Abstract
Immunotherapeutic approaches, based on the generation of tumor-specific cytotoxic T-lymphocytes (CTL), are currently emerging as promising strategies of anti-tumor therapy. The potential use of attenuated bacteria as engineered vectors for vaccine development offers several advantages, including the stimulation of innate immunity. We developed an attenuated live bacterial vector using the type III secretion system (TTSS) of Pseudomonas aeruginosa to deliver in vivo tumor antigens. Using an inducible and rapid expression plasmid, vaccination with several antigens of different length and epitope composition, including TRP-2, gp100 and MUC18, was evaluated against glioma tumor cells. We observed similar CTL immunity and T-cell receptor (TCR) repertoire diversity with the vaccines, TRP2125-243, TRP2L125-376 and TRP2S291-376. However, only immunization with TRP2L125-376 induced significant anti-tumor immunity. Taken together, our data indicate the importance of the epitopes composition and/or peptide length of these peptides for inducing cytotoxic T-lymphocyte (CTL) mediated immunity. Characteristics that consistently improved anti-tumor immunity include: long peptides with immunodominant and cryptic CD8+ epitopes, and strong CD4+ Th epitopes. Our bacterial vector is versatile, easy-to-use and quick to produce. This vector is suitable for rapide screening and evaluation of antigens of varying length and epitope composition.
Introduction
Immunotherapeutic approaches are gaining importance for the treatment of different diseases, including cancer and infectious diseases. For the former, immunotherapy is envisaged in combination with radiotherapy and/or chemotherapy. The main goal of antitumor immunotherapy is the generation of tumor-specific CTLs. The success of this approach relies on the identification of target tumor antigens and the antigen delivery system. Current immunotherapies involve vaccination with free peptides, the loading of dendritic cells (DCs) with peptides or tumor lysates, and DNA-based vaccine.Citation1 In most cases, a specific immune response to a given antigen is obtained but the response rate of the disease to the therapy is usually low. Several factors, including low immunogenicity of the antigen and/or the efficacy of the delivery vector contribute to the low anti-tumor immunity. The most successful approach remains adoptive cell therapy. However, the procedure is arduous and complicated and does not allow the evaluation of multiple target antigens by high-throughput screening. For this reason, there is an urgent need for new, versatile and easy-to-use vectors capable of delivering the antigen-to-antigen presenting cells (APCs), while stimulating the innate immune system. Indeed activating APCs is an essential step for the subsequent generation of tumor-specific CTLs.
The potential of live, attenuated bacteria as vaccines and antigen delivery vectors has long been recognized and offers several advantages. Already more than 100 years ago, bacteria were already used in the treatment of cancer. Indeed tumor regression in patients with sarcomas was occasionally observed after stimulation of the innate immune system with a preparation of killed Streptococcus pyogenes and Serratia marcescens (Coley's toxin).Citation2 In addition bacille Calmette-Guerin (BCG) is routinely used for the treatment of bladder cancer.Citation3 Others have taken advantage of the observation that some bacteria such as Salmonella and Clostridium preferentially migrate and replicate within hypoxic region of solid tumors.Citation4 Cancer cells death then results as a consequence of competition for limiting nutrients with the bacteria and by the non-specific innate immune response. Bacterial toxins such as Diphtheria toxin (DT) and Pseudomonas exotoxin A (PE) are currently evaluated in clinical trial as fusion proteins with respectively anti-IL2 (ONTAK)Citation5 and CD25 Fv fragment (LMB2).Citation6 More recently, the type III secretion system (TTSS) from Gramnegative bacteria has been used for antigen delivery. These highly sophisticated natural injection device allows bacteria to deliver effector proteins into the cytoplasm of eukaryotic cells in order to modulate host cellular functions. The TTSS also known as the needle complex is a protein secretion apparatus composed of more than 20 proteins forming a basal body anchored in the bacterial membranes and an injection needle through which effectors travel to reach target cell cytoplasm.Citation7 We and others have previously shown that protein or peptides-antigens can be delivered in this way to APCs in vivo to generate a specific CTL response against tumors expressing the same antigen.Citation8,Citation9
The advantages of bacterial vectors are their ability to stimulate the innate immune system, their simple administration route and their flexibility to deliver a broad variety of proteins. Importantly, large proteins are correctly secreted and translocated by the TTSS of Pseudomonas aeruginosa.Citation10 This property means that immunization protocols with whole antigenic proteins are feasible, simultaneously providing a panel of putative antigenic epitopes. This approach offers the possibility of a broad spectrum of applications for vaccination, bypassing the limitations associated with the use of short peptides which are restricted to particular major histocompatibility complex (MHC) class I alleles. To develop a fast screening platform for candidate antigens evaluation, we engineered an attenuated Pseudomonas aeruginosa strain and an inducible expression plasmid. Here we show that this approach can be used to rapidly screen for tumor-associated antigens in a glioma tumor model. Our results accentuate the importance of the peptide length and/or epitope content for (1) protection against tumor development and (2) CTL generation.
Results
P. aeruginosa type III secretion system supports recombinant protein transfer to dendritic cells in vivo.
We engineered a bacterial vector using the type III secretion system (TTSS) of Pseudomonas aeruginosa to deliver in-vivo tumor antigens to antigen presenting cells (APCs). The region of P. aeruginosa TTSS toxin Exoenzyme S (ExoS) required for the efficient secretion and translocation was determined and included the N-terminal 54 amino acidsCitation11 (). To control fusion protein expression in P. aeruginosa, the exsA gene encoding the ExoS transcriptional activatorCitation13 was cloned under the control of an IPTG-inducible promoter.Citation14 Addition of IPTG to the culture resulted in an increase of intrabacterial and secreted S54-fusion proteins. Finally, the cytotoxicity of the strain was reduced by deleting the sequences encoding the two TTSS toxin ExoS and ExoT. After subcutaneous injection of this vector, fusion proteins are translocated into different cell types including APCs, which appear in large quantity in the intradermal and subcutaneous area.
