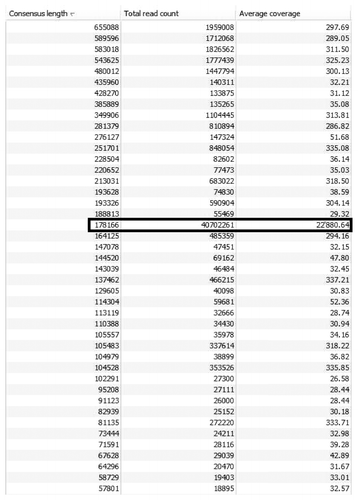
The Illumina HiSeq2000 sequencer uses the sequencing-by-synthesis technology and delivers an unrivalled number of short sequencing reads (i.e., up to 6 billion paired-end reads, equaling 600 Gb of sequence information in one ten day run using 2 flow cells). Generally speaking, the Illumina technology delivers an incredible amount of data, which is too much for the typical laboratory workstation computer to assemble; thus, necessitating the use of high throughput computing grids or cloud services. Also, because of the sheer volume of data, several (incorrect) variants of the phage genome can be constructed with good confidence from the bulk of read data. Such an example is best illustrated in the case of sequencing of Cronobacter phage A19 in which 599,997,144 100 bp paired-end reads were generated. Results generated by de novo assembly are depicted in . A total of 219 large contigs could be produced, each with reliable coverage of > 15-fold. However, the 178 kb contig stands out, because the coverage is by far higher than for any of the other contigs. This is the actual A19 genome, as confirmed by restriction profiling and partial re-sequencing. The large number smaller “chaff” contigs likely represent reads with sequencing errors, variants within the population and contaminants that only show up due to the large number of reads. Assemblers such as NewblerCitation46 have trouble with this large volume of read data and hence artificially fragment a genome that is completely represented (e.g., mapping assembly generates in one piece yet the de novo assembly is fragmented). Various labs have been working on methods to reduce redundant reads, reduce sequence error and flatten pile-up regions to try to address these issues. This is particularly a problem with using random-primed amplification to generate libraries. For example, SISPA can generate pile-up regions due to partial matches to the bar-code sequence within the genome.
Figure 2. De novo assembly of approximately 60 million Illumina reads generated for a 178 kb Cronobacter phage. 219 large contigs were produced and at least 20 of them are of similar size or larger than the actual phage genome, which sticks out because of the unusual high sequence coverage of 22,880-fold. Several other assemblies also feature reliable coverage when viewed separately from the rest.
Illumina sequencing generates a large amount of data but with very short read lengths and is therefore problematic when phage genomes contain repetitive sequence stretches. The maximum distance that can be bridged, using the HiSeq 2500/1500 instruments announced for mid-2012 and paired-end library prep is 2x150 bp (2x 100 bp on current model instruments) plus a variable 200–500 bp pair distance, which equals 800 bp on the HiSeq 2500/1500 and 700 bp on the current models HiSeq2000/1000 (http://www.illumina.com/systems/hiseq_systems.ilmn). Also, two or more genes with similar sequence in a single phage genome can lead to assembly mistakes. Such short read data are also insufficient to resolve genome end repeats, which are present in a large fraction of bacteriophages. In our opinion, Illumina sequencing has only limited usefulness for de novo assembly of phage genomes, but is rather the method of choice for re-sequencing of existing genomes, for increasing depth of coverage and/or in combination with a certain amount of sequences with longer read lengths (i.e., Sanger or PacBio). Error-correction of PacBio reads by using Illumina reads and the Celera Assembler is a particularly attractive possibility.Citation48
Pacific Biosciences RS
PacBio is currently one of the newest and most discussed of the next-generation sequencing technologies. The RS device is capable of single-molecule sequencing omitting amplification steps. Also, the expected read length is by far greater than that of any of the other sequencing technologies. Individual read lengths of up to 23 kb have been generated,Citation49 which makes the RS results the ultimate resource for genome scaffolding and supposedly speeding up assembly by several orders of magnitude. However, due to the high error-rate of the polymerase-based sequencing, some researchers have so far refrained from using this technology at all. In making such a decision it is vitally important to understand the technical