Abstract
The promoter is a key element in gene transcription and regulation. We previously reported that artificial sequences rich in the dinucleotide CpG are sufficient to drive expression in vitro in mammalian cell lines, without requiring canonical binding sites for transcription factor proteins. Here, we report that introducing a promoter organization that alternates in CpGs and regions rich in A and T further increases expression strength, as well as how insertion of specific binding sites makes such sequences respond to induced levels of the transcription factor NFκB. Our findings further contribute to the mechanistic understanding of promoters, as well as how these sequences might be shaped by evolutionary pressure in living organisms.
Introduction
The transcription and regulation of genes is a complex process that, in its basic form, requires the binding of transcription factors (TF) to a promoter region, a sequence of variable length located upstream of the transcription start site of a gene. Once bound, these TFs subsequently recruit the transcriptional machinery, leading to the production of primary transcripts. Consequently, the interaction of the promoter and respective TFs initially determines the strength and timing of gene expression, and understanding the molecular basis for this interaction has therefore been subject of much discussion and research.Citation1,Citation2 Previous work suggests that the binding of TFs to the promoter may be controlled by short, but defined sequence motifs.Citation3,Citation4 However, while transcription factors do appear to exhibit binding preferences for certain sequences, experimental data was able to show that they can bind to a variety of different motifs, and might even show strong affinity to more than one theme.Citation5,Citation6 This apparent promiscuity is reflected in the challenges encountered when trying to develop computational methods aiming at predicting transcription factor binding sites from nucleotide sequences, which remain prone to producing a high number of false positive candidates.Citation7 We may therefore ask ourselves, how exactly do TFs achieve high specificity in recognizing short binding regions out of billions of nucleotides? Is it the sequence, the constellation of critical motifs or may an even wider context be needed, meaning that short sequences in isolation carry little information to answer this question?
CpG Distribution May Determine Expression Strength Potential
We previously reported that artificial DNA sequences do not have to share homology with existing promoters to drive strong expression of a reporter gene in vitro,Citation8 as long as these sequences are rich in the di-nucleotide CpG, even in the absence of canonical transcription factor binding sites (TFBS). Such sequences, we found, could be bound by the generic transcription factors TFIIB and TFIID, which would then recruit the polymerase II complex and initiate transcription. Interestingly, short binding sequences, such as the TATA-Box, can inhibit expression if situated in certain spots in the promoter (i.e., in close proximity to the transcription start site), indicating that there is a highly localized component to TF binding, and that spacing and overall sequence arrangement might play an important role for specificity in TF/promoter interaction.Citation9
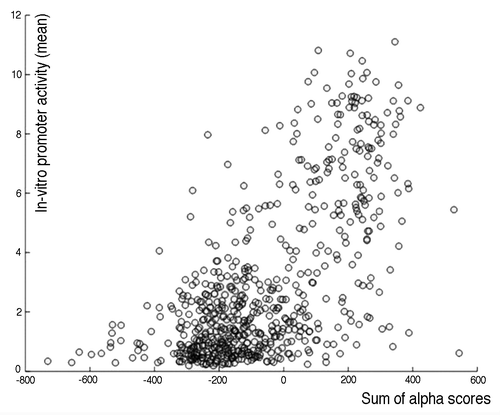
Sequences that are rich in CpGs are found in the vast majority of active human promoters.Citation10 The abundance of CpGs, however, appears not to be a direct predictor for expression strength potential. A total of 614 selected human sequences have been tested for in vitro activity as part of the ENCODE projectCitation11 and while there is a statistically significant correlation between the fraction of CpGs and transcriptional activity on the entire data set (ρ = 0.60, p = 7.6 × 10−61), it has not been possible to derive reliable predictions for in vitro strength based on CpG richness for individual sequences. Case in point, a number of regions with very few CpGs constitute fairly strong promoters, while others rich in CpGs show little activity (); the former suggests that there are non-CpG-mediated transcriptional mechanisms. Motivated by these findings, we set out to investigate how a changed number of CpGs, leading to altered CpG spacing, contributes to the latter.
