Abstract
Epithelial cell adhesion molecule EpCAM is expressed on a subset of normal epithelia and overexpressed on malignant cells from a variety of different tumor entities. This overexpression is even more pronounced on so-called tumor-initiating cells (TICs) of many carcinomas. Taking this rather ubiquitous expression of EpCAM in carcinomas and TICs into account, the question arises how EpCAM can serve as a reliable marker for tumor-initiating cells and what might be the advantage for TICs to express this molecule. Furthermore, several approaches for therapeutic strategies targeting exclusively EpCAM on cancer cells were undertaken over the past decades and have recently been transferred to pre-clinical attempts to eradicate TICs. In the present review, we will depict potential functions of EpCAM in tumor cells with a special focus on TICs and therapeutic implications.
Keywords: :
Introduction
Two basic models attempt to explain the initiation and development of tumors in vivo. According to the stochastic or clonal evolution model an accumulation of mutations in somatic cells leads to an uncontrolled proliferation and finally to transformation into cancer cells.Citation1 The second model assumes the existence of a hierachical arrangement of tumor cells, which implies that only a subpopulation of cells is able to induce tumor formation.Citation2,Citation3 These cells are characterized by two main features, which position them at the apex of the cancer cascade: self-renewal and multipotency. Several pieces of evidence, which primarily originated from the field of leukemia, support the idea that various types of stem cells might themselves be the targets of transformation.Citation4,Citation5 First of all, there is no requirement for an ectopic activation of self-renewal and multipotency programs in these cells and, second, the longer life span increases the probability of accumulating mutations that would uncouple these cells from controlled homeostasis.Citation6 In addition, pathways, which are known to be important in cancer progression, like the Wnt, the Hedgehog and the Notch signaling pathway are also involved in maintaining stemness and self-renewal.Citation7-Citation11
It must be noted that all assumptions and theories can at best be challenged and eventually verified in animal models. Accordingly, xenograft transplantations remain at present the gold standard for the identification and characterization of TICs in vivo. Purified human tumor cells are transferred into immunocompromised mice in varying concentrations and tumor formation is observed over time. Obviously, with the usage of such animal models initial steps of transformation and major changes in cellular homeostasis occurring in vivo and leading to the development of malignancies in humans are still inaccessible to our understanding. Nonetheless, these xenogeneic transplantation models reveal the formation of heterogenous tumors reflecting the tumorigenic and differentiation capacity of these cells. Cells with the ability to induce new tumors are known as TICs, tumor-initiating cells, or cancer stem cells (CSCs). Interestingly, TICs show a stem-like phenotype including traits of self-renewal and multipotency. It is however important to keep in mind that the observed heterogeneity can be also explained by the stochastic or clonal evolution model,Citation2 for example owing to differences in the microenvironment that determine the diversification of various cells of the whole tumor.Citation12,Citation13
Isolation and identification of TICs are commonly achieved with the use of markers, which revealed more or less selective. Even if there is no universal marker for TICs of all types of cancer, some molecules are most frequently shared across entities.Citation14 These proteins are: CD20, CD24, CD34, CD44, CD90, CD117, CD133, CD166, ALDH, nestin and finally EpCAM.Citation14,Citation15 Noteworthy, some of these proteins, such as EpCAM or CD44, are expressed in an almost ubiquitous manner on most carcinoma cells and even on selected normal adult tissues, and differ primarily in their expression amplitude in TICs. Thus, combinations of several markers are more reliable to characterize cells as TICs. For instance, high expression of EpCAM combined with