To investigate TTSS-mediated protein delivery to DCs in vivo, β-lactamase was expressed in P. aeruginosa strain CHA-OST as a C-terminal fusion with the N-terminal 54 amino acids of the P. aeruginosa toxin EXO-S, a requirement for TTSS-mediated transport.Citation11 β-lactamase in combination with its membranepermeable fluorescent substrate (CCF2-AM) allows highly sensitive visual monitoring of protein delivery in live cells.Citation15 Upon cleavage of CCF2-AM by β-lactamase, FRET was disrupted and blue fluorescence was emitted. C57BL/6 mice were injected subcutaneously with the recombinant bacterial strain. At different times post-injection, the mice were sacrificed, and β-lactamase activity was assessed in DCs purified from the spleen. At 6 h post-injection, no reporter protein was detected (data not shown). The first β-lactamase-positive DCs were observed 16 h after injection in a minority of the purified cells (), in contrast to the control mice injected with the empty CHA-OST strain (EI, ). shows the DCs purified from the β-lactamase injected mouse loaded with CCF2 substrate and from the control mouse. Sixteen h corresponded approximately to the time required for the migration of the DCs from the periphery to the spleen. The reduction in β-lactamase activity that was observed in cells recovered 24 h post-injection (data not shown) may have been due to β-lactamase processing in the cytoplasm of DCs.Citation16 This result suggests that P. aeruginosa is able to deliver in vivo protein into DCs, which are able to migrate to the spleen. Thus, this live bacterial vector can be used for immunization against a wide spectrum of antigens to elicit tumor-specific CTL response.
In vitro evaluation of P. aeruginosa vaccine strains expressing different antigenic epitopes.
Various tumor-associated antigens were investigated using a reverse immunology-based approach starting with the in silico screening of immunogenic epitopes (). The tumor-associated antigens evaluated included TRP-2,Citation17 MUC18,Citation18 survivinCitation19 and gp100.Citation20 The prediction of peptide binding motifs for H2-b MHC I molecules were made using the SYFPEITHI,Citation21 BIMAS,Citation22 ProPred-ICitation23 and RANKPEPCitation24 algorithms. Since significant differences in the predictions were observed, each candidate epitope listed in was validated by at least two other algorithms than SYFPEITHI. Although some epitopes had a low scoring with SYFPEITHI, they were still retained because of higher scoring with one of the other algorithms. For each antigen, 2 or 3 epitope-containing regions were tested in vivo. Each was expressed in P. aeruginosa as a fusion protein with EXO-S as described above. Three different constructions were made for TRP-2 (TRP2125–243, TRP2L125–376, TRP2S291–376) and two for MUC18 (MUC18N100–300 and MUC18C400–560). The mouse epitope EGSRNQDWL in hgp100 (gp10021–150) was replaced with the human epitope KVPRNQDWL by site-directed mutagenesis. Full length survivin protein was expressed. To verify if each fusion protein was produced and secreted, we took advantage of the activation of the P. aeruginosa TTSS in vitro by depletion of calcium from the medium.Citation25 The activation of the TTSS was confirmed by the presence of popB and popD in the medium (). The secretion of each fusion protein was confirmed after 3 h of induction by EGTA and IPTG (). Survivin fusion protein was secreted at very low level (data not shown).
In vivo evaluation of P. aeruginosa vaccine strains expressing different antigenic epitopes.
To screen the candidate constructs for immunotherapy of glioma, C57BL/6 mice were subcutaneously injected twice in the right flank with 5 × 106 bacteria at two weeks and one week before the tumor challenge. The tumor was induced by subcutaneous injection of 1 × 105 exponentially growing gl26 cells on the left flank. The control mice were vaccinated with the empty bacteria (EI). The median survival for mice vaccinated with the control strain was 26 days (). The mice vaccinated with CHA-OST expressing epitopes from gp100, hgp100 or survivin had median survival times of 30, 28 and 29 days, respectively (). The mice injected with bacteria expressing different TRP-2 epitopes had median survival times of 29 days (TRP2), 30 days (TRP2S) and 44 days (TRP2L; p < 0.0001) (). The mice immunized with bacteria expressing MUC18N and MUC18C had median survival times of 31 and 29 days, respectively (). The mice immunized with MUC18N had a slight retarded delay of tumor appearance compared to the control mice (p = 0.065, data not shown). These data provide evidence that the live bacterial vector allows TTSS-mediated delivery of different antigens and for TRP-2 the efficacy of the tumor specific CTL response is dependant of the length and/or epitope containing peptide.
Influence of the different TRP-2 vaccines on the TCR repertoire and CTLs.