limitations vs. the advantages of a platform such as PacBio. The PacBio technology utilizes so-called SMRT-cells, which produce about 40,000–50,000 reads each. Each such SMRT cell is patterned with 150000 zero mode waveguides, basically small cavities containing an immobilized DNA polymerase which sequences DNA by synthesis.Citation50 Briefly, SMRT-bell hairpin adaptersCitation51 are ligated to fragmented DNA and this is sequenced on one strand, and in ideal cases, the polymerase passing the template multiple times, generating plus- and minus-strand reads. A good approach for de novo genome sequencing would be to combine a short insert library (250 bp) that is passed multiple times [circular consensus sequencing (CCS)] and which can be sequenced with 100% accuracy, with a larger insert library (2 kb or 10 kb) which is sequenced with 85–87% accuracy (post-filter). Alternatively, PacBio data can be error-corrected with other data from short-but-high-coverage sequencing technologies, i.e., Illumina.Citation48 The drawback is that such a combination of technologies requires at least two cost-intensive sequencing runs.
Pacific Biosciences has recently released the C2 upgrade, consisting of changed chemistry, SMRT-cells and software, which is aimed at improving accuracy and read length. From our experience, the new SMRTAnalysis software trades number of valid reads for accuracy with some assembly settings. The number of reads (and post-filter bases) in our data sets has nearly dropped to half the number from version 1.2.2 to 1.3 using protocol RS_Assembly.1, whereas the read accuracy went up 2.5%. In extreme cases, less than 10% of the actual sequencing reads pass the quality-filtering step (). For bacteriophage genomes, we believe that PacBio is a very valuable technology to generate a sequence assembly scaffold and provide an orientation for contig alignment. The bulk of sequence data should be generated with a small-insert library and CCS sequencing or a complementary technology, e.g., 454 or Illumina, where 10 or more phage genomes can be run on one sequencing plate.
Table 3. Results from PacBio RS sequencing of bacteriophage CP-51 (Bacillus) and P70 (Listeria) DNA using SMRTanalysis version 1.3
We have so far sequenced two bacterial genomes (approximately 4.5 Mbp each) and two bacteriophage genomes by this method (67 kb Listeria phage P70 and 140 kb Bacillus phage CP-51) (). The overall single-pass sequence accuracy (2 kb insert libraries) did not exceed 87.5%, meaning 12.5% of the base calls were incorrect. Assemblies of reads with this magnitude of error can be computed by an assembly algorithm with tolerant settings (i.e., low insertion and gap penalties, low overlap identity) against a good reference genome, but requires a large amount of redundant sequence information for de novo sequencing projects, either from a small-insert PacBio library or from a second sequencing technology. However, the very long PacBio reads (on average, a good proportion of a large insert library yields 2–3 kb long individual reads) make a good starting point for genome scaffolding (i.e., in cases where other, amplification-based sequencing technologies fail to sequence a certain genomic region because of amplification biases). Also, for small viral genomes, a single SMRT-cell output provides enough data for multiple-fold genome coverage and therefore massively reduced error rate. The average PacBio read length is far greater than on any other current technology, which considerably speeds up genome assembly.
Summary and Conclusion
Although next-generation sequencing technology moves at an incredible pace, most researchers, especially in the phage biology field, have barely adopted the first generation of the new sequencing technologies due to technical difficulties or lack of funding. Furthermore, researchers are faced with large amounts of raw data with limited bioinformatics support. Commercial solutions to phage sequencing are virtually nonexistent, as most sequencing companies focus on genomes of bacteria or eukaryotes and have little expertise for the specific requirements of virus genome sequencing. Commercial sequencing is also expensive and the wait-time for results is rather long. University-based sequencing centers have filled this niche and many research groups have also acquired their own sequencing infrastructure.
