Abstract
Intravital microscopy is an extremely powerful tool that enables imaging several biological processes in live animals. Recently, the ability to image subcellular structures in several organs combined with the development of sophisticated genetic tools has made possible extending this approach to investigate several aspects of cell biology. Here we provide a general overview of intravital microscopy with the goal of highlighting its potential and challenges. Specifically, this review is geared toward researchers that are new to intravital microscopy and focuses on practical aspects of carrying out imaging in live animals. Here we share the know-how that comes from first-hand experience, including topics such as choosing the right imaging platform and modality, surgery and stabilization techniques, anesthesia and temperature control. Moreover, we highlight some of the approaches that facilitate subcellular imaging in live animals by providing numerous examples of imaging selected organelles and the actin cytoskeleton in multiple organs.
Introduction
In the past two decades the development of fluorescent protein technology and live cell microscopy resulted in an explosion of information on every cellular process imaginable, leading to tremendous advancements in our understanding of cell biology.Citation1,Citation2 Most of this knowledge has been acquired by employing cell culture models, which are extremely powerful when it comes to experimental manipulations. However, they do not often provide a true representation of the biology of complex multicellular organisms. A first step toward addressing this issue has been taken by the development of imaging approaches in C. elegans, D. melongaster and D. rerio which offer the possibility to easily perform genetic manipulations and thus to unravel the molecular basis of several processes involved in tissue development.Citation3-Citation8 More recently, light microscopy-based techniques have been extended to live mammalian models leading to the development of a new imaging approach called intravital microscopy (IVM).Citation9,Citation10 Rapidly, IVM has become an essential tool in several areas, such as neurobiology, immunology and tumor biology.Citation11-Citation16 In addition, in the last five years the achievement of subcellular imaging in live rodents has opened the door to study cell biology at a molecular level and in physiological conditions.Citation17-Citation22 These advancements have been made possible by the improvement of microscopes, optics and software for imaging analysis, and by novel genetic tools that enable controlling the expression of a given gene both temporally and spatially. In particular, several knock-in mice expressing fluorescently labeled proteins have become available at relatively low costs enabling scientists to address several key scientific questions.
The goal of this review is to introduce the readers to the practical aspects of IVM by providing basic information and guidelines to begin imaging live rodents. First, we will describe how to choose the most appropriate imaging modality and settings that are crucial to properly perform IVM. We want to emphasize that a detailed description of the principles on which the various imaging techniques are based is beyond the goal of this paper, and we will direct the readers to more specialized literature. Next, we will provide a general description of the strategies to minimize motion artifacts and maintain proper physiological conditions. These are general guidelines and some adjustments in the procedures described here will be necessary to fit the specific needs and instrumentation of each investigator. Finally, we will discuss the current tools that are available to image cellular, subcellular structures and some of the components of the actin cytoskeleton, at different levels of resolution, providing several examples from our own work.
Imaging Modality and Depth
IVM has been performed by using various light microscopy techniques, such as widefield fluorescence, laser scanning confocal, multiphoton and spinning disk microscopyCitation9,Citation18,Citation19,Citation23-Citation25 (). Although other techniques such as harmonic generations (SHG and THG), fluorescence life time microscopy (FLIM), coherent anti-stokes Raman microscopy (CARS) and optical frequency domain imaging (OFDI) have been successfully used for IVM they have not been applied extensively to subcellular imaging in live animals and will not be discussed further.Citation26-Citation34 The most important consideration in choosing the appropriate imaging modality is the depth that has to be reached. In order to form an image by light microscopy, the excitation beam has to collide with the target molecules and excite a transition between two energy levels (). After a thermal decay due to vibrational and rotational motions, the light emitted upon relaxation travels toward the detection system where it is collected and transduced into the signal that ultimately generates the final image (). In the case of solid tissues, the main limiting factor is the light scattering that affects both the excitation beam and the emitted light (). If the imaging area is located more than 50–100 µm below the surface of the organ the obligatory choice is multiphoton microscopy (MPM) ().Citation35-Citation37 MPM is based on the simultaneous absorption (within few femtoseconds) of two or more photons with wavelengths in the near infrared (NIR) or infrared (IR) (). Light in this region of the spectrum has an intrinsic deeper penetration than visible or UV light.Citation37 The maximum depth that can be imaged depends on both the optical properties of the tissue and the experimental set up (i.e., optics, excitation wavelength, power of the beam and detection system). As for the “optical transparency” of a given organ, it strongly depends on its homogeneity. Indeed, light undergoes scattering when crossing the interface between component of the tissues with different refractive indexes.Citation38,Citation39 It is also generally true that highly vascularized tissues are less optically transparent due to the light absorption and scattering of the red blood cells. Although a rigorous side by side comparison of the various organs has never been performed, it has been empirically determined that tissues such as the brain are optically more suitable for IVM than skeletal muscle, skin, liver or kidney. Notably, in the last couple of years several approaches have been used to “clear” tissues by using special solutions that either reduce the difference between the refractive index in the various tissue layersCitation38,Citation39 or make them optically transparent.Citation40 This approach has enabled imaging neurons labeled with genetically encoded fluorescent proteins up to 8 mm below the surface with an unprecedented resolution ().Citation40 Other tissues, such as skeletal muscle, benefit from the “clearing” approach, although not to the same extent as the brain (). Although the use of clearing agents has been limited to fixed tissue, it should be worthwhile to invest the effort in developing modified formulations for applications in live animals. It is also important to mention that in the brain the imaging depth has been extended by the use of particular tools such as gradient-index (GRIN) lenses, micro prisms or the installation of ports of observation.Citation41-Citation45 GRIN lenses are small cylindrical lenses (350 µm in diameter) () that can be inserted into the brain after removal of the skull, enabling the visualization of areas, such as the hippocampus, located 1.5 mm below the cortex.Citation42,Citation46 Micro prisms are also inserted into the cortex although they work with a different principle.Citation43,Citation44 The hypotenuse of a micro prism has an enhanced silver reflective coating that translates the rastering pattern of the excitation beam from an xy plane to an xz plane (). This approach has made possible extending the imaging depth up to the length of the prism (1–2 mm) without loss of resolution. Finally, optically clear guides have been permanently implanted in the mouse brain serving as a chronic port of observation to perform longitudinal studies.Citation41 Although these approaches have been originally applied to the brain, some of them have been successfully extended to other organs such as the kidney in mice or skeletal muscle in humans.Citation45,Citation47
Table 1. Imaging techniques used for intravital microscopy
Figure 1. Imaging depth in intravital microscopy. (A) Jablonski diagram of fluorophore excitation by single and multiphoton microscopy. The energy gap between two energy levels (ground state, E0 and excited state, E*) can be filled by one, two or three photons. After thermal decay, the system relaxes to the ground state by emitting a photon. (B) All the photons generated by the imaging area are emitted, and collected by the detection system. (C) When the imaging area is located in solid tissue both the excitation beam (blue arrows) and the emitted photons (green arrows) are scattered. The deeper the targeted area the lower is the probability to excite a transition or to detect the emitted photons. (D) Diagram illustrating imaging modality, resolution and optics as a function of imaging depth. (E) Diagram illustrating alternative approaches to extend the imaging depth by MPM. (F) Diagram illustrating GRIN lenses and micro prisms inserted in the tissue to image up to 1–2 mm in depth. (G) Effect of the clearing agent scale/e. A portion of the quadriceps was excised from a mouse expressing soluble GFP, fixed in 2% formaldehyde and incubated for 3 d in the clearing agent scale/e.Citation40 A z-scan was acquired by two-photon microscopy (excitation wavelength 930 nm) using water immersion 25x lens (XLPL25XWMP, from Olympus). Notably, cleared tissue was imaged up to 1.2 mm and the muscle fibers were nicely resolved, whereas usually non-cleared tissue cannot be imaged beyond 500 µm.