As described above a statistically significant protection against the development of subcutaneously implanted glioma tumor cells was observed with TRP2L and an intermediate level of protection was observed with TRP2 and TRP2S. TRP2 included the published TRP2180–189 epitope,Citation17 TRP2S included the strongest in silico predicted epitope (TRP2363–372) and TRP2L contained both epitopes (). To understand why the TRP2L vaccination raised a better protection against subcutaneously implanted tumors, different immuno-monitoring approaches were used. First, we investigated whether the immunization with a given TRP-2 construction resulted in a variation of the TCR repertoire diversity. The V-J combinatorial diversity of the TCRβ chain was investigated in lymphocytes from the draining lymph nodes (DLN) 1 week after the second immunization using a newly developed multiplex genomic approach and analyzed by real time PCR. The vaccination with the control bacteria resulted in a 6% reduction of the diversity (). A Tukey multiple comparison of means was performed to analyze the difference in the repertoire diversity. The TRP2- and TRP2L-immunized mice showed a statistically significant reduction of their repertoire diversity compared to the naïve mice with a mean diversity of 63% for both and p of 0.017 and 0.010, respectively (). These results compared with the two controls: (1) non-vaccinated mice (mean diversity of 79%) and (2) mice vaccinated with the control vector (mean diversity of 73%) indicated that there was an increase of a specific T cell population. The difference between the repertoire diversity of naïve and TRP2S-immunized mice was not significant (p = 0.054). Immunization with OVA248–376, which previously resulted in 80% tumor-free mice after B16 OVA challenge,Citation8 yielded the greatest decrease in repertoire diversity (59% and p = 0.012). Immunization of mice against the three TRP-2 constructs elicited a CTL response (). This experiment shows that GL26 cells are susceptible to lysis by the TRP-2 vaccine-induced CTL and the magnitude of the TRP-2 CTL response was comparable for TRP2L and TRP2S (). Cytotoxicity was analyzed by multiway analysis of variance showing an overall very significant difference between epitopes (p < 0.0001). Fisher's Least Significant Difference post hoc test was then used to determine the significant differences between group means. At a threshold of 5% we observed three significantly distinct groups with increased cytotoxicity: (1) naïve mice, (2) TRP2 vaccine and (3) TRP2L and TRP2S vaccine.
Addition of the PADRE epitope to TRP2S results in improved protection against tumor development.
It is known that CD4+ T-helper (Th) cells play a critical role in the development of an efficient anti-tumor immune response and are involved in the generation of CD8+ T effector and memory T-cell populations.Citation26 In contrast to CD8+ T cells, CD4+ T cells recognize exogenous antigens in the context of MHC class II. Presently, several MHC class II restricted peptides have been identified that may be helpful for the development of vaccines including pan-HLA-DR-binding epitope (PADRE, AKFVAAWTLKAAA).Citation27 The TRP2S construct contains the strong in silico predicted epitope TRP-2363–372. To evaluate whether vaccination with TRP2S resulted in a more potent protection against tumor development, the PADRE epitope was expressed as a fusion protein either between the EXO-S domain and TRP2S or the C-terminus of the EXO-S-TRP2S fusion protein. Only the first fusion protein resulted in an improved protection against GL26 tumor challenge. The median survival of the mice immunized against TRP2S was 28 days. In contrast, the immunization with PADRE-TRP2S resulted in a median survival of 39 days (). Immunization with PADRE alone showed comparable median survival as for the control strain EI (data not shown). The induction of the TRP-2-specific CTL response was comparable for the mice immunized with TRP2S or with PADRE-TRP2S (). Thus, tumor specific CTL response can be induced with the CD8+ epitopes contained on the TRP2S construct.
Discussion
Considerable progress has been made in the last decade to induce tumor-specific immunity. One bottleneck in this progress is the identification of tumor-specific antigens. The ideal target antigen should be highly immunogenic and abundant in the tumor, but scarce or non-existent in normal tissues. In addition, target antigens must be processed by tumor cells into peptides that have a high affinity for MHC-I molecules. These peptides are usually between 8–10 amino acids residues. Different studies reported that several CTL raised against high-affinity binding peptides did not recognize tumor cells expressing the targeted antigen, mainly because their protein processing machinery did not display the peptides.Citation28–Citation30 Furthermore, TCRs that have a high avidity for their peptides are often eliminated. Thus, there is an increasing interest in generating a low avidity TCR repertoire for improved tumor-specific immunity. A vector that permits in vivo and/or ex vivo evaluation of different TSA or TAA should: (1) deliver a broad variety of antigen candidates with different CD4+ and CD8+ epitopes to APCs; (2) stimulate the innate immune system; and (3) be versatile and easy to produce. The live bacterial vector, CHA-OST, was engineered to: (1) reduce its toxicity while maintaining its ability to stimulate DCsCitation8 and (2) allow the easy and rapid expression of different proteins.Citation11 Furthermore, the production of the vector is very simple, since 1 ml of bacteria culture is sufficient to vaccinate up to 200 mice. This approach allows the rapid construction and production of numerous vaccine strains with different antigen/epitope compositions for rapid antigen/epitope screening. We investigated the capacity of this vector to produce various antigens of different length, immunize mice before subcutaneous glioma challenge, elicit TRP-2-specific CD8+ T cell responses, and identify the portion of the TRP-2 antigen that induces efficient tumor-specific immunity.
We have shown that different antigen fragments from TRP-2, gp100 and MUC18 can be efficiently produced and transported through the TTSS of our live bacterial vector. Variations in the level of secreted protein were most likely due to the amino acids sequence and/or the tertiary structure of the proteins. The structure of the needle complex, isolated from Salmonella typhimurium, suggests that it serves as a hollow tube through which the exported proteins are actively transferred to the target cell.Citation31 Additionally, the TTSS includes components that may be involved in proteins trafficking to the channel. Little is known about the actual export mechanism, but recognition of a signal sequence on the mRNA or on the exported protein itself may be involved. In addition, specific chaperones may be directly involved in protein export or in stabilizing mature proteins prior to export.Citation32–Citation34 Whether the exported proteins travel folded or unfolded through the needle complex has not yet been determined; however, given the size of the channel, it is likely they travel at least partially unfolded. A broad variety of proteins were previously expressed as fusion proteins with the N-terminal part of ExoS and secreted through the TTSS.Citation11 From our experience, proteins secreted at the level observed in the SDS gel in , are also translocated in cells. Here we demonstrate the injection of active β-lactamase (36 kDa) in vivo in DCs after subcutaneous injection of bacteria. Nevertheless, some small and non-hydrophobic proteins, such as survivin, can interfere with the TTSS channel and may not be secreted and/or injected. Our future investigations seek to predict the effectiveness of TTSS mediated transport to enhance antigen screening procedures.