Here we report our experiences with different commercial and university/institute-based sequencing facilities and describe some of the rather extreme hurdles which a modern-day phage biologist might face. It must be stated that most bacteriophage genomes from Gram-positive and Gram-negative organisms are fairly easy to sequence and require no specific bioinformatics expertise. Usually, 454 sequencing is used and the vendor software does a satisfying job of genome assembly. Only a few error-corrections, usually done by PCR-amplification and Sanger sequencing, are needed to obtain a complete sequence of the virus genome with good depth of coverage in all parts of the sequence.
Large phage genomes, the presence of modified bases, extensive DNA secondary structures, DNA-attached proteins or segmented genomes present a different story and require the application of a combination of sequencing technologies. A scaffold for read placement and contig orientation and alignment are the critical features in these cases. Currently, only three technologies, Sanger, PacBio and 454 (only with long insert mate-pair libraries), deliver the required read length for scaffolding phage genomes. Gap-filling, increasing depth of coverage and/or error correction (i.e., in case of PacBio data) can be done with a second technology that generates accurate, but rather short reads, such as Illumina or IonTorrent.
The sheer variety of commercial and freely-available software for processing sequencing data is overwhelming. Commercial software solutions are available for a variety of operating systems, usually integrated with a central database with a graphical user interface and intuitive handling (point-and-click). However, the software licenses are normally rather expensive and often coupled to maintenance contracts which add follow-up costs. Most open-source tools and most vendor software have to be installed under Linux (sometimes requiring a specific Linux distribution to run) and are mostly command-line operated. This may be routine for experienced bioinformaticians, but may be a problem for biologists.
Generally speaking, the best software for homogenous data sets from one technological source is the vendor software, i.e., gsAssembler (a.k.a. Newbler) from Roche or SMRT-Portal from PacBio. Other than that, large commercial software suites, such as DNAStar or CLC Bio products, as well as open-source products such as Mira,Citation52 VelvetCitation53 or wgs-Assember (Celera Assembler),Citation47 offer the possibility to integrate data from various sources and assemble them together or as separate data sets. However, manufacturer software assembling only one source of data might lead to hard-to-resolve ambiguities and mis-assemblies in the final genome draft. Thus, assembly should be attempted using different tools or iterative steps in two or more software tools. It is not the aim of this article to provide an overview of software options. The reader is kindly referred to reviews specifically dealing with this topic.Citation54
Another problem for individual labs performing next-generation sequencing is data storage. Results of NGS sequencing runs can easily add up to several hundred gigabases of raw sequence and large assemblies files. A reliable, redundant data storage option is mandatory to ensure data safety and consistency. Also, powerful computer workstations and computing grids are needed for data processing and result visualization.
In conclusion, next generation sequencing technologies offer a thrilling variety of methods to obtain bacteriophage genome sequences quickly, reliably and rather inexpensively. While traditional costs of shotgun library preparation and Sanger sequencing roughly amounts to several thousand USD per genome, modern technologies can sequence an individual phage genome for $800 USD or less if several samples are combined into one run. However, not all bacteriophage genomes sequence effortlessly and potential obstacles to sequencing, such as DNA structure, sequence repeats and problems due to DNA methylation must be taken into account. We propose a blended approach of a long-read technology for scaffolding purposes combined with a large number of short reads from a second technology for efficient DNA sequencing of bacteriophage genomes.
Acknowledgments
The authors would like to acknowledge Richard Calendar, UC Berkeley, USA for some phage stock preparation and Kimberley Bishop-Lilly, NMRC, Ft. Detrick, USA for editorial assistance. We would like to thank Andrea Patrignani, Stefan Neuenschwander and Weihong Qi of Functional Genomics Center Zurich for their technical assistance and helpful suggestions.