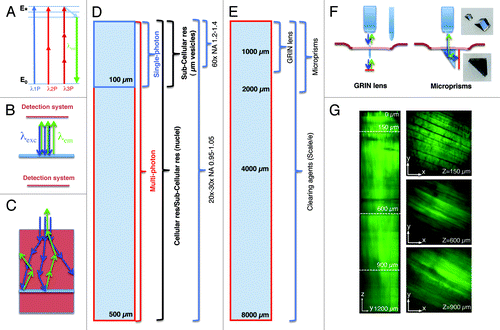
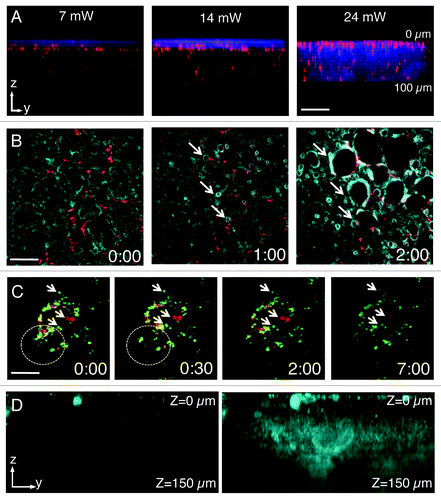
As for the experimental set-up, the properties of the laser are important to successfully perform deep tissue imaging. MP excitations are typically achieved by using tunable Ti:Sapphire lasers which emit NIR light (wavelengths between 680–1080 nm) in very short pulses (in the order of 80–140 fs) and at high repetition rates (80–100 MHz). It has been shown that photons with lower energy (and longer wavelengths) are subjected to less scattering and reach deeper areas.Citation13,Citation48,Citation49 In this respect, the ability to extend the range of the tunable lasers up to 1600 nm via optical parametric oscillators (OPO) has enabled reaching greater depths and image solid tumors up to 600 µm.Citation13,Citation34,Citation49 In addition, regenerative amplifiers have been used in the brain to reach depths up to 1 mm.Citation50 Although increasing the power of the excitation beam results in increasing the imaging depth () the reader should be aware that above certain thresholds (5–20 mW) tissue damage can occur (). In some instances, tissue damage is easily detectable, due to the appearance of a large damaged area (; Vid. S1), whereas in other circumstances, the damage is more subtle to detect and may result in sudden halting of intracellular organelle motility (; Vid. S2). As a general rule, bleaching of a fluorescent probe would indicate that the power should be reduced (, right panel). We should mention that the power effects are mitigated by the depth of the imaging plane in the tissue, so generally higher power can be used to image deeper areas of the tissue. Imaging depth is also limited by the resolution that has to be achieved. For example, in order to perform subcellular imaging, high magnification lenses (i.e., 60x or higher) with high numerical apertures (NA) are used. However, their working distances are restricted to 200–250 µm (). Objectives with lower power (i.e., 20x or 30x) have lower NAs but longer working distances (up to 2 mm), which enable imaging single cells at greater depth in the tissue ().
Figure 2. Laser power and detector sensitivity in intravital two-photon microscopy. Anesthetized rats were injected with Hoechst (blue) and Texas red-dextran (red) (A), Texas red–dextran alone (B), Texas Red- and Alexa 488-Dextran (green) (C) or not injected. After 2 h, the salivary glands were exposed and imaged by two-photon microscopy (excitation 750 nm) using a 60x water immersion objective (NA 1.2, Olympus). (A) Salivary glands were imaged by using 3 different laser powers (7 mW, 14mW and 24 mW, as measured at the objective). Dextran was internalized by stromal cells (red) and Hoechst labeled the nuclei (blue). Bar, 50 µm. (B and C) Salivary glands were imaged using two laser powers: 30 mW (B) and 15 mW (C). (B) Excitation of endogenous fluorescence reveals the parenchyma (cyan) whereas stromal cells are highlighted by internalized dextran (red). After 1 min, the tissue shows signs of photodamage (arrows) and around 2 min is completely disrupted (Vid. S1) Bar, 40 µm. (C) Internalized dextrans are localized in two different endosomal populations within stromal cells and exhibit high mobility within the first 30 sec (see area within dotted circles). Endosomal mobility is suddenly stopped after 1–2 min from the beginning of the imaging session (Vid. S2) Note also that photobleaching occurred (right panel). Bar 10 µm. (D) Z-scan of unstained salivary glands by using a conventional multialkali detector (left panel) or GaAS detector (right panel).
MPM also has other advantages when compared with other imaging techniques. MP excitation requires a high number of photons concentrated in space and time, therefore the absorption and the emission occur in a very small volume (1 fl).Citation36,Citation37,Citation51 This reduces significantly both phototoxicity and photobleaching. Moreover, point excitation avoids the issue of off-focus emission, which in confocal microscopy is blocked through the use of a pinhole. This implies that all the light collected from the specimen comes from the focal point and contributes to the image. In MPM microscopy it is essential to maintain the shortest light path from the specimen to the detection system in order to gather as much photons as possible. Typically, multialkali detectors are routinely used, although in the last few years GaAs detectors have become very popular due to their exceptional signal to noise ratio. This makes them ideal for detection of very low signals and sampling deeper areas (). Another important feature of MP excitation is that fluorophore absorption spectra are much broader than in single photon excitation. This facilitates imaging multiple fluorophores using a single excitation wavelength.Citation10,Citation52,Citation53 Finally, MPM provides a way to visualize endogenous fluorescence or collagen fibers by second harmonic generation, which allows observing the tissue without any exogenous labels.Citation10,Citation26,Citation54
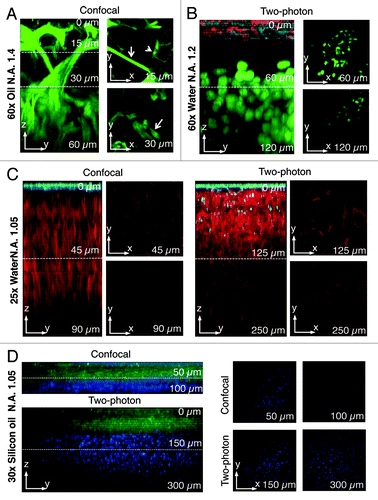
Even though MPM has become the most common way to perform IVM, we want to emphasize that other imaging modalities can be used as successfully. For example, if the target organ is very homogenous and the biological process under investigation occurs within the first 50–100 µm from the surface, confocal or widefield microscopy can be used (). Our group has recently shown that exocytosis and the actin cytoskeleton can be studied at a subcellular resolution in the salivary glands of live mice and rats by using intravital confocal microscopy.Citation18,Citation55-Citation57 Indeed the salivary glands are a very homogenous tissue and the first two layers of acinar cells are located within 30 µm from the surface.Citation18 One of the main advantages of confocal over MP microscopy is a better spatial resolution that enables a better definition of the subcellular structures. Indeed, in MP modality the optical slice (between 1 and 1.5 µm) depends on the instrument and the optics.Citation37,Citation51 Under these conditions subcellular organelles that are in the submicron range cannot be completely resolved. On the other hand, in confocal modality the optical slice can be tuned by changing the size of the pinhole to match the size of the imaged organelles.Citation18 In addition, single photon excitation allows performing other techniques such as FRAP, FRET and photoactivation in a more effective way than in multiphoton modality.Citation58-Citation60 Finally, it is important to emphasize that confocal IVM requires a bright specimen that can be imaged with reduced laser power in order to minimize photobleaching and photodamage.