Prins et al. showed that the melanoma associated antigens, (MAA) TRP-2 and gp100, are expressed in murine glioma and presented in a MHC class-I context.Citation35 TRP-2 is widely used as a tumor associated antigen in different mouse and human tumor models. These studies typically used the TRP-2180–188 (SVYDFFVWL) peptide for MHC class I-presentation on CD8+ T cells.Citation17 We produced and evaluated three constructs of this MAA. TRP2 and TRP2L contain the TRP-2180–188 CD8+ epitope. The construct TRP2S and TRP2L contain epitopes that include H2-Kb restricted TRP-2218–220 (TWHRYHLL) and H2-Db restricted TRP-2363–372 (SQVMNLHNL) and are predicted to have strong binding capacity. Survival analysis showed protection after vaccination with CHA-OST expressing TRP2L but not TRP2 or TRP2S. Neither the TCR repertoire diversity nor the CTL responses showed a significant difference between the three constructs. Although there was a significant change in CTLs the ability of TRP2S vaccine to protect against tumor development was low and comparable to the TRP2 construct. It has been demonstrated that the C-terminal flanking residues determine the efficiency of epitope liberation by the proteasome. 36 More recently, Textoris-Taube et al. showed that the N-terminal flanking region of TRP–2 determines whether the proteasome activator, PA28, will process the epitope.Citation37 Therefore one explanation for the lower efficiency of TRP2S, compared to TRP2L, could be the length and/or the N- or C-terminal flanking region of the construct. On the other hand, different CD8+ epitopes, including TRP-2180–188 (SVYDFFVWL) are not contained in TRP2S. With the addition of the widely-used MHC class II-presented CD4+ T-helper epitope, PADRE, we have shown that CD8+ epitopes on the TRP2S construct elicit a potent tumor-specific CTL response. Therefore, we hypothesize that TRP2L may contain a strong tumor-specific CD4+ epitope that stimulates Th CD4+ cells. Indeed, Robbins et al. identified a human leukocyte antigen (HLA)-DR15 restricted epitope on human TRP-2 (80% homology to mouse TRP-2) at the position 241–250 (ALPYWNFATG).Citation38 Our TRP2L contains the same sequence and is also predicted to bind to I-Ab molecules. This T helper epitope on TRP2L could, in part, explain its superior anti-tumor protection. The high homology between human and mouse TRP-2 sequences allows a comparison of our results to human applications. The two predicted epitopes TRP-2218–220 (TWHRYHLL) and TRP-2363–372 (SQVMNLHNL) are very similar to two identified HLA-I restricted immunodominant epitopes TRP-2197–205 (LLGPGRPYR)Citation39 and TRP-2360–368 (TLDSQVMSL).Citation40 Therefore, we believe that optimizing the peptide length and including different immunodominant and cryptic CD8+ and CD4+ epitopes could improve the efficiency of anti-tumor vaccination, especially for new antigen targets where specific epitopes have not been identified.
In this work, we evaluated two other MAA, including MUC18 and gp100, as well as the universal TAA, survivin. MUC18 is known as a melanoma cell adhesion molecule and is expressed by advanced and metastatic melanomas but not in normal melanocytes.Citation41 Recently Leslie et al. showed efficient CD8+ T-cell immune responses against MUC18 expressing melanomas in response to vaccination with an a-virus based DNA plasmid.Citation18 In our experiment, the candidate vaccines did not effectively protect mice from challenge with glioma. However, the appearance of tumors for MUC18N was slightly delayed compared to the control (p = 0.065), indicating that optimizing the amino acid sequences and/or length could improve protection against glioma cells. For the gp100 and hgp100 vaccines, only three amino acids from the original gp100 sequence were included at the N-terminus. As previously described for TRP-2, the epitope sequence environment for our gp100 and hgp100 vaccines may not be optimized for epitope processing and presentation. These results highlight the optimization required to develop a long peptide or small protein vaccine. Since the TTSS mediated in vitro secretion of survivin was very low, the delivery of the antigen in vivo was probably not sufficient to elicit CD8+ T cell immunity against survivin-expressing tumor cells.
Immunization with single or multiple epitopes from one tumor-specific antigen often results in the outgrowth of antigen loss variants.Citation42 It is not clear how the same antigen can stimulate CD4+ helper cells and/or regulatory T (Treg) cells, which mediate immune suppression. It is likely that future approaches to immunotherapy will benefit from immunizing with long peptides or even small protein vaccines that contain multiple HLA-class I epitopes from a variety of target antigens and different tumor-specific MHC II epitopes. Therefore the live bacterial vaccine presented in this study is an ideal vector for in vivo and ex vivo optimization and evaluation of each candidate antigen. The development of humanized mouse models will allow antigen screening and optimization to be translated into relevant human therapies.
Material and Methods
Bacterial strain and expression plasmid.
The attenuated P. aeruginosa strain CHA-OST ΔexoSΔexoT and the expression plasmid pEAI-S54 were described previously.Citation11
Cells.
C57BL/6 syngenic glioma cell line GL26 was kindly provided by Dr. F. Berger (INSERM U318, Grenoble, France). The cells were grown in DMEM containing 10% FCS and 1% Penicillin-Streptomycin.