References
- Summers WC. Bacteriophage therapy. Annu Rev Microbiol 2001; 55:437 - 51; http://dx.doi.org/10.1146/annurev.micro.55.1.437; PMID: 11544363
- Sulakvelidze A. Phage therapy: an attractive option for dealing with antibiotic-resistant bacterial infections. Drug Discov Today 2005; 10:807 - 9; http://dx.doi.org/10.1016/S1359-6446(05)03441-0; PMID: 15970258
- Sulakvelidze A, Alavidze Z, Morris JG Jr.. Bacteriophage therapy. Antimicrob Agents Chemother 2001; 45:649 - 59; http://dx.doi.org/10.1128/AAC.45.3.649-659.2001; PMID: 11181338
- Fischetti VA. Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int J Med Microbiol 2010; 300:357 - 62; http://dx.doi.org/10.1016/j.ijmm.2010.04.002; PMID: 20452280
- Loessner MJ. Bacteriophage endolysins--current state of research and applications. Curr Opin Microbiol 2005; 8:480 - 7; http://dx.doi.org/10.1016/j.mib.2005.06.002; PMID: 15979390
- Summers W. Bacteriophage research: early history. In: Kutter E, Sulakvelidze A, eds. Bacteriophages: Biology and Applications. Boca Raton, FL: CRC Press, 2005:5-27.
- Projan S. Phage-inspired antibiotics?. Nat Biotechnol 2004; 22:167 - 8; http://dx.doi.org/10.1038/nbt0204-167; PMID: 14755287
- Anany H, Chen W, Pelton R, Griffiths MW. Biocontrol of Listeria monocytogenes and Escherichia coli O157:H7 in meat by using phages immobilized on modified cellulose membranes. Appl Environ Microbiol 2011; 77:6379 - 87; http://dx.doi.org/10.1128/AEM.05493-11; PMID: 21803890
- Callaway TR, Edrington TS, Brabban AD, Anderson RC, Rossman ML, Engler MJ, et al. Bacteriophage isolated from feedlot cattle can reduce Escherichia coli O157:H7 populations in ruminant gastrointestinal tracts. Foodborne Pathog Dis 2008; 5:183 - 91; http://dx.doi.org/10.1089/fpd.2007.0057; PMID: 18407757
- Guenther S, Herzig O, Fieseler L, Klumpp J, Loessner MJ. Biocontrol of Salmonella Typhimurium in RTE foods with the virulent bacteriophage FO1-E2. Int J Food Microbiol 2012; 154:66 - 72; http://dx.doi.org/10.1016/j.ijfoodmicro.2011.12.023; PMID: 22244192
- Guenther S, Huwyler D, Richard S, Loessner MJ. Virulent bacteriophage for efficient biocontrol of Listeria monocytogenes in ready-to-eat foods. Appl Environ Microbiol 2009; 75:93 - 100; http://dx.doi.org/10.1128/AEM.01711-08; PMID: 19011076
- Hooton SP, Atterbury RJ, Connerton IF. Application of a bacteriophage cocktail to reduce Salmonella Typhimurium U288 contamination on pig skin. Int J Food Microbiol 2011; 151:157 - 63; http://dx.doi.org/10.1016/j.ijfoodmicro.2011.08.015; PMID: 21899907
- Patel J, Sharma M, Millner P, Calaway T, Singh M. Inactivation of Escherichia coli O157:H7 attached to spinach harvester blade using bacteriophage. Foodborne Pathog Dis 2011; 8:541 - 6; http://dx.doi.org/10.1089/fpd.2010.0734; PMID: 21453119
- Callewaert L, Walmagh M, Michiels CW, Lavigne R. Food applications of bacterial cell wall hydrolases. Curr Opin Biotechnol 2011; 22:164 - 71; http://dx.doi.org/10.1016/j.copbio.2010.10.012; PMID: 21093250
- Fenton M, Ross P, McAuliffe O, O’Mahony J, Coffey A. Recombinant bacteriophage lysins as antibacterials. Bioeng Bugs 2010; 1:9 - 16; http://dx.doi.org/10.4161/bbug.1.1.9818; PMID: 21327123
- Pierce CL, Rees JC, Fernández FM, Barr JR. Detection of Staphylococcus aureus using 15N-labeled bacteriophage amplification coupled with matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Anal Chem 2011; 83:2286 - 93; http://dx.doi.org/10.1021/ac103024m; PMID: 21341703
- Rees JC, Voorhees KJ. Simultaneous detection of two bacterial pathogens using bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2005; 19:2757 - 61; http://dx.doi.org/10.1002/rcm.2107; PMID: 16136521
- Griffiths MW. Phage-Based Methods for the Detection of Bacterial Pathogens. In: Sabour PM, Griffiths MW, eds. Bacteriophage in the control of Food- and Waterborne Pathogens. Washington, DC: ASM Press, 2010:31-61.