Figure 3. Comparison between two-photon and confocal microscopy at different magnifications. (A) The salivary glands of a transgenic mouse expressing Lifeact-GFP were imaged by confocal microscopy (excitation 488 nm) using an oil immersion lens (NA 1.4, Olympus). A z-scan was acquired reaching a maximum depth of 60 nm (zy view). In the xy views, myoepithelial cells (arrows) and the apical plasma membrane (arrowheads) are highlighted. (B) Human head and neck squamous carcinoma cells (HN12) were engineered to express GFP-histone 2B and injected in the back of an immunocompromised mouse. After 2 weeks, Texas red-dextran was injected systemically, the primary tumor was surgically exposed and imaged by intravital two-photon microscopy (excitation 930 nm) with a 60x water immersion lens (NA 1.2, Olympus). A z-scan shows that two-photon microscopy extend the imaging depth to 120 µm (see xy views). The surface of the tumor was determined by visualizing collagen fibers that are excited at this wavelength (cyan) and superficial stromal cells (red) that internalize dextran. The tumor mass is highlighted by the nuclear staining (green). (C) A mouse ubiquitously expressing a membrane targeted peptide fused to the tandem tomato (m-tomato) was engineered to express a membrane targeted peptide fused to GFP (m-GFP) in keratin 14-expressing cells as previously described.Citation83 The liver was excised and imaged by either confocal (left panels, excitation 488nm and 561 nm) or two-photon (excitation 930 nm) microscopy with a 25x water lens (NA 1.05, Olympus). GFP expressing cells are confined to the surface of the liver (cyan) whereas the hepatocytes were imaged up to 90 µm (confocal) or 250 µm (two-photon). (D) The mouse described in (C) was injected with Hoechst, and the tongue was imaged by either confocal (excitation 350 nm and 488 nm) or two-photon (excitation 820 nm) using a 30x silicon lens (NA 1.05, Olympus). A z-scan shows that nuclei (blue) can be imaged up to 100 µm (confocal) or 300 µm (two-photon).
In terms of temporal resolution, both confocal and MPM microscopy suffer from inherently slow imaging rates that reach at best a few frames per seconds. Although, recent microscopes are equipped with resonant scanners that acquire up to 30 frames/second,Citation61 other techniques such as spinning disk confocal microscopy are superior in terms of both acquisition speed and limited photobleaching. Indeed, spinning disk has been successfully applied in D. rerio and C. elegans to perform 4D imaging and it should be extended to those mammalian systems that do not require deep imaging.Citation62-Citation64
Minimizing the Motion Artifacts
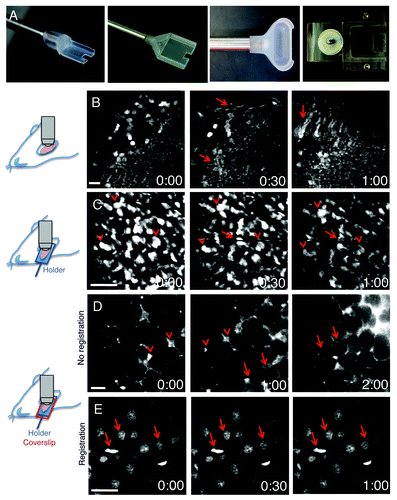
The main challenge to successfully perform IVM is to minimize the motion artifacts that are generated by heartbeat, respiration and peristaltic movements. This is especially true for applications that require imaging at high magnification, such as dynamics of subcellular structures that are affected by small shifts of a few microns in every direction (). The first action to take in order to minimize motion artifacts is to devise specific surgical procedures and to properly position the exposed organs. These procedures are determined primarily by the configuration of the microscope (i.e., upright vs. inverted). Most of the organs can be imaged in either configuration, with the exception of the brain that is exclusively imaged using upright microscopes due to the use of stereotactic devices to immobilize the head of the rodents. To image in the upright configuration, the main strategy is to surgically expose the organs and to immobilize them by using either custom-made holders or microstage devices. For example, holders have been used for salivary glands, the kidney and the tongue (),Citation19,Citation65 whereas abdominal organs have been imaged by using custom-made microstages.Citation17,Citation66 Although, these approaches significantly minimize the motion artifacts, particular care has to be taken to ensure that the physiology of the organ is properly maintained. Pressing an organ to increase stabilization can lead to reduction of the blood flow, hypoxia and tissue damage. Imaging in the inverted configuration, generally results in better stabilization and better preservation of the physiology of the organ but requires more extensive surgical procedures. In this case, the surgery is not limited to expose the organs but is aimed at isolating them from the rest of the body without damaging tissue, nerves or causing major bleeding. For example, removal of a ligament or the surrounding connective tissue may significantly reduce motion artifacts. Moreover, the body of the animal and areas such as the thorax should be coupled to the stage without interfering with the vital functions. Finally, a series of tools such as bars, spacers, threads have to be used as needed to immobilize specific areas of the body.Citation67
Figure 4. Minimization of motion artifacts. (A) Custom-made holders were built to accommodate mice and rat salivary glands (left panels), rat kidney (center right panel) or mouse tongue (right panel). (B–E) Anesthetized rats were either injected with Hoechst (B, C and E) or the salivary glands were exposed and bathed with Texas Red-dextran to label the stroma (D). Time-lapse imaging was performed by two-photon microscopy (excitation 800 nm for B, C and E; 930 nm for D). Salivary glands were exposed and imaged directly with a 60x water immersion objective (NA 1.2, Olympus) (B), or exposed and placed in a custom made holder without (C) or with a coverglass to minimize the motion (D and E). In (B), motion artifacts create distortions within the frames (arrows), shifts in the xy plane and changes in the focal plane. In (C), although several structures are still in register (arrowheads) distortions within the frames are observed (arrows). In (D), only shifts in the xy plane are observed and can be corrected by using software such as ImageJ (Stack-ref plug ins) (E). Bars, 20 µm.
The upright configuration permits the use of dipping lenses that can be coupled directly onto the organ via aqueous buffers, optical coupling gels having the appropriate refractive index or physiological fluids, such as artificial cerebrospinal fluid that has been used for brain imaging.Citation19,Citation68 Although dipping lenses provide better image quality, often it is necessary to secure a coverslip on top of the organ to further reduce the motion artifacts and/or to flatten its surface in order to properly position the objective (). Normally high NA objectives require use of coverslips and an immersion media such as oil or silicon oil, although there are water immersion objectives available that have coverslip correction collars, which facilitate their dual use. To avoid optical aberrations coverslips of appropriate thickness should be used. It is important to point out that having access only to one configuration does not necessarily limit the choice of the procedure. Indeed, devices called objective inverters (LSM Tech, www.lsmtech.com) have made possible converting inverted microscopes into upright and vice versa with minimal loss of signals (). These very helpful tools add tremendous flexibility to any imaging set up since they allow rotating the objectives and reach areas of the body that could not be easily accessed otherwise (). Moreover, these devices can be directly connected to non-descanned detectors shortening the light path, and increasing the sensitivity dramatically ().
Figure 5. The objective inverter. (A) Objective inverter from LSM tech to convert a microscope from an inverted into an upright configuration. (B) The inverter is equipped with a rotating hinge that allows tilting the objective in order to image inclined surfaces. (C) Objective inverter to convert a microscope from an upright into an inverted configuration. (D) Objective inverter with a multialkali detector installed on top of the lens to shorten the light path and increase the efficiency of light collection.