Cloning of the mouse antigens.
Protein sequences were analyzed with different MHC class I binding algorithms including SYFPEITHI (http://www.syfpeithi.de), BIMAS (http://bimas.dcrt.nih.gov/molbio/hla_bind), ProPred-I (http://www.imtech.res.in/raghava/propred1) and RANKPEP (http://bio.dfci.harvard.edu/RANKPEP). The different antigen fragments were amplified by reverse transcription PCR (RT-PCR) from GL26 tumor cells with the primers listed in . The PCR products were cloned as BamHI/SphI fragments into BamHI/SphI-digested pEAI-S54. The murine epitope EGSRNQDWL was mutated to a human epitope KVPRNQDWL with the Quick Change site-directed mutagenesis kit (Strategene, La Jolla, CA) using the following primers 5′-AAG TCG ACA AGG TGC CGA GGA ATC AGG ACT GGC TTG GT-3′ and 5′-AAA AGC TTT TAA GAT GGG CAG GGT CCA CCG-3′.
TTSS assay.
A single colony from Pseudomonas isolation agar was inoculated into 1.5 ml Luria-Bertani (LB) medium supplemented with 300 µg/ml carbenicillin and grown overnight with agitation at 37°C. After harvesting by centrifugation, the bacteria were washed and resuspended in fresh LB medium containing 300 µg/ml carbenicillin and 20 mM MgCl2 at an optical density (OD600) of 0.2 and grown to an OD600 between 1.291–376.8. To induce TTSS-protein expressions 0.5 mM IPTG was added. TTSS-mediated protein secretion was induced by addition of 5 mM EGTA. Bacterial cultures were centrifuged at 17,000 g for 15 min, and the supernatant was recovered. Proteins were precipitated from the supernatant with 15% perchloric acid at 4°C overnight. The precipitated protein fraction was pelleted at 17,000 g, washed two times with acetone, dried at room temperature, and resuspended in 60 µl denaturation buffer (0.2 M Tris-HCl pH 6.8, 2% dithiothreitiol, 10% SDS, 10% glycerol, 0.02% bromophenol blue) prior to analysis by denaturing SDS-PAGE.
Animal immunization.
Female C57BL/6 mice (5–6 weeks of age) were obtained from the Elevage Janvier (Le Genest St. Isle, France). They were kept under pathogen-free conditions in the animal facility of the University Joseph Fourier (Grenoble, France). All animal experiments were approved by the Animal Experiment Committee of the Region and were performed in accordance with institutional and national guidelines. Animals were immunized twice (2 weeks and 1 week before tumor challenge) by injection of 5 × 106 bacteria in 100 µl PBS subcutaneously in the right flank. GL26 cells were harvested, washed with PBS, and adjusted to 1 × 105 cells in 100 µl. The tumor cells were implanted subcutaneously in the left flank of each mouse. When mice had tumors >10 mm in the greatest dimension, or when skin ulceration occurred, they were sacrificed.
In vivo TTSS-mediated protein translocation into DCs cells.
The β-lactamase gene was amplified from pUC19 (Invitrogen Paisley, UK) with the following primers 5′-GAC CCG GGC AGA AAC GCT GGT GAA AG-3′ (forward primer) and 5′-CGA CCG GTC CAA TGC TTA ATC AGT GA-3′ (reverse primer) and cloned as a BamHI/SphI fragment into BamHI/SphI-digested pEAI-S54 plasmid. The plasmid was electroporated into P. aeruginosa strain CHA-OST to generate strain CHA-OST bla. For mice experiments, a single subcutaneous injection of 5 × 106 CHA-OST bla in 100 µl PBS were made in the right flank. The splenocytes were recovered and DCs were isolated by magnetic cell sorting with CD11c (N418) MicroBeads (Miltenyi Biotec, Bergish Gladbach, Germany). Dendritic cells were incubated with CCF2-AM (Invitrogen, Paisley, UK) in the dark at 37°C during 1 h. CCF2-AM is cephalosporin with two attached fluorophores (coumarin and fluorescein) that exhibit fluorescence resonance energy transfer (FRET). Excitation of coumarin (409 nm) results in green fluorescence emission from fluorescein (520 nm). The fluorescence of the substrate changes from green to blue upon cleavage by β-lactamase. The DCs were observed on an inverted fluorescent Nikon Eclipse TE2000-E microscope (Nikon, Champigny sur Marne, France) with DAPI (340- to 380 nm excitation and 435- to 485 nm emission) and FITC (465- to 495 nm excitation and 515- to 555 nm emission) filter set.
Generation of tumor-specific lymphocytes (CTL).
C57BL/6 mice were immunized subcutaneously two times with 5 × 106 bacteria expressing the target antigen. Splenocytes were harvested 7 days after the second immunization and depleted of RBCs on a ficoll density gradient. Splenocytes (8 × 106) were cultivated with 2 × 105 stimulator cells (mitomycin c-treated GL26 tumor cell) in RPMI 1640 medium with 10% FBS, 100 U/ml penicillin, 100 µ g/ml streptomycin and 30 U/ml IL-2. Cells were cultured for 6 days at 37°C and 5% CO2. Non-adherent effectors cells were harvested and plated with target GL26 tumor cells at different effector/target ratios (E:T) ranging from 10:1 to 100:1. After 4 h incubation in RPMI 1640 medium with 1% FBS, cytolytic activity was determined by measuring lactate dehydrogenase (LDH) release with a cytotoxicity detection kit (Roche, Indianapolis, IN). Spontaneous (negative control) and maximum release were determined from wells containing either medium alone or 1% Triton X100. The percentage of lysis was calculated as (experimental spontaneous release-negative control)/(maximum release-negative control) * 100.