Figure 1. Weak correlation between CpG richness and in vitro promoter activity. Shown is a scatterplot between CpG richness, as measured by the α score,Citation8 and in vitro promoter strength for the Stanford ENCODE Promoter data set. While the correlation is statistically significant (Spearman’s ρ = 0.60, p = 7.6 × 10−61), prediction power for individual promoter sequences based on CpG counts is very limited.
We previously described promoter ArS232, a 232 nucleotide long artificial sequence that is rich in CpG and devoid of canonical binding sites, which acts as a promoter in vitro in several mammalian cell lines.Citation8 The CpGs are fairly evenly spaced, with seven nucleotides being the largest distance between two consecutive instances. To determine the possible role of these motifs, we decided to alter this sequence’s organization. We replaced three or four regions of 20–25 nucleotides, roughly apart by the same distance, with nucleotide sequence devoid of CpGs (randomly chosen from the “genomic concatomer”) and more abundant in A and T (). The ArS232d2 parental sequence and the two mutated constructs ArS232d2_3inh and ArS232d2_4inh, cloned into the reporter vector pGL3-Basic (Promega) upstream of a firefly luciferase gene, were transfected into HEK293 cells and measured for bioluminescence 24 h post transfection. Vector pGL3-Promoter (Promega), containing the SV40 promoter, served as positive control, whereas the promoterless pGL3-Basic vector was used to determine the background level. Quantitative reporter assays were done via co-transfection with the Renilla luciferase vector pRL-SV40 (Promega), used as internal standard to normalize for experimental variations.
Figure 2. Modified artificial promoter constructs. We edited promoter ArS232-d2Citation8 by replacing three (ArS232-d2_3inh) and four (ArS232-d2_4inh) CpG-rich regions in local areas (25, 20, 20, and 25 nucleotides in size) with CpG-less sequences (A). This results in more than 3-fold increased in vitro activity for ArS232-d2_3inh, while ArS232-d2_4inh reduces activity to the original level. Shown are also activity of promoterless expression (-) and the SV40 core promoter (+). All relative luciferase activities were normalized to Renilla luciferase activity (B).
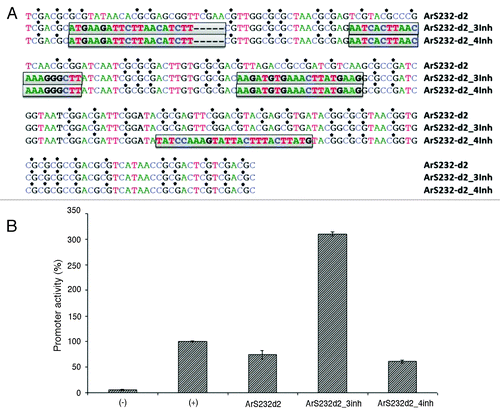
Unexpectedly, the removal of 14 CG di-nucleotides in construct ArS232-d2–3Inh increased expression levels markedly as compared with the construct ArS232-d2 (). Deletion of another 6 CG nucleotides in construct ArS232-d2-4inh decreased expression levels below the ones of ArS232-d2. By deleting said CpGs another spatial setting is creating longer intervals between adjacent CpGs. Thus, not only the total number was decreased but also the spatial arrangements. We note that the distribution of CpGs in the derived promoters more closely resembles that of existing promoters (e.g., CTAG1A, which we previously used to modulate promoter strength), suggesting that, in fact, local nucleotide changes involving CpGs can both increase and decrease activity. By changing the overall CpG content, flexibility and melting temperature also change the accessibility for DNA binding proteins and thus may cause the difference in transcription levels.