Surgical procedures and proper positioning of the organs may not result in a complete elimination of the motion artifacts. In this case, additional procedures can be adopted. For example, the animal can be intubated, connected to either a ventilator or a device to monitor the heartbeat, which can be both connected and synchronized to the scanning device.Citation69,Citation70 In this way, the imaging frames are acquired in between the heartbeat and/or each inspiration/expiration cycle.Citation69,Citation70 Alternatively, the frames can be acquired at the fastest speed (without compromising the image quality). Finally, some motion artifacts can be mitigated during or post-acquisition. For example, shifts in the xy plane or in the z direction can be compensated during the acquisition by manually moving the objective or changing the focal plane. In addition, xy shifts can be corrected by software such as ImageJ (Stack-reg plug-ins, W. Rasband, National Institutes of Health, Bethesda, MD), whereas more severe distortions within a frame can be corrected by a novel algorithm that has been recently developed.Citation71
Anesthetics and Temperature Control
Another important factor to take in consideration is the duration of the imaging session. For some applications, such as tracking subcellular structures, imaging for 30–60 min may be sufficient. In this case, the most commonly used anesthetic for mice and rats is a mixture of ketamine (100 – 200 mg/kg) and xylazine (10 – 40 mg/kg) administered by either intramuscular or intraperitoneal injection. This combination ensures anesthesia for up to one hour and allows a full recovery of the animal that may undergo additional imaging sessions. Re-administration upon appearance of the first signs of wakefulness can be applied to further extend the anesthesia. Specific dosage and re-administration protocols should be determined individually, as it can vary depending on the species, age and weight of the animal. Other choices for anesthetics include pentobarbital (30–60 mg/kg), inactin (100 mg/kg) and urethane (2 g/kg) that ensure longer anesthesia but are not suggested if recovery is required. For longer imaging sessions (6–12 h), inhalation of Isofluorane is recommended. This must be coupled with procedures for monitoring and maintaining normal physiological parameters, and in particular, breathing and heart rate, blood-oxygenation levels and vascular distension.Citation72 Moreover, animals should be supplemented with warm saline injections to prevent dehydration. Anesthesia induces hypothermia, which can adversely affect animal health, survival and reproducibility of experiments. Therefore, the body temperature should be monitored and heat should be provided to the animal, as needed. The rectal temperature probe is the most common way to continuously monitor the body temperature of an anesthetized animal. There are several ways to stabilize the heat of the animal and the tissue being imaged. The preferred way is to have a full microscope enclosure and to provide humidified circulating air at 37°C. Such environmental chambers would warm not only the animal but also the microscope stage and the objectives. These setups usually result in better image stability and easier management of animal health under anesthesia. It is very important to make sure that the objectives are at physiological temperature, as they tend to disperse a considerable amount of heat away from the tissue through the immersion media. This is a major issue for microscopes without enclosures and it is usually addressed by stand-alone objective heaters. In this case, the temperature should be monitored by a miniature probe at the interface between the coverslip and the tissue. In the absence of an environmental chamber the animal must be warmed via other means, such as chemical heat pads, circulating water blankets or electrical blankets.Citation19,Citation68
Imaging Cellular and Subcellular Structures in Live Animals
Until recently, the main applications of IVM were aimed at imaging biological processes at the level of the tissue and individual cells.Citation9,Citation10,Citation12,Citation73 The development of probes and approaches to specifically label sub cellular structures combined with the efforts to minimize motion artifacts have made possible imaging the dynamics of intracellular organelles in several organs.Citation10,Citation17,Citation18,Citation20-Citation22,Citation68 In this section, we want to provide the reader with a basic understanding of the strategies available to image subcellular organelles and the actin cytoskeleton in live rodents.
In order to label cellular and subcellular structures three major strategies can be followed: (1) injection or administration of fluorescently labeled probes that target specific cellular structures, (2) transient expression of fluorescently labeled molecules and (3) transgenic animals that express selected fluorescent molecules.
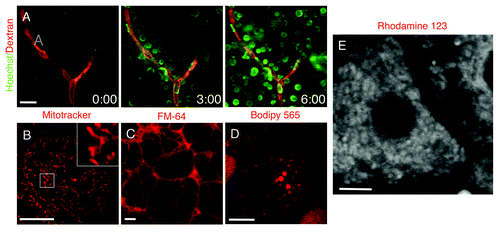
The repertoire of fluorescent molecules that can be used for IVM by systemic injections have increased substantially in the last few years. One of the most commonly used probes is the nuclear label Hoechst (or the equivalent DAPI) that very rapidly penetrate cells and label nuclei in vivo (; Vid. S3). This can be coupled to injections of high molecular weight dextrans to highlight the vasculature providing thus a good reference point in the imaged tissue (; Vid. S3). Systemic injections usually require relatively large amounts of probe and in some cases this approach can be expensive or limited by the available amount of the probe (seeCitation21,Citation67 for a more detailed list of probes and experimental conditions). Moreover, with this approach it may be hard to estimate the amount of the probe that reaches the target cells. As an alternative, some molecules can be applied directly onto the exposed organs by bathing them in the probe dissolved in saline. This route can be used for dyes such as mitotracker and lysotracker that label mitochondria and lysosomes respectively (), lipophilic FM dyes that are incorporated very rapidly into the plasma membranes () or Bodipy 493, Nile red and Bodipy 665 that are very good markers for lipid droplets (). Other probes may be used to study specific cellular functions, such as calcium sensors or dyes sensitive to membrane potential (; Vid. S4). In addition, small molecules can also be delivered to inhibit or stimulate cellular processes and provide information at a molecular level.Citation18,Citation19,Citation22,Citation67,Citation74 Although, small molecules are relatively easy to use and deliver, their major drawback is the fact that they cannot be used for prolonged periods of time due to their toxicity or because they are rapidly excreted from the body. The latter case can be remedied by designing a constant perfusion through the surface of the organ that is being imaged, thus exposing the cells to a constant concentration of the agent.
Figure 6. Fluorescent probes administered systemically or topically on the exposed organ. (A) The salivary glands of an anesthetized rat were exposed, and 70 kDa Texas Red-dextran was injected systemically. The glands were imaged in time lapse by two-photon microscopy (excitation 800 nm) using a 60x water immersion lens (NA 1.2, Olympus) and Hoechst was injected. The dye rapidly diffused from the vasculature (red) and labeled the nuclei of the adjacent cells (green) (Vid. S3). (B–D) The salivary glands (B and C) and the mammary glands (D) of anesthetized rats were exposed and bathed in mitotracker (B), FM-64 (C) and bodipy 665 (D) to label for mitochondria, plasma membrane and lipid droplets, respectively. The organs were exposed and imaged by confocal microscopy (excitation 561 nm) using a 60x water immersion lens (NA 1.2, Olympus). Bars, 10 µm. (E) The liver of an anesthetized rat was exposed, bathed with Rhodamine 123 and imaged by two-photon microscopy (excitation 750 nm) using a 60x water immersion lens (NA 1.2, Olympus) (Vid. S4). Bar, 5 µm.
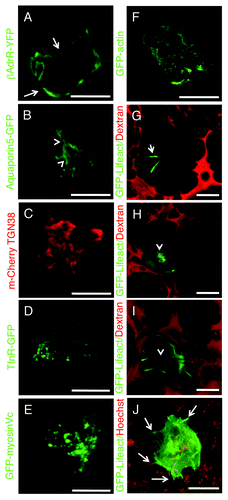
Alternatively, genetically encoded fluorescently labeled molecules can be expressed transiently in live animals. To this aim two main strategies have been used: viral and non-viral gene delivery. Viral-based delivery relies on adeno-, adeno associated- (AAV) or lenti-viral expression systems. Although they have a very high efficiency of transduction, they often induce an immune response and may affect the architecture of the intracellular organelles. For this reason, several groups have developed non-viral mediated approaches that rely on the use of “naked DNA.” Naked DNA has been pioneered in skeletal muscle and more recently has been used in rat salivary glands to target specific subpopulations of the salivary glands epithelium.Citation74,Citation75 However, the efficiency of naked DNA delivery may be quite low in some tissues/species of rodents and in this case researchers may need to resort to viral delivery. Several constructs are currently available to effectively label cellular compartments. For example the GFP-labeled histone 2B has been extensively used to study nuclear dynamics in tumor cellsCitation11,Citation13,Citation20,Citation34 and the last 21 aminoacids of K-Ras which contain a CAAX box (GFP-Farnesyl) is a very effective probe to label the plasma membrane that we have recently used to study exocytosis in the salivary glands of live rats.Citation18 Markers to specifically label domains of the plasma membranes in epithelial cells are also available, such as Aquaporin 5 () and the β-adrenergic receptors (), which are targeted to the apical and the basolateral poles respectively. Smaller intracellular organelles such as the Golgi apparatus (TGN-38, ),Citation75 endosomes (Transferrin receptor, TfnR, ) and constitutive secretory vesicles (Myosin Vc, )Citation76 can also be easily detected.
