Abstract
A key challenge in the area of determining how the microbiome communicates with the host's karyome is deciding which microbial functions should be studied. Ideally we would wish to look at functions which are not only important to the microbial host, but which also play roles in host physiology. Selecting the key microbial functions is essential to developing robust strategies to either promote or demote them, with the aim to enhancing host health. This commentary argues that the bottom-up approach is not providing the necessary gene-set from which we can start to develop a robust core microbiome and in fact we should adopt a top-down strategy in order to indentify the functions that are important and need further study.
Introduction
The current interest in the microbiota of the mammalian gut has been driven by the realisation that, what we thought were the major bacterial members of the community are actually those that are easily culturedCitation1 and the gut microbiota may act as an extension of the host's karyome and potentially plays a role in maintaining a healthy hostCitation2,Citation3 and possibly initiating diseases not only of the gut, but external to this organ.Citation4 The first point has led to the implementation of culture-independent methods, which were mainly developed to explore the roles that microbes play in non-animal ecosystem function, while the second has resulted in a “gold-rush” to try and find microbes responsible for many of the developed countries' diseases, such as cardio-vascular disease,Citation5 colorectal cancer,Citation6 obesity,Citation7 atopic diseasesCitation8 (asthma and eczema) and some behavioral diseasesCitation9 (stress and depression). The overall aim is to determine which functions are assets and which are liabilities and to maximise and minimise them respectively. In respect of this strategy we have quite a comprehensive knowledge base for the members of the gut microbiota responsible for synthesising butyrate and other short chain fatty acids,Citation10–Citation12 but beyond this role we have a limited understanding of the other functions which may be important to the host.
The Current Approaches Used to Map the Bacterial Landscape of the Gut
Many of the initial forays which aimed to measure gut bacterial diversity were based on community “profiling” methods such as denaturing/temperature gradient gel electrophoresisCitation13 which provided an insight, albeit at low resolution, into the community structure of the gut microbiota. Surprisingly the gut microbiota were relatively stable over time and host specific, which made life difficult when comparing samples, as an individual generally became their own control, since inter-individual comparisons were almost meaningless when using species to discriminate between samples. The obvious next step was to develop inventories of the species present, by using the 16S rRNA gene as a proxy for a cultured isolate. While the technology was available (dideoxy based sequencing chemistry or 1st generation sequencing technology), the costs, both time and financial, were prohibitive for large scale “deep” sequencing. However, small cohort projects were undertaken and one of the most cited (>1,000 citations in Web of Science) was able to capture the diversity in the colon. Eckburg and co-workersCitation14 created inventories of 16S rRNA genes from 6 mucosal sites in the colon (cecum, ascending colon, transverse colon, descending colon, sigmoid colon and rectum) and a fecal sample from each of the 3 volunteers. In total they sequenced 13,355 partial genes products (average numbers of reads was 563, range 355–1,060) which provided the most in depth analysis of the distal gut and showed that this system is predominated by members of the Bacteria, predominantly from two phyla, Firmicutes and Bacteroidetes () and principally one species of Archaea, namely Methanobrevibacter smithii. This distribution of the main phyletic groups in the distal colon has been accepted as a consensus for the general adult large intestine and regardless of the starting conditions (birth mode, feeding regimes, etc.,) the majority of infants will, after the first 18 months, show signs of reaching this climax community.Citation15,Citation16 Once we had established the range of phyla in the distal gut, which happened to be quite limited, it was hypothesised that there might be a set of bacterial species or core microbiota (which should be called the bacteriota, as no viral or micro-eukaryotic components were included in the census) which is shared by all humans. Several studies have attempted to determine what the members of this core would be and have reported mixed resultsCitation17,Citation18 while reports using 2nd generation sequencing platforms (analysis of 16S rRNA genes primarily on the Roche 454 platform) have also addressed this topic,Citation7,Citation19–Citation22 but with mixed conclusions. The consensus of opinion is that the core microbiota exists at a higher taxonomic levels, for example at the phylum or class, but at the lower levels the core is absent. So some researchers have started to move away from determining species and moved instead to looking at functions in the gut. The aim would be to determine whether there is a core microbiome rather than a core microbiota/bacteriota.