- Smartt AE, Ripp S. Bacteriophage reporter technology for sensing and detecting microbial targets. Anal Bioanal Chem 2011; 400:991 - 1007; http://dx.doi.org/10.1007/s00216-010-4561-3; PMID: 21165607
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376 - 80; PMID: 16056220
- Rasko DA, Altherr MR, Han CS, Ravel J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol Rev 2005; 29:303 - 29; PMID: 15808746
- Hagens S, Loessner MJ. Bacteriophage for biocontrol of foodborne pathogens: calculations and considerations. Curr Pharm Biotechnol 2010; 11:58 - 68; http://dx.doi.org/10.2174/138920110790725429; PMID: 20214608
- Merabishvili M, Pirnay JP, Verbeken G, Chanishvili N, Tediashvili M, Lashkhi N, et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS One 2009; 4:e4944; http://dx.doi.org/10.1371/journal.pone.0004944; PMID: 19300511
- Carlton RM, Noordman WH, Biswas B, de Meester ED, Loessner MJ. Bacteriophage P100 for control of Listeria monocytogenes in foods: genome sequence, bioinformatic analyses, oral toxicity study, and application. Regul Toxicol Pharmacol 2005; 43:301 - 12; http://dx.doi.org/10.1016/j.yrtph.2005.08.005; PMID: 16188359
- Buckingham SD. Next Generation Data Explosion. Lab Times 2010; 1:52 - 3
- Eisenstein M. Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotechnol 2012; 30:295 - 6; http://dx.doi.org/10.1038/nbt0412-295; PMID: 22491260
- Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, et al. Genomics. Genome project standards in a new era of sequencing. Science 2009; 326:236 - 7; http://dx.doi.org/10.1126/science.1180614; PMID: 19815760
- Hatfull GF. Bacteriophage genomics. Curr Opin Microbiol 2008; 11:447 - 53; http://dx.doi.org/10.1016/j.mib.2008.09.004; PMID: 18824125
- Casjens S, Gilcrease EB. Determining DNA Packaging Stragety by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. In: Clokie MRJ, Kropinski A, eds. Bacteriophages - Methods and Protocols Volume 2: Molecular and Applied Aspects. New York: Humana Press, 2009:91-111.
- Klumpp J, Dorscht J, Lurz R, Bielmann R, Wieland M, Zimmer M, et al. The terminally redundant, nonpermuted genome of Listeria bacteriophage A511: a model for the SPO1-like myoviruses of gram-positive bacteria. J Bacteriol 2008; 190:5753 - 65; http://dx.doi.org/10.1128/JB.00461-08; PMID: 18567664
- Hendrix RW, Hatfull GF, Smith MC. Bacteriophages with tails: chasing their origins and evolution. Res Microbiol 2003; 154:253 - 7; http://dx.doi.org/10.1016/S0923-2508(03)00068-8; PMID: 12798229
- Sambrook J, Russell DW. Molecular Cloning - A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press, 2001.