Figure 7. Transfections of naked DNA in live animals. (A–I) Rats were anesthetized, two fine polyethylene tubing inserted in the salivary duct and naked DNA injected as previously described.Citation18,Citation75 Specifically, DNAs encoding the following probes were injected: β-adrenergic receptor-GFP (A) and Aquaporin 5-GFP (B), markers for the basolateral (arrows) and apical (arrowheads) plasma membrane respectively; mCherry-TGN38 (C) a marker for the trans-Golgi network; GFP-transferrin receptor, a marker for early and recycling endosomes (D); GFP-myosin Vc, a marker for constitutive secretory vesicles (E); GFP-actin (F); GFP-lifeact (G and H). The glands were exposed, and imaged by two-photon microscopy (A–F, excitation 930 nm) or bathed with Texas Red-dextran to highlight the acinar structures.Citation18 The animals were injected with either saline (G and I) or isoproterenol (H) to stimulate exocytosis, and imaged as described above. GFP-lifeact is expressed in acinar cells (G and H) and localized at the apical plasma membrane (G, arrow). Upon stimulation with isoproterenol, GFP lifeact is recruited onto secretory granules (H, arrowheads). In (I), GFP lifeact is expressed in myoepithelial cells. (J) Human squamous carcinoma cells (Hela-O3) were engineered to express GFP-lifeact and injected in the tongue of immunocompromised mice. After 5 d the mouse was injected with Hoechst (red) and imaged by two-photon microscopy (excitation 930 nm) using a 25x water immersion lens (NA 1.05, Olympus). F-actin is enriched in the protrusions at the cell surface of the tumor cells (Vid. S5). Bars, 20 µm.
As for the actin cytoskeleton, so far GFP-actin has been the main tool utilized for both viral-mediatedCitation77 and non-viral expressionCitation78 (). Recently, the 17 aa of Abp140, a yeast protein not expressed in eukaryotes that specifically binds to F-actin, has been used as a novel probe to image the dynamics of the actin cytoskeleton in live cells (Lifeact).Citation79 When fluorescently-labeled lifeact is transiently transfected in the salivary glands of live rats, it targets either acinar () or myoepithelial cells (). In acinar cells, lifeact localizes at the apical plasma membrane (), and upon stimulation with isoproterenol translocates onto the secretory granules (). This tool has permitted us to perform for the first time a detailed kinetic analysis of the assembly on F-actin on membranes in a living multicellular organism.Citation18 Another powerful application for lifeact is to investigate the dynamics of the actin cytoskeleton during metastasis in immunocompromised mice. To this aim, we engineered highly metastatic tumor cell lines to stably express GFP-Lifeact and injected them in the tongue of immunocompromised mice. This has revealed a series of highly dynamic actin-rich structures that formed during the metastatic process (; Vid. S5). This probe clearly shows an enormous potential for in vivo imaging and no sign of interference with the function of the actin cytoskeleton or cellular toxicity.
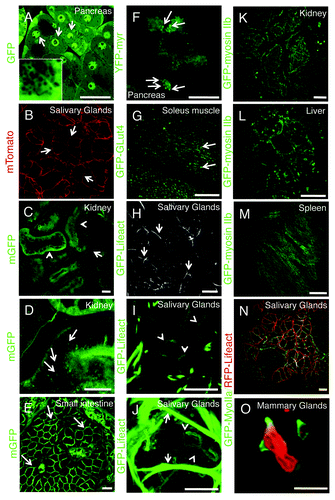
Although transient transfections in live animals are a very powerful tool, they suffer from the same limitations observed in cell culture. Indeed, both the yield of transfection and the levels of expression cannot be easily controlled. These issues can be overcome by the use of transgenic mice expressing selected fluorescent molecules under specific promoters that ensure endogenous levels of expression, specific tissue targeting and precise temporal regulation. Several transgenic lines have been made available at reasonable costs opening the door to a new series of investigations. One of these models is the FVB-GFP mouse, which ubiquitously expresses cytoplasmic GFP (). In these mice, single cells and nuclei are highlighted, and very interestingly large vesicular structures, such as secretory granules, exclude the GFP and hence can be easily detected (, inset) providing a powerful model to study regulated exocytosis in live animals.Citation56,Citation67 Another powerful mouse model ubiquitously expresses a peptide derived from the myristoylated alanine-rich C-kinase substrate (MARCKS) that possesses two palmitoylation sites and is fused with the tandem-tomato (mTomato mouse).Citation80 This probe is almost exclusively targeted to the plasma membrane and is a very useful tool to study processes at the cell surface such as endocytosis, exocytosis and membrane mobility in several organs such as salivary glands (),Citation56,Citation67 kidney (; Vid. S6), gut (; Vid. S7) and liver. Similar mice expressing fluorescently tagged molecules fused with a GPI anchor (not shown) or a myristoylated peptide () are also commercially available and can be very useful to study several aspect of membrane trafficking. Another transgenic mouse expresses in skeletal muscle the glucose transporter 4 (GLUT4) fused with GFP and can potentially provide a robust tool to study insulin-dependent exocytosis of GLUT4-containing vesicles (; Vid. S8).
Figure 8. Transgenic mice models currently used for intravital microscopy. (A–O) Selected transgenic mice were anesthetized, the organs indicated in the panels exposed, and imaged by confocal microscopy (excitation 488 nm, A and C–M; excitation 561, B; excitation 488 and 561, N and O) at a depth of approximately 30 µm by using a 60x oil immersion lens (NA 1.4, Olympus). (A) GFP-FVB mouse expressing soluble GFP. Nuclei (arrows) and secretory granules (inset) are highlighted in the pancreas. (B–E) Mice expressing a membrane targeted peptide fused with tandem Tomato (B) or GFP (C–E) (Vids. S6 and S7). In the salivary glands the mTomato probe is localized at the plasma membrane (B, arrows), in the kidney the mGFP probe highlights the microvasculature (C, arrows), both proximal and distal tubuli (B, arrowheads), and a series of small endocytic vesicles (D, arrows; Vid. S6); in the small intestine, mGFP is localized at the plasma membrane of the enterocytes (F, arrows; Vid. S7). (F) Mouse expressing GFP fused to a myristoylated peptide (GFP-myr). In exocrine pancreas, GFP-myr is localized onto the secretory granules (arrows). (G) Mouse expressing GFP-GLUT4 in the skeletal muscle. GFP-Glut4 is localized in small vesicles (G, arrows; Vid. S8). (H–J) In the salivary glands, GFP-lifeact is expressed in both myoepithelial (H and J, arrows) and at the apical pole of acinar cells (H and J, arrowheads; Vid. S9). (K–M) GFP-myosin IIb is expressed in the kidney in several intracellular structures (K, arrows), in liver hepatocytes, both at the plasma membrane and small vesicles (H, arrows), and in the spleen in long filaments (M). (N and O) Mice expressing GFP-myosin IIb and RFP-lifeact. In salivary glands both GFP and myosin IIb are localized at the plasma membrane (N), whereas in neutrophils in mammary glands, GFP-myosin IIb is localized in protrusive edges (O; Vid. S10). Bars, 20 µm.