The experiment demonstrated that the activity of a promoter can be modulated by changing the number and at the same time the intervals of existing CpGs. Further mutations will serve to elucidate the optimal spatial arrangements and/or interactions of CpG islands. However it was demonstrated that for recombinant protein expression small artificial promoters are sufficient and can be modulated in terms of activity. This might further be useful since shorter promoter sequences allow for smaller plasmids to be transfected in vitro (cell line) and in vivo (e.g., in gene therapy), thereby increasing transfection efficiency.
Specificity through Canonical Binding Sites: NFκB
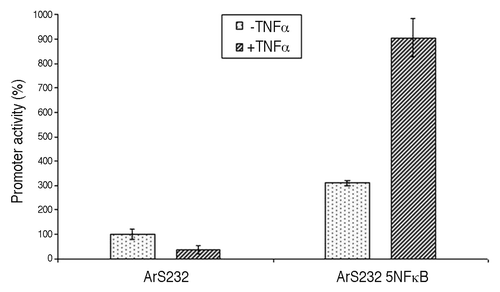
Many genes commonly interact in concert with other genes, making necessary a finely tuned system for their collective expression and regulation. Therefore, in order for a gene to be part of such a regulatory network, its promoter needs to react to the presence of specific transcription factors to alter expression levels. In order to test whether insertion of canonical sites creates a dependency on specific transcription factors, we chose the NFκB protein, which is well studied in mammals and whose binding site consensus has been described previously.Citation12 Upon insertion of five binding sites in tandem constellation, separated by CpGs, into construct ArS232, we measured in vitro activity under normal conditions as well as after induction of NFκB. HEK293 cells were transfected with the promoter constructs ArS232 and ArS232 5NFkB, inserted into the reporter vector pGL3 basic (Promega), and incubated for 12 h before induction of NFκB with recombinant human TNFα (Invitrogen) at a final concentration of 20 ng/ml. Luciferase activity was determined 6 h post induction. For standardization, the relative light units (RLU) of each promoter construct were expressed as the ratio of firefly and Renilla luciferase activity. While we note that many more experiments will be needed to explore the complexity of regulation through transcription factor binding, including determining the precise effects of spatial organization of binding sites and CpGs, initial results show that expression of construct ArS232 with binding sites increases 2.9-fold in presence of NFκB upon induction, while the expression level of the original construct, which lacks binding sites, remained roughly the same (). We note that we could also observe an increase in expression for the uninduced ArS232 5NFkB construct as compared with the original ArS232 construct, probably due to background levels of NFκB. Yet, it was clearly shown that induction was provided by the insertion of these transcription factor binding sites and transcription could be regulated upon external stimuli. The implications of these findings are quite remarkable, as it indicates that it should be possible to build artificial promoters that are driven by targeted TFs and can as such become part of a pathway.
Figure 3. Luciferase activity of promoter constructs controlled by NFκB. Shown are the in vitro activity of ArS232, and a derived construct ArS2325NFkB that contains five canonical NFκB binding sites (GGGGACTTTCC) in tandem constellation in HEK293 cells. In-vitro activity, shown in relative light units (RLU), of the sequence ArS2325NFkB increases 2.9 fold upon induction with 20 ng/ml TNFalpha, in contrast to sequence ArS232 which shows little change. All relative luciferase activities were normalized to Renilla luciferase activity.
Conclusions and Future Directions
Transcriptional expression and regulation is a complex process, with the promoter being only one part of the regulatory machinery within living cells (for examples and recent reviews see refs. Citation13–Citation15). Our in vitro results, however, show that it is possible to make predictions for transcriptional expression based on sequence features beyond the exact nucleotide sequence. We previously demonstrated that different nucleotide sequences can act in similar ways, as long as they are rich in CpGs. Here, we touch upon how CpG richness affects expression levels leading us to the assumption that possibly also the spatial relationship of these features determines promoter activity. Further we studied how canonical binding sites embedded in CpG rich sequences can add regulation through specific transcription factors, leading us to speculate that it is the combination of both features that provides the specificity required for this regulation. On one hand, such artificially designed promoters have great potential for industrial use, e.g., for the recombinant production of antibodies, with the ability to both optimize promoter strength statically to desired levels, as well as to control them dynamically through outside stimuli. By introducing specific transcription factor binding sites and combining several of these promoter elements, it becomes possible to design artificial regulation circuits in any desired manner, e.g., to synthetically engineer monitoring systems or mechanisms that trigger cell differentiation upon certain stimuli.