Multiplex PCR assay for TCRβ repertoire analysis.
Genomic DNA was extracted and multiplex PCR was performed as describedCitation12 using a forward primer specific for all functional members of a given V family and a reverse primer specific for a given J segment. This assay allowed the simultaneous detection of several V-J recombinants in the same reaction. All V-J1, J2, J3, J4, Jn products were separated as a function of size with the maximum length being approximately 5 kbp. The multiplex PCR strategy could have amplified DNA fragments corresponding to the rearrangement of a Vx + 1 gene 3′ to that targeted by the Vx-specific primer. Constel'ID software (developed by ImmunIDtechnologies, Grenoble, France) excluded these products. Amplifications were performed with long-range Expand High Fidelity PCR System (Roche, Meylan, France). The reactions were stopped during exponential amplification to perform semi-quantitative analysis. In order to monitor the TCR selection induced by vaccination, PCR conditions to only detect major V-J combinations were used. Each reaction was normalized to the level of the actin gene in the sample. Negative PCR controls were included. Amplicons were separated on 1% agarose gels and stained with SybrGreen I. DNA levels were quantified using a CCD camera with BIO-1D software (Vilbert Lourmart, France) in conjunction with Constel'ID software. The combinatorial diversity was given in % and was calculated by counting the number of different V-J combinations detected in the assay divided by the theoretical V-J number. The theoretical V-J number was determined by the multiplication of the 23 BV functional families ×13 BJ functional genes.
Statistical analysis.
Kaplan-Meyer survival curves were analyzed using the logrank test. Two-way ANOVA followed by a multiple comparison procedure for means at a level of confidence of 95% were used to analyze CTL. The differences in the repertoire diversity was assessed with a Tukey multiple comparison of means (95% family-wise confidence level). A p value less than 0.5 was considered significant.
Abbreviations
| APC | = | antigen presenting cell |
| CTL | = | cytotoxic T-lymphocyte |
| DC | = | dendritic cell |
| DLN | = | draining lymph node |
| HLA | = | human leukocyte antigen |
| LDH | = | lactate dehydrogenase |
| MAA | = | melanoma associated antigen |
| MHC | = | major histocompatibility complex |
| TAA | = | tumor associated antigen |
| TCR | = | T-cell receptor |
| Th cell | = | T-helper cell |
| Treg cell | = | regulatory T cell |
| TSA | = | tumor specific antig |
Figures and Tables
Figure 1 Schematic representation of the TTSS.
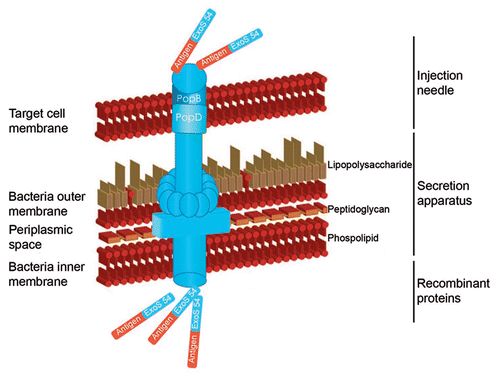
Figure 2 Type III-mediated injection into DCs in vivo. Mice were injected in the right flank with 5 × 106 OST-CHA cells transformed with either pEAI-S54 plasmid without an antigen (control) or with the β-lactamase. Sixteen h after injection, DCs were isolated from the splenocytes and incubated with CCF2-AM. β-lactamase activity is revealed by the blue fluorescence emitted by the cleaved CCF2 product in DCs purified from β-lactamase-injected mouse (A) and control mouse (B). The uncleaved CCF2 emits in green fluorescence in DCs purified from β-lactamase-injected mouse (C) and control mouse (D). Live cells were observed by fluorescence microscopy. The scale bar is equivalent to 100 µm.
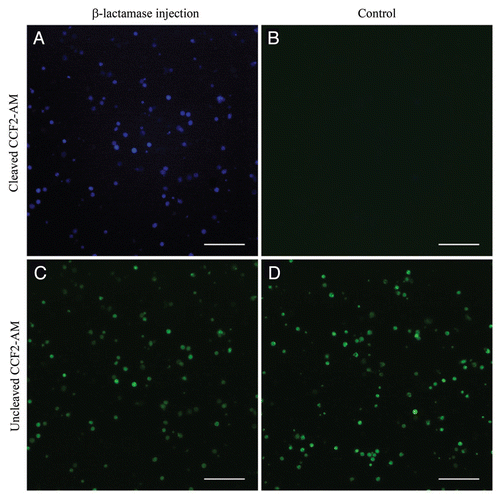
Figure 3 Screening for CTL epitopes. (A) A schematic diagram of the experimental design is shown. In parallel, 5 different antigens including TRP2, MUC18, survivin, gp100 and hgp100 were evaluated for different CTL epitopes in 42 days. (B) TTSS-mediated protein secretion was evaluated by SDS-PAGE (E and F). The positions of popB (bottom arrow) and popD (top arrow) are marked with arrows on the right. The positions of the EXO-S fusion proteins are marked with stars within the gel.