Recently, transgenic mice expressing fluorescently tagged lifeact mice have been generated by the Wedlich-Soldner’s group.Citation81 These mice represent an exceptionally powerful tool, as shown by the fact that in the salivary glands lifeact is properly targeted to the acinar and the myoepithelial cells (; Vid. S9). Although this tool has not been fully exploited for IVM, it is going to provide novel and compelling information on several aspects of epithelial cell biology, membrane trafficking, cell migration and more.
Two complementary models are the knock-in mice expressing GFP-myosin IIa and IIb generated by Robert Adeslstein’s group.Citation82 These mice enable the dynamic imaging of these actin based-motors in several organs, and to elucidate the role of myosin during membrane trafficking (). Notably, we have recently crossed these mice with those expressing RFP-lifeact creating even more powerful tools to study at the same time actin and myosin dynamics in vivo (), as shown by preliminary experiments where the dynamics of the actin cytoskeleton was monitored in neutrophils during migration in mammary glands (; Vid. S10).
Finally, we want to emphasize that this list of mice is not exhaustive and new mice models are constantly being developed. They will provide very powerful tools to address biological questions and to further develop the field of IVM.
Conclusions
Growing evidence has highlighted that many biological processes are regulated by the coordination of multiple cellular events and are strongly influenced by the architecture of the experimental model. Reductionist approaches based on the use of simplified models offer the advantages of a better control of the experimental conditions and the opportunity to study processes at a molecular level. However, certain aspects of the physiology of an organ, that may have unanticipated effects on cellular functions cannot be recapitulated in in vitro systems and require the use of in vivo models. In this respect, IVM has become a formidable tool to provide valuable information on the dynamics of biological processes in vivo. Whereas fields such as neurobiology, immunology and tumor biology have adopted IVM as a well-established tool, other areas such as cell biology are still not up to speed. However, three main factors are attracting the attention of investigators to this new opportunities: (1) the pioneering work of a few groups that have developed the technologies to perform high resolution imaging in vivo, (2) the availability of more sophisticated microscopes specifically designed to perform IVM, and (3) the development of novel genetic tools, such as mice expressing various fluorescently tagged markers, libraries of knockout and conditional knockout animals and relatively novel procedures to perform protein ablation in vivo via siRNA or shRNA.
IVM has a huge potential to uncover cellular mechanisms that regulate physiological and pathological events and we envision that this approach is going to be one of the main tools to study cell biology in the near future.
Additional material
Download Zip (53.6 MB)Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research.
Reference
- Lippincott-Schwartz J. Bridging structure and process in developmental biology through new imaging technologies. Dev Cell 2011; 21:5 - 10; http://dx.doi.org/10.1016/j.devcel.2011.06.030; PMID: 21763598
- Lippincott-Schwartz J. Emerging in vivo analyses of cell function using fluorescence imaging (*). Annu Rev Biochem 2011; 80:327 - 32; http://dx.doi.org/10.1146/annurev-biochem-121010-125553; PMID: 21513458
- Hove JR, Craig MP. High-speed confocal imaging of zebrafish heart development. Methods Mol Biol 2012; 843:309 - 28; http://dx.doi.org/10.1007/978-1-61779-523-7_26; PMID: 22222541
- Rauzi M, Lenne PF, Lecuit T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature 2010; 468:1110 - 4; http://dx.doi.org/10.1038/nature09566; PMID: 21068726
- Rohde CB, Yanik MF. Subcellular in vivo time-lapse imaging and optical manipulation of Caenorhabditis elegans in standard multiwell plates. Nat Commun 2011; 2:271; http://dx.doi.org/10.1038/ncomms1266; PMID: 21468023
- Tomer R, Khairy K, Amat F, Keller PJ. Quantitative high-speed imaging of entire developing embryos with simultaneous multiview light-sheet microscopy. Nat Methods 2012; 9:755 - 63; http://dx.doi.org/10.1038/nmeth.2062; PMID: 22660741
- Tserevelakis GJ, Filippidis G, Megalou EV, Fotakis C, Tavernarakis N. Cell tracking in live Caenorhabditis elegans embryos via third harmonic generation imaging microscopy measurements. J Biomed Opt 2011; 16:046019; http://dx.doi.org/10.1117/1.3569615; PMID: 21529088
- Yaniv K, Isogai S, Castranova D, Dye L, Hitomi J, Weinstein BM. Live imaging of lymphatic development in the zebrafish. Nat Med 2006; 12:711 - 6; http://dx.doi.org/10.1038/nm1427; PMID: 16732279
- Pittet MJ, Weissleder R. Intravital imaging. Cell 2011; 147:983 - 91; http://dx.doi.org/10.1016/j.cell.2011.11.004; PMID: 22118457
- Weigert R, Sramkova M, Parente L, Amornphimoltham P, Masedunskas A. Intravital microscopy: a novel tool to study cell biology in living animals. Histochem Cell Biol 2010; 133:481 - 91; http://dx.doi.org/10.1007/s00418-010-0692-z; PMID: 20372919
- Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol 2008; 130:1147 - 54; http://dx.doi.org/10.1007/s00418-008-0529-1; PMID: 18987875
- Amornphimoltham P, Masedunskas A, Weigert R. Intravital microscopy as a tool to study drug delivery in preclinical studies. Adv Drug Deliv Rev 2011; 63:119 - 28; http://dx.doi.org/10.1016/j.addr.2010.09.009; PMID: 20933026
- Andresen V, Alexander S, Heupel WM, Hirschberg M, Hoffman RM, Friedl P. Infrared multiphoton microscopy: subcellular-resolved deep tissue imaging. Curr Opin Biotechnol 2009; 20:54 - 62; http://dx.doi.org/10.1016/j.copbio.2009.02.008; PMID: 19324541
- Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods 2008; 5:1019 - 21; http://dx.doi.org/10.1038/nmeth.1269; PMID: 18997781
- Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 2002; 296:1873 - 6; http://dx.doi.org/10.1126/science.1071065; PMID: 12052961
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 2006; 50:823 - 39; http://dx.doi.org/10.1016/j.neuron.2006.05.019; PMID: 16772166
- Cao L, Kobayakawa S, Yoshiki A, Abe K. High resolution intravital imaging of subcellular structures of mouse abdominal organs using a microstage device. PLoS One 2012; 7:e33876; http://dx.doi.org/10.1371/journal.pone.0033876; PMID: 22479464
- Masedunskas A, Sramkova M, Parente L, Sales KU, Amornphimoltham P, Bugge TH, et al. Role for the actomyosin complex in regulated exocytosis revealed by intravital microscopy. Proc Natl Acad Sci U S A 2011; 108:13552 - 7; http://dx.doi.org/10.1073/pnas.1016778108; PMID: 21808006