Functional Analysis of the Gut Microbiome
Determination of the functions in the gut can be assessed in one of two ways, assuming a culture independent approach is used. Either by sequencing random fragments of genomic DNA (gDNA) or by functionally screening a clone library of semi-random fragments of gDNA (mainly through heterologous expression of the gDNA from a surrogate host). Each method has its advantages and disadvantages which are outlined in and will not be discussed any further in this commentary. Approaches based on sequencing have recently been used to undertake a comprehensive analysis of the distal gut microbiomeCitation23 and have shown that there exists a huge repertoire of unique genes in the distal gut, approximately 3.3 million and around 536,000 per individual gut. The authors concluded that the core microbiome consisted of over 6,300 functional orthologous groups which were found in all subjects analysed, however, we need to be clear on what was measured, as this is really the bacteriome and not the microbiome, no viral components or micro-eukaryotes were targeted. Interestingly they were able to determine that the shared species between all 124 subjects was very small, 18 species, and were all members of the Firmicutes or Bacteroidetes, thus supporting the notion that each person's gut is host-specific and a core bacteriota does not exist at the species level. However, the authors do stress that the depth at which the gut is sequenced sampled can strongly influence this set, so if groups are willing to spend the time and monies, we may answer this question in the near future. In the meantime the more interesting aspect of this study was the aim to establish the core functions in the distal gut, both at the individual bacterial genome and metagenomic levels. To this end the authors were able to show that a significant proportion of the functions they identified were involved in housekeeping roles and gut specific functions, such as adhesins and sugar harvesting, were noted, but this list was limited. In addition there were a significant number of genes which were unknown or hypothetical in the minimal gut genome and minimal gut metagenome, 73% and 80% respectively. This data, while of great value, does raise the central issue of this commentary, are these approaches actually addressing the issue of what microbial functions are important to both hosts? The short list of gut specific functions shows the difficulty in determining what is important to both hosts in this ecosystem. In respect of this it is suggested that we need to re-address the issue of what constitutes a core function. If we now view a core function as one that must benefit the bacterial host as well as interacting with the human host and is present in the majority of healthy distal guts (>50%), many simple housekeeping functions are not included. The need to undertake DNA replication, protein synthesis, amino acid metabolism or nucleotide metabolism are bacterial core functions, irrespective of where you find the bacterium. However, the need to process host molecules such as primary bile saltsCitation24 and metabolise exogenous and endogenous β-glucuronidesCitation25 are definitely core gut functions, since they benefit the bacterial host and interact with the host's physiology/biochemistry. While these two functions are ubiquitous in the gut bacteriome () it can be difficult to determine the full repertoire of what should be considered in the list of core functions of the gut bacteriome. These two functions were established by analysis of cultured isolates and the diversity expanded using functional metagenomics. Similar studies have been undertaken to investigate the genes involved in butyrate synthesisCitation26 and show a large degree of diversity and dispersion. However, it becomes difficult if no genes are identified, but there is a clear case for a microbial function which is gut specific and influences the host, for example being involved in cardio vascular disease or behaviour.Citation5,Citation27 In the example shown in genes have been chosen which have been clearly implicated in bacterial host function and at the same time eukaryotic host physiology, so some genes are considered to be involved in pathogenesis, e.g., genes from the Afa operon of the diffusely adhering E. coli,Citation28 while the butyrate synthesising genes are mutualistic and are essential for the production of butyrate which is used by colonocytes in preference to any other energy sourceCitation29 and the disposal of electrons by the bacterial hostCitation30 and thus fulfils all the criteria for being a core function. From this simple analysis it can be seen that we can start to define foundations of a core bacteriome, one that includes short chain fatty acid synthesis (proxies used were β subunit of CO dehydrogenase/acetyl-CoA synthase complex, butyryl-CoA:acetate CoA-transferase and formate-tetrahydrofolate ligase), bile processitivity (bile salt hydrolases) and β-glucuronide processing (H11G11 like β-D-glucuronidases). Moreover, genes involved in pathogenesis, e.g., from the AFA operon, are infrequent and not widely disseminated, which is to be expected. The key to establishing this set of genes is what should we include and what should we omit. Clearly a bottom-up approach does not help as we are swamped with house-keeping genes and unknown/hypotheticals. Hence a top-down approach needs to be implemented which guides us to the core bacteriome/microbiome.