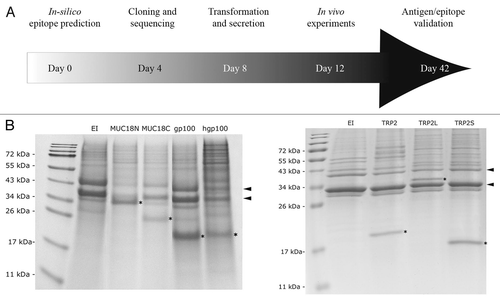
Figure 4 Survival of C57BL/6 mice with subcutaneously implanted GL26 tumor cells. Within 2-week mice received in the right flank 2 injections of CHA-OST expressing a target antigen. EI was the negative control CHA-OST strain transformed with pEAI-S54 plasmid without an antigen. Tumor cells were implanted 7 days after the last immunization. Kaplan-Meier curves displayed survival data from groups of 12 mice. Cumulative results of two independent experiments conducted with the same methodology are shown. Statistical analysis: TRP2L versus EI (p < 0.0001), TRP2L versus TRP2 (p = 0.002), and TRP2L versus TRP2S (p = 0.018). The vaccinations were performed with CHA-OST expressing (A) gp100, hgp100 and survivin, (B) TRP2, TRP2L and TRP2S, (C) MUC18N and MUC18C.
Figure 5 Characterization of the immune response induced by different TRP-2 constructs. (A) Schematic representation of TRP-2FL with different predicted and experimentally determined epitopes binding to H2-Kb (grey) and H2-Kd (black) MHC class I molecules. The TRP2125–243, TRP2S291–376 and TRP2L125–376 segments of the full-length proteins are represented below TRP-2FL. (B) TCR V-J combinatorial diversity analysis. TCR repertoire of draining lymph nodes (DLN) from naïve mice (n = 7) or from mice after two vaccinations with CHA-OST control cells (n = 8) or from mice injected with CHA-OST expressing OVA (n = 3), TRP2 (n = 6), TRP2L (n = 7), or TRP2S (n = 8) are shown. −+ A diversity of 100% corresponded to all theoretical TRB V-J recombinations considering all functional TRB V and BJ genes. (C) Induction of tumor-specific CTL in mice vaccinated against different TRP-2 constructions. Splenocytes from naïve or vaccinated mice were harvested 7 days after the last vaccination and restimulated for 6 days on mitomycin C-treated GL26 cells. LDH release was measured after 4 h of incubation at 37°C at different E:T ratios. Data represented the cytotoxicity of pooled splenic T lymphocytes from three mice.
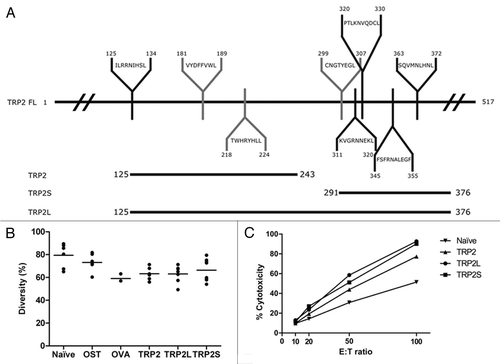
Figure 6 Combination of PADRE epitope with TRP2S produced better tumor immunity. (A) Mice received 2 injections in the right flank at a 1-week interval with CHA-OST cells expressing either TRP2S or PADRE-TRP2S. Tumor cells were implanted 7 days after the last immunization. Kaplan-Meier curves displayed survival data from groups of 10 mice (cumulative results of two independent experiments conducted with the same methodology). Statistical analysis: PADRE-TRP2S versus PBS control p = 0.003. (B) Induction of tumor-specific CTL in mice vaccinated against TRP2S and PADRE-TRP2S. Splenocytes from naïve or vaccinated mice were harvested 7 days after the last vaccination and restimulated for 6 days on mitomycin C-treated GL26 cells. LDH release was measured after 4 h of incubation at 37°C with different E:T ratios. Data represented the mean value +/− SD from three animals per group.
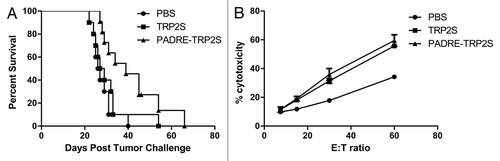
Table 1 Primers
Table 2 Antigens and epitopes evaluated
Acknowledgements
This work was supported by a grant from the French National Agency for Research. The first author was supported by the Swiss National Science Foundation. The authors thank David Hacker for critical review of the manuscript and Bruno Marques for preparation of the figures.