- Masedunskas A, Weigert R. Intravital two-photon microscopy for studying the uptake and trafficking of fluorescently conjugated molecules in live rodents. Traffic 2008; 9:1801 - 10; http://dx.doi.org/10.1111/j.1600-0854.2008.00798.x; PMID: 18647170
- Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res 2011; 71:4608 - 16; http://dx.doi.org/10.1158/0008-5472.CAN-11-0412; PMID: 21712408
- Dunn KW, Sandoval RM, Kelly KJ, Dagher PC, Tanner GA, Atkinson SJ, et al. Functional studies of the kidney of living animals using multicolor two-photon microscopy. Am J Physiol Cell Physiol 2002; 283:C905 - 16; PMID: 12176747
- Sandoval RM, Kennedy MD, Low PS, Molitoris BA. Uptake and trafficking of fluorescent conjugates of folic acid in intact kidney determined using intravital two-photon microscopy. Am J Physiol Cell Physiol 2004; 287:C517 - 26; http://dx.doi.org/10.1152/ajpcell.00006.2004; PMID: 15102609
- Bagher P, Segal SS. The mouse cremaster muscle preparation for intravital imaging of the microcirculation. J Vis Exp 2011; 52:2874; http://dx.doi.org/10.3791/2874; PMID: 21694690
- Guan Y, Worrell RT, Pritts TA, Montrose MH. Intestinal ischemia-reperfusion injury: reversible and irreversible damage imaged in vivo. Am J Physiol Gastrointest Liver Physiol 2009; 297:G187 - 96; http://dx.doi.org/10.1152/ajpgi.90595.2008; PMID: 19407214
- Jenne CN, Wong CH, Petri B, Kubes P. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PLoS One 2011; 6:e25109; http://dx.doi.org/10.1371/journal.pone.0025109; PMID: 21949865
- Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol 2003; 21:1356 - 60; http://dx.doi.org/10.1038/nbt894; PMID: 14595363
- Vakoc BJ, Fukumura D, Jain RK, Bouma BE. Cancer imaging by optical coherence tomography: preclinical progress and clinical potential. Nat Rev Cancer 2012; 12:363 - 8; http://dx.doi.org/10.1038/nrc3235; PMID: 22475930
- Vakoc BJ, Tearney GJ, Bouma BE. Statistical properties of phase-decorrelation in phase-resolved Doppler optical coherence tomography. IEEE Trans Med Imaging 2009; 28:814 - 21; http://dx.doi.org/10.1109/TMI.2009.2012891; PMID: 19164078
- Fu Y, Huff TB, Wang HW, Wang H, Cheng JX. Ex vivo and in vivo imaging of myelin fibers in mouse brain by coherent anti-Stokes Raman scattering microscopy. Opt Express 2008; 16:19396 - 409; http://dx.doi.org/10.1364/OE.16.019396; PMID: 19030027
- Mahou P, Olivier N, Labroille G, Duloquin L, Sintes JM, Peyriéras N, et al. Combined third-harmonic generation and four-wave mixing microscopy of tissues and embryos. Biomed Opt Express 2011; 2:2837 - 49; http://dx.doi.org/10.1364/BOE.2.002837; PMID: 22025988
- Provenzano PP, Eliceiri KW, Keely PJ. Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis 2009; 26:357 - 70; http://dx.doi.org/10.1007/s10585-008-9204-0; PMID: 18766302
- Roberts MS, Roberts MJ, Robertson TA, Sanchez W, Thörling C, Zou Y, et al. In vitro and in vivo imaging of xenobiotic transport in human skin and in the rat liver. J Biophotonics 2008; 1:478 - 93; http://dx.doi.org/10.1002/jbio.200810058; PMID: 19343674
- Wang HW, Fu Y, Huff TB, Le TT, Wang H, Cheng JX. Chasing lipids in health and diseases by coherent anti-Stokes Raman scattering microscopy. Vib Spectrosc 2009; 50:160 - 7; http://dx.doi.org/10.1016/j.vibspec.2008.11.007; PMID: 19763281
- Weigelin B, Bakker G, Friedl P. Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Intravital 2012; 1:1 - 12; http://dx.doi.org/10.4161/intv.21223
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science 1990; 248:73 - 6; http://dx.doi.org/10.1126/science.2321027; PMID: 2321027
- Mertz J. Nonlinear microscopy: new techniques and applications. Curr Opin Neurobiol 2004; 14:610 - 6; http://dx.doi.org/10.1016/j.conb.2004.08.013; PMID: 15464895
- Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 2003; 21:1369 - 77; http://dx.doi.org/10.1038/nbt899; PMID: 14595365
- Young PA, Clendenon SG, Byars JM, Decca RS, Dunn KW. The effects of spherical aberration on multiphoton fluorescence excitation microscopy. J Microsc 2011; 242:157 - 65; http://dx.doi.org/10.1111/j.1365-2818.2010.03449.x; PMID: 21118240
- Young PA, Clendenon SG, Byars JM, Dunn KW. The effects of refractive index heterogeneity within kidney tissue on multiphoton fluorescence excitation microscopy. J Microsc 2011; 242:148 - 56; http://dx.doi.org/10.1111/j.1365-2818.2010.03448.x; PMID: 21118239
- Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci 2011; 14:1481 - 8; http://dx.doi.org/10.1038/nn.2928; PMID: 21878933
- Barretto RP, Ko TH, Jung JC, Wang TJ, Capps G, Waters AC, et al. Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat Med 2011; 17:223 - 8; http://dx.doi.org/10.1038/nm.2292; PMID: 21240263
- Barretto RP, Messerschmidt B, Schnitzer MJ. In vivo fluorescence imaging with high-resolution microlenses. Nat Methods 2009; 6:511 - 2; http://dx.doi.org/10.1038/nmeth.1339; PMID: 19525959
- Chia TH, Levene MJ. In vivo imaging of deep cortical layers using a microprism. J Vis Exp 2009; http://dx.doi.org/10.3791/1509; PMID: 19713883
- Chia TH, Levene MJ. Microprisms for in vivo multilayer cortical imaging. J Neurophysiol 2009; 102:1310 - 4; http://dx.doi.org/10.1152/jn.91208.2008; PMID: 19494189
- Llewellyn ME, Barretto RP, Delp SL, Schnitzer MJ. Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans. Nature 2008; 454:784 - 8; PMID: 18600262
- Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW. In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol 2004; 91:1908 - 12; http://dx.doi.org/10.1152/jn.01007.2003; PMID: 14668300
- Li X, Yu W. Deep Tissue Microscopic Imaging of the Kidney with a Gradient-Index Lens System. Opt Commun 2008; 281:1833 - 40; http://dx.doi.org/10.1016/j.optcom.2007.08.074; PMID: 19572038
- Kobat D, Durst ME, Nishimura N, Wong AW, Schaffer CB, Xu C. Deep tissue multiphoton microscopy using longer wavelength excitation. Opt Express 2009; 17:13354 - 64; http://dx.doi.org/10.1364/OE.17.013354; PMID: 19654740
- Kobat D, Horton NG, Xu C. In vivo two-photon microscopy to 1.6-mm depth in mouse cortex. J Biomed Opt 2011; 16:106014; http://dx.doi.org/10.1117/1.3646209; PMID: 22029361
- Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett 2003; 28:1022 - 4; http://dx.doi.org/10.1364/OL.28.001022; PMID: 12836766
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods 2005; 2:932 - 40; http://dx.doi.org/10.1038/nmeth818; PMID: 16299478
- Bestvater F, Spiess E, Stobrawa G, Hacker M, Feurer T, Porwol T, et al. Two-photon fluorescence absorption and emission spectra of dyes relevant for cell imaging. J Microsc 2002; 208:108 - 15; http://dx.doi.org/10.1046/j.1365-2818.2002.01074.x; PMID: 12423261