Shifting to a Top-Down Approach to Help Define the Core Bacteriome
Current approaches work from a bottom-up, a gene to product information flow, which results in a bias in a metagenomic strategy towards genes which are common, but not necessarily true core functions. Working from the top of the information pile may deliver more relevant candidates which we can test as being members of the core microbiome. In this instance the information we are dealing with are the metabolites being produced by the microbes in the gut and which can be shown to be bioactive, which can either be small molecules such as the SCFA, proteins (BSHs, H11G11 like β-glucuronidases) or carbohydrates/polysaccharide.Citation31 In order to determine these we need metabonomic and metabolomic approaches to determine the core metabolites in the gut and those that are due to diet, those that are variable in the population and those produced by the microbiota. Good examples of these lead metabolites are those derived from the metabolism of tryptophan metabolism,Citation27,Citation32 phosphatidylcholine,Citation5 primary bile salts,Citation33 amino acidsCitation5,Citation34 and plant polyphenols.Citation35 A list of candidate metabolites has been already been developed () which would act as a starting point.Citation36 A more comprehensive list of metabolites needs to be constructed, which are found in all guts studied and especially which are not found in humans, but are present in experimental animal models. For some members of the list we will have candidate genes and for others we will need to determine the functional genes responsible using a variety of approaches that are available and these will include culture-dependent as well culture-independent methods. A notable strategy to start and find organisms responsible for synthesising these metabolites would be to construct heat maps showing species or OTUs which correlate with specific metabolitesCitation37 and use this information to guide the culturing or metagenomic analysis.
Conclusions
The area of gut microbiology has come a long way since the early forays into this system using culturing, but the application of 2nd and now 3rd generation sequencing platforms has started to produce huge arrays of data and trying to determine what's important in the context of gut microbiology is daunting, we are struggling to see the wood for the trees. The metabolic phenotype of an individual, while very complex, contains valuable information that can be used to determine the core microbiome. In this commentary it has been suggested that the concept of a gut core function be restricted and that we should start to be more selective in what we call a core function. In addition we should use the metabolic data to inform us as to which functions we should be investigating and those to which we should be paying attention.
Figures and Tables
Figure 1 (A) Phylogenetic tree based on the combined human intestinal 16S rRNA gene sequence data set, the label for each clade includes, in order, the total number of recovered sequences, phylotypes and novel phylotypes (in parentheses; adapted from ref. Citation14). (B) Relative abundance of sequences from stool and pooled mucosal samples per subject: ■-other groups represents the Fusobacteria, Actinobacteria and unclassified near Cyanobacteria phyla, ■-Verrumicrobia and □-Proteobacteria; “M” denotes pooled mucosal sequences per subject and “S” refers to stool sample (adapted from ref. Citation16).
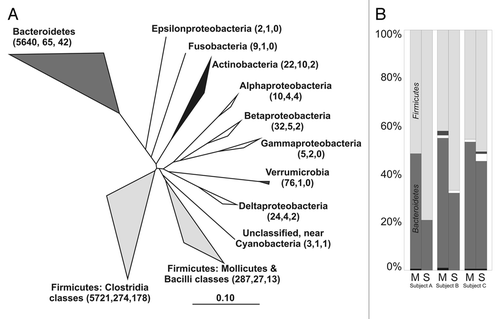
Figure 2 Tblastn analysis of the MetaHIT healthy dataset (n = 85) with functional genes from the distal gut (e ≤ 1 × 10−5). (A) Mb DNA screened for one “hit” to the target gene and (B) Percentage of samples in which the gene was found. Gene codes were: McrA-α subunit of methyl coenzyme M reductase from Methanobrevibacter smithii ATCC 35061 (YP_001273475); AFaB and AFaC; two genes from the Afa operon involved in adhesion of pathogenic diffusely adhering E. coli;Citation28 DsrA-α subunit of dissimilatory sulfite reductase from Desulfovibrio piger ATCC 29098 (AAL57441); Acs-β subunit of CO dehydrogenase/acetyl-CoA synthase complex from Blautia hansenii DSM 20583 (EEX21742); Lipo-putative lipoprotein from fosmid clone 52B7 (ADR74178);Citation38 But-Coa-butyryl-CoA:acetate CoA-transferase from Clostridium sp. SS2/1 (ZP_02439482);Citation26 Gluc-β-D-glucuronidase from fosmid clone H11G11 (CBJ55485);Citation25 Fhs-formate-tetrahydrofolate ligase from Bacteroides thetaiotaomicron VPI-5482 (AAO75844) and Bsh-bile salt hydrolase from Alistipes sp. HGB5 (ZP_08513987).