References
- Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, et al. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest 2004; 113:1515 - 1525
- Coley W. The treatment of malignant tumors by repeated inoculation of erysipelas. Report of ten original cases. Am J Med Sci 1893;
- Alexandroff AB, Jackson AM,, O'Donnell MA, James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet 1999; 353:1689 - 1694
- Sznol M, Lin SL, Bermudes D, Zheng LM, King I. Use of preferentially replicating bacteria for the treatment of cancer. J Clin Invest 2000; 105:1027 - 1030
- Frankel AE, Fleming DR, Powell BL, Gartenhaus R. DAB389IL2 (ONTAK) fusion protein therapy of chronic lymphocytic leukaemia. Expert Opin Biol Ther 2003; 3:179 - 186
- Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol 2000; 18:1622 - 1636
- Mueller CA, Broz P, Cornelis GR. The type III secretion system tip complex and translocon. Mol Microbiol 2008; 68:1085 - 1095
- Epaulard O, Toussaint B, Quenee L, Derouazi M, Bosco N, Villiers C, et al. Anti-tumor immunotherapy via antigen delivery from a live attenuated genetically engineered Pseudomonas aeruginosa type III secretion system-based vector. Mol Ther 2006; 14:656 - 661
- Nishikawa H, Sato E, Briones G, Chen LM, Matsuo M, Nagata Y,, et al. In vivo antigen delivery by a Salmonella typhimurium type III secretion system for therapeutic cancer vaccines. J Clin Invest 2006; 1
- Polack B, Vergnaud S, Paclet MH, Lamotte D, Toussaint B, Morel F. Protein delivery by Pseudomonas type III secretion system: Ex vivo complementation of p67(phox)-deficient chronic granulomatous disease. Biochem Biophys Res Commun 2000; 275:854 - 858
- Derouazi M, Toussaint B, Quenee L, Epaulard O, Guillaume M, Marlu R, et al. High-yield production of secreted active proteins by the Pseudomonas aeruginosa type III secretion system. Appl Environ Microbiol 2008; 74:3601 - 3604
- Pasqual N, Gallagher M, Aude-Garcia C, Loiodice M, Thuderoz F, Demongeot J, et al. Quantitative and qualitative changes in V-J alpha rearrangements during mouse thymocytes differentiation: implication for a limited T cell receptor alpha chain repertoire. J Exp Med 2002; 196::163 - 173
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 1998; 62::379 - 433
- Filopon D, Merieau A, Bernot G, Comet JP, Leberre R, Guery B, et al. Epigenetic acquisition of inducibility of type III cytotoxicity in P. aeruginosa. BMC Bioinformatics 2006; 7:272
- Moore JT, Davis ST, Dev IK. The development of beta-lactamase as a highly versatile genetic reporter for eukaryotic cells. Anal Biochem 1997; 247:203 - 209
- Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, et al. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 1998; 279:84 - 88
- Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med 1997; 185:453 - 459
- Leslie MC, Zhao YJ, Lachman LB, Hwu P, Wu GJ, Bar-Eli M. Immunization against MUC18/MCAM, a novel antigen that drives melanoma invasion and metastasis. Gene Ther 2007; 14:316 - 323
- Andersen MH, Pedersen LO, Becker JC, Straten PT. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res 2001; 61:869 - 872
- Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med 1998; 188:277 - 286
- Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999; 50:213 - 219
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 1994; 152:163 - 175
- Singh H, Raghava GP. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003; 19:1009 1014
- Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 2004; 56:405 - 419
- Yahr TL, Mende-Mueller LM, Friese MB, Frank DW. Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme S regulon. J Bacteriol 1997; 179:7165 - 7168
- Rocha B, Tanchot C. Towards a cellular definition of CD8+ T-cell memory: the role of CD4+ T-cell help in CD8+ T-cell responses. Curr Opin Immunol 2004; 16:259 - 263
- Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity 1994; 1:751 - 761
- Nijman HW, Van der Burg SH, Vierboom MP, Houbiers JG, Kast WM, Melief CJ. p53, a potential target for tumor-directed T cells. Immunol Lett 1994; 40:171 - 178
- Pelte C, Cherepnev G, Wang Y, Schoenemann C, Volk HD, Kern F. Random screening of proteins for HLAA* 0201-binding nine-amino acid peptides is not sufficient for identifying CD8 T cell epitopes recognized in the context of HLA-A*0201. J Immunol 2004; 172:6783 - 6789
- Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369–377 from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res 1998; 58:4902 - 4908
- Kubori T, Matsushima Y, Nakamura D, Uralil J, Lara-Tejero M, Sukhan A, et al. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 1998; 280:602 - 605
- Lloyd SA, Norman M, Rosqvist R, Wolf-Watz H. Yersinia YopE is targeted for type III secretion by N-terminal, not mRNA, signals. Mol Microbiol 2001; 39:520 - 531
- Shen DK, Quenee L, Bonnet M, Kuhn L, Derouazi M, Lamotte D, et al. Orf1/SpcS chaperones ExoS for type three secretion by Pseudomonas aeruginosa. Biomed Environ Sci 2008; 21:103 - 109
- Woestyn S, Sory MP, Boland A, Lequenne O, Cornelis GR. The cytosolic SycE and SycH chaperones of Yersinia protect the region of YopE and YopH involved in translocation across eukaryotic cell membranes. Mol Microbiol 1996; 20:1261 - 1271
- Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res 2003; 63:8487 - 8491
- Seifert U, Liermann H, Racanelli V, Halenius A, Wiese M, Wedemeyer H, et al. Hepatitis C virus mutation affects proteasomal epitope processing. J Clin Invest 2004; 114:250 - 259
- Textoris-Taube K, Henklein P, Pollmann S, Bergann T, Weisshoff H, Seifert U, et al. The N-terminal flanking region of the TRP2360–368 melanoma antigen determines proteasome activator PA28 requirement for epitope liberation. J Biol Chem 2007; 282:12749 - 12754
- Robbins PF, El-Gamil M, Li YF, Zeng G, Dudley M, Rosenberg SA. Multiple HLA class II-restricted melanocyte differentiation antigens are recognized by tumor-infiltrating lymphocytes from a patient with melanoma. J Immunol 2002; 169:6036 - 6047
- Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med 1996; 184:2207 - 2216
- Noppen C, Levy F, Burri L, Zajac P, Remmel E, Schaefer C, et al. Naturally processed and concealed HLA-A2.1-restricted epitopes from tumor-associated antigen tyrosinase-related protein-2. Int J Cancer 2000; 87:241 - 246
- Lehmann JM, Holzmann B, Breitbart EW, Schmiegelow P, Riethmuller G, Johnson JP. Discrimination between benign and malignant cells of melanocytic lineage by two novel antigens, a glycoprotein with a molecular weight of 113,000 and a protein with a molecular weight of 76,000. Cancer Res 1987; 47:841 - 845
- Slingluff CL Jr, Colella TA, Thompson L, Graham DD, Skipper JC, Caldwell J, et al. Melanomas with concordant loss of multiple melanocytic differentiation proteins: immune escape that may be overcome by targeting unique or undefined antigens. Cancer Immunol Immunother 2000; 48:661 - 672