- Spiess E, Bestvater F, Heckel-Pompey A, Toth K, Hacker M, Stobrawa G, et al. Two-photon excitation and emission spectra of the green fluorescent protein variants ECFP, EGFP and EYFP. J Microsc 2005; 217:200 - 4; http://dx.doi.org/10.1111/j.1365-2818.2005.01437.x; PMID: 15725123
- Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci U S A 2003; 100:7075 - 80; http://dx.doi.org/10.1073/pnas.0832308100; PMID: 12756303
- Masedunskas A, Porat-Shliom N, Weigert R. Linking differences in membrane tension with the requirement for a contractile actomyosin scaffold during exocytosis in salivary glands. Commun Integr Biol 2012; 5:84 - 7; http://dx.doi.org/10.4161/cib.18258; PMID: 22482019
- Masedunskas A, Porat-Shliom N, Weigert R. Regulated exocytosis: novel insights from intravital microscopy. Traffic 2012; 13:627 - 34; http://dx.doi.org/10.1111/j.1600-0854.2012.01328.x; PMID: 22243493
- Masedunskas A, Sramkova M, Weigert R. Homeostasis of the apical plasma membrane during regulated exocytosis in the salivary glands of live rodents. Bioarchitecture 2011; 1:225 - 9; http://dx.doi.org/10.4161/bioa.18405; PMID: 22754613
- Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods 2003; 29:58 - 73; http://dx.doi.org/10.1016/S1046-2023(02)00283-9; PMID: 12543072
- Testa I, Garrè M, Parazzoli D, Barozzi S, Ponzanelli I, Mazza D, et al. Photoactivation of pa-GFP in 3D: optical tools for spatial confinement. Eur Biophys J 2008; 37:1219 - 27; http://dx.doi.org/10.1007/s00249-008-0317-9; PMID: 18379772
- Waharte F, Brown CM, Coscoy S, Coudrier E, Amblard F. A two-photon FRAP analysis of the cytoskeleton dynamics in the microvilli of intestinal cells. Biophys J 2005; 88:1467 - 78; http://dx.doi.org/10.1529/biophysj.104.049619; PMID: 15596489
- Kirkpatrick ND, Chung E, Cook DC, Han X, Gruionu G, Liao S, et al. Video-rate resonant scanning multiphoton microscopy: An emerging technique for intravital imaging of the tumor microenvironment. Intravital 2012; In Press
- Boyd L, Hajjar C, O’Connell K. Time-lapse microscopy of early embryogenesis in Caenorhabditis elegans. J Vis Exp 2011; http://dx.doi.org/10.3791/2852; PMID: 21897352
- Thiberge S, Blazquez S, Baldacci P, Renaud O, Shorte S, Ménard R, et al. In vivo imaging of malaria parasites in the murine liver. Nat Protoc 2007; 2:1811 - 8; http://dx.doi.org/10.1038/nprot.2007.257; PMID: 17641649
- Swoger J, Muzzopappa M, López-Schier H, Sharpe J. 4D retrospective lineage tracing using SPIM for zebrafish organogenesis studies. J Biophotonics 2011; 4:122 - 34; http://dx.doi.org/10.1002/jbio.201000087; PMID: 20925108
- Dunn KW, Sutton TA, Sandoval RM. Live-animal imaging of renal function by multiphoton microscopy. Curr Protoc Cytom 2007; Chapter 12:Unit12 9.
- Klinger A, Orzekowsky-Schroeder R, von Smolinski D, Blessenohl M, Schueth A, Koop N, et al. Complex morphology and functional dynamics of vital murine intestinal mucosa revealed by autofluorescence 2-photon microscopy. Histochem Cell Biol 2012; 137:269 - 78; http://dx.doi.org/10.1007/s00418-011-0905-0; PMID: 22227801
- Masedunskas A, Sramkova M, Parente L, Weigert R. Intravital Microscopy to Image Membrane Trafficking in Live Rats. Cell Imaging techniques: Springer, 2012.
- Masedunskas A, Weigert R.. Internalization of fluorescent dextrans in the submandibular salivary glands of live animals: a study combining intravital two-photon microscopy and second harmonic generation. SPIE 2008; 6860; http://dx.doi.org/10.1117/12.768051
- Li W, Nava RG, Bribriesco AC, Zinselmeyer BH, Spahn JH, Gelman AE, et al. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest 2012; 122:2499 - 508; http://dx.doi.org/10.1172/JCI62970; PMID: 22706307
- Presson RG Jr., Brown MB, Fisher AJ, Sandoval RM, Dunn KW, Lorenz KS, et al. Two-photon imaging within the murine thorax without respiratory and cardiac motion artifact. Am J Pathol 2011; 179:75 - 82; http://dx.doi.org/10.1016/j.ajpath.2011.03.048; PMID: 21703395
- Lorenz KS, Salama P, Dunn KW, Delp EJ. Digital correction of motion artefacts in microscopy image sequences collected from living animals using rigid and nonrigid registration. J Microsc 2012; 245:148 - 60; http://dx.doi.org/10.1111/j.1365-2818.2011.03557.x; PMID: 22092443
- Ewald AJ, Werb Z, Egeblad M. Monitoring of vital signs for long-term survival of mice under anesthesia. Cold Spring Harb Protoc 2011; 2011: pdb.prot5563; http://dx.doi.org/10.1101/pdb.prot5563; PMID: 21285263
- Ritsma L, Ponsioen B, van Rheenen J. Intravital imaging of cell signaling in mice. Intravital 2012; 1:1 - 9; http://dx.doi.org/10.4161/intv.20802
- Sramkova M, Masedunskas A, Weigert R. Plasmid DNA is internalized from the apical plasma membrane of the salivary gland epithelium in live animals. Histochem Cell Biol 2012; 138:201 - 13; http://dx.doi.org/10.1007/s00418-012-0959-7; PMID: 22544351
- Sramkova M, Masedunskas A, Parente L, Molinolo A, Weigert R. Expression of plasmid DNA in the salivary gland epithelium: novel approaches to study dynamic cellular processes in live animals. Am J Physiol Cell Physiol 2009; 297:C1347 - 57; http://dx.doi.org/10.1152/ajpcell.00262.2009; PMID: 19794147
- Jacobs DT, Weigert R, Grode KD, Donaldson JG, Cheney RE. Myosin Vc is a molecular motor that functions in secretory granule trafficking. Mol Biol Cell 2009; 20:4471 - 88; http://dx.doi.org/10.1091/mbc.E08-08-0865; PMID: 19741097
- Tanner GA, Sandoval RM, Molitoris BA, Bamburg JR, Ashworth SL. Micropuncture gene delivery and intravital two-photon visualization of protein expression in rat kidney. Am J Physiol Renal Physiol 2005; 289:F638 - 43; http://dx.doi.org/10.1152/ajprenal.00059.2005; PMID: 15886277
- Sramkova M, Porat-Shliom N, Masedunskas A, Wigand T, Amornphimoltham P, Weigert R. Salivary Glands: A Powerful Experimental System to Study Cell Biology in Live Animals by Intravital Microscopy. In: Najman S, ed. Current Frontiers and Perspectives in Cell Biology: Intech, 2012.
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods 2008; 5:605 - 7; http://dx.doi.org/10.1038/nmeth.1220; PMID: 18536722
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007; 45:593 - 605; http://dx.doi.org/10.1002/dvg.20335; PMID: 17868096
- Riedl J, Flynn KC, Raducanu A, Gärtner F, Beck G, Bösl M, et al. Lifeact mice for studying F-actin dynamics. Nat Methods 2010; 7:168 - 9; http://dx.doi.org/10.1038/nmeth0310-168; PMID: 20195247
- Bao J, Ma X, Liu C, Adelstein RS. Replacement of nonmuscle myosin II-B with II-A rescues brain but not cardiac defects in mice. J Biol Chem 2007; 282:22102 - 11; http://dx.doi.org/10.1074/jbc.M702731200; PMID: 17519229
- Wu XS, Masedunskas A, Weigert R, Copeland NG, Jenkins NA, Hammer JA. Melanoregulin regulates a shedding mechanism that drives melanosome transfer from melanocytes to keratinocytes. Proc Natl Acad Sci U S A 2012; 109:E2101 - 9; http://dx.doi.org/10.1073/pnas.1209397109; PMID: 22753477