- Lech K. Preparing Lambda DNA from phage lysates. In: Ausubel FM, Brent R, Kingston RE, Moore DD, G. SJ, Smith JA, eds. Current Protocols in Molecular Biology. New York: John Wiley & Sons Inc., 1990.
- Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 2001; 11:1095 - 9; http://dx.doi.org/10.1101/gr.180501; PMID: 11381035
- Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, et al. Viral genome sequencing by random priming methods. BMC Genomics 2008; 9:5; http://dx.doi.org/10.1186/1471-2164-9-5; PMID: 18179705
- Reyes GR, Kim JP. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol Cell Probes 1991; 5:473 - 81; http://dx.doi.org/10.1016/S0890-8508(05)80020-9; PMID: 1664049
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae.. Science 2001; 293:498 - 506; http://dx.doi.org/10.1126/science.1061217; PMID: 11463916
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998; 8:186 - 94; PMID: 9521922
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998; 8:175 - 85; PMID: 9521921
- Li M, Nordborg M, Li LM. Adjust quality scores from alignment and improve sequencing accuracy. Nucleic Acids Res 2004; 32:5183 - 91; http://dx.doi.org/10.1093/nar/gkh850; PMID: 15459287
- Fouts DE, Rasko DA, Cer RZ, Jiang L, Fedorova NB, Shvartsbeyn A, et al. Sequencing Bacillus anthracis typing phages gamma and cherry reveals a common ancestry. J Bacteriol 2006; 188:3402 - 8; http://dx.doi.org/10.1128/JB.188.9.3402-3408.2006; PMID: 16621835
- Dorscht J, Klumpp J, Bielmann R, Schmelcher M, Born Y, Zimmer M, et al. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J Bacteriol 2009; 191:7206 - 15; http://dx.doi.org/10.1128/JB.01041-09; PMID: 19783628
- Klumpp J, Calendar R, Loessner MJ. Complete Nucleotide Sequence and Molecular Characterization of Bacillus Phage TP21 and its Relatedness to Other Phages with the Same Name. Viruses 2010; 2:961 - 71; http://dx.doi.org/10.3390/v2040961; PMID: 21994663
- Stevens RH, Ektefaie MR, Fouts DE. The annotated complete DNA sequence of Enterococcus faecalis bacteriophage φEf11 and its comparison with all available phage and predicted prophage genomes. FEMS Microbiol Lett 2011; 317:9 - 26; http://dx.doi.org/10.1111/j.1574-6968.2010.02203.x; PMID: 21204936
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 2008; 5:235 - 7; http://dx.doi.org/10.1038/nmeth.1184; PMID: 18264105
- Kumar S, Blaxter ML. Comparing de novo assemblers for 454 transcriptome data. BMC Genomics 2010; 11:571; http://dx.doi.org/10.1186/1471-2164-11-571; PMID: 20950480
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al. A whole-genome assembly of Drosophila.. Science 2000; 287:2196 - 204; http://dx.doi.org/10.1126/science.287.5461.2196; PMID: 10731133
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 2012; 30:693 - 700; http://dx.doi.org/10.1038/nbt.2280; PMID: 22750884
- Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 2011; 365:709 - 17; http://dx.doi.org/10.1056/NEJMoa1106920; PMID: 21793740
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science 2009; 323:133 - 8; http://dx.doi.org/10.1126/science.1162986; PMID: 19023044
- Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 2010; 38:e159; http://dx.doi.org/10.1093/nar/gkq543; PMID: 20571086
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 2004; 14:1147 - 59; http://dx.doi.org/10.1101/gr.1917404; PMID: 15140833
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821 - 9; http://dx.doi.org/10.1101/gr.074492.107; PMID: 18349386
- Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics 2010; 95:315 - 27; http://dx.doi.org/10.1016/j.ygeno.2010.03.001; PMID: 20211242
- Schmuki MM, Erne D, Loessner MJ, Klumpp J. Bacteriophage P70: Unique morphology and unrelatedness to other Listeria bacteriophages.. J Virol 2012; 86