On the other hand, we hypothesize that gene expression levels could be gradually modulated up or down by very local mutations in living organisms. Adaptive evolution may act on such malleable regions to adjust gene expression to optimal levels in presence of selective pressure, and that changes in the local arrangements of short sequence features allow for increasing or decreasing a promoter’s response to specific transcription factors and pathways. Likewise, near neutral changes in the affinity to specific TFs may, over time, profoundly change the expression profile of entire gene networks, giving rise to new phenotypes or even species.
References
- Hochheimer A. Tjian. Diversified transcription initiation complexes expand promoter selectivity and tissue-specific gene expression. Genes Dev 2003; 17:1309 - 20; http://dx.doi.org/10.1101/gad.1099903; PMID: 12782648
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol 2006; 41:105 - 78; http://dx.doi.org/10.1080/10409230600648736; PMID: 16858867
- Juven-Gershon T, Hsu JY, Theisen JW, Kadonaga JT. The RNA polymerase II core promoter – the gateway to transcription. Curr Opin Cell Biol 2008; 20:253 - 9; http://dx.doi.org/10.1016/j.ceb.2008.03.003; PMID: 18436437
- Butler JE, Kadonaga J. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 2002; 16:2583 - 92; http://dx.doi.org/10.1101/gad.1026202; PMID: 12381658
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol 2006; 24:1429 - 35; http://dx.doi.org/10.1038/nbt1246; PMID: 16998473
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, et al. Diversity and Complexity in DNA Recognition by Transcription Factors. Science 2009; 324:1720 - 3; http://dx.doi.org/10.1126/science.1162327; PMID: 19443739
- Xie X, Rigor P, Baldi P. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics 2009; 25:167 - 74; http://dx.doi.org/10.1093/bioinformatics/btn605; PMID: 19017655
- Grabherr MG, Pontiller J, Mauceli E, Ernst W, Baumann M, Biagi T, et al. Exploiting nucleotide composition to engineer promoters. PLoS ONE 2011; 6:e20136; http://dx.doi.org/10.1371/journal.pone.0020136; PMID: 21625601
- Georges AB, Benayoun BA, Caburet S, Veitia RA. Generic binding sites, generic DNA-binding domains: where does specific promoter recognition come from?. FASEB J 2010; 24:346 - 56; http://dx.doi.org/10.1096/fj.09-142117; PMID: 19762556
- Kim TH, Barrera L, Zhang M, Qu C. Singer M, Richmond T, Wu Y, Green R, Ren B. A high-resolution map of active promoters in the human genome. Nature 2005; 436:876 - 80; http://dx.doi.org/10.1038/nature03877; PMID: 15988478
- Cooper SJ, Trinklein N, Anton E, Nguyen L, Myers R. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res 2006; 16:1 - 10; http://dx.doi.org/10.1101/gr.4222606; PMID: 16344566
- Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappa B signaling module. Oncogene 2006; 25:6706 - 16; http://dx.doi.org/10.1038/sj.onc.1209933
- Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol 2010; 220:126 - 39; http://dx.doi.org/10.1002/path.2638; PMID: 19882673
- Carthew RW, Sonetheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009; 136:642 - 55; http://dx.doi.org/10.1016/j.cell.2009.01.035; PMID: 19239886
- Muro EM, Mah N, Andrade-Navarro MA. Functional evidence of post-transcriptional regulation by pseudogenes. Biochimie http://dx.doi.org/10.1016/j.biochi.2011.07.024; PMID: 21816204