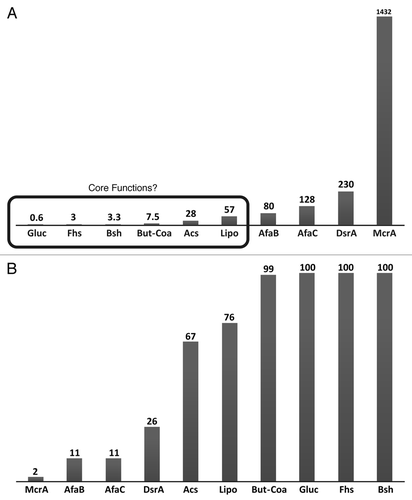
Table 1 Comparison of the pros and cons of the two metagenomics methods used to study the functions in an ecosystem
Table 2 List of common metabolites found in human, mouse and rat feacal water
References
- Savage DC. Microbial ecology of the gastrointestinal tract. Ann Rev Microbiol 1977; 31:107 - 133
- Shanahan F. The colonic microflora and probiotic therapy in health and disease. Curr Opin Gastroenterol 2011; 27:61 - 65
- O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 2006; 7:688 - 693
- Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut Microbiota in Health and Disease. Physiol Rev 2010; 90:859 - 904
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011; 472:57 - 63
- Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011; 6:16393
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457:480 - 484
- Huffnagle GB. The microbiota and allergies/asthma. PLoS Pathogens 2010; 6
- Heijtz RD, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 2011; 108:3047 - 3052
- Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 2010; 12:304 - 314
- Bugaut M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp Biochem Physiol Part B: Comp Biochem 1987; 86:439 - 472
- Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 2006; 40:235 - 243
- Zoetendal EG, Akkermans AL, deVos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol 1998; 64:3854 - 3859
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the Human Intestinal Microbial Flora. Science 2005; 308:1635 - 1638
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the Human Infant Intestinal Microbiota. PLoS Biology 2007; 5:177
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 2011; 108:4578 - 4585
- Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 2009; 6:6
- Rajilic-Stojanovic M, Heilig HGHJ, Molenaar D, Kajander K, Surakka A, Smidt H, et al. Development and application of the human intestinal tract chip, a phylogenetic microarray: Analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environmental Microbiology 2009; 11:1736 - 1751
- Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 2011; 108:4586 - 4591
- Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative Analysis of Human Gut Microbiota by Barcoded Pyrosequencing. PLoS ONE 2008; 3:2836
- Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, et al. Organismal, genetic and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA 2010; 107:7503 - 7508
- Gillevet P, Sikaroodi M, Keshavarzian A, Mutlu EA. Quantitative Assessment of the Human Gut Microbiome Using Multitag Pyrosequencing. Chem Biodivers 2010; 7:1065 - 1075
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59 - 65
- Jones BV, Begley M, Hill C, Gahan CGM, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA 2008; 105:13580 - 13585
- Gloux K, Berteau O, El Oumami H, Beguet F, Leclerc M, Dore J. A metagenomic beta-glucuronidase uncovers a core adaptive function of the human intestinal microbiome. Proc Natl Acad Sci USA 2011; 108:4539 - 4546
- Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol 2010; 12:304 - 314
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 2009;
- Servin AL. Pathogenesis of Afa/Dr diffusely adhering Escherichia coli. Clin Microbiol Rev 2005; 18:264 - 292
- Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterol 1982; 83:424 - 429
- Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc 2003; 62:67 - 72
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005; 122:107 - 118
- Swann JR, Tuohy KM, Lindfors P, Brown DT, Gibson GR, Wilson ID, et al. Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. J Proteom Res 2011; Epub
- Bernstein C, Holubec H, Bhattacharyya A, Nguyen H, Payne C, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 2011; 1 - 9
- Broadley KJ, Akhtar Anwar M, Herbert AA, Fehler M, Jones EM, Davies WE, et al. Effects of dietary amines on the gut and its vasculature. Brit J Nut 2009; 101:1645 - 1652
- Kutschera M, Engst W, Blaut M, Braune A. Isolation of catechin-converting human intestinal bacteria. J Appl Microbiol 2011; 111:165 - 175
- Saric J, Wang Y, Li J, Coen M, Utzinger J, Marchesi JR, et al. Species variation in the fecal metabolome gives insight into differential gastrointestinal function. J Proteome Res 2008; 7:352 - 360
- Li JV, Ashrafian H, Bueter M, Kinross J, Sands C, le Roux CW, et al. Metabolic surgery profoundly influences gut microbial-host metabolic cross-talk. Gut 2011;
- Lakhdari O, Cultrone A, Tap J, Gloux K, Bernard F, Ehrlich SD, et al. Functional metagenomics: a high throughput screening method to decipher microbiota-driven NFκB modulation in the human gut. PLoS ONE 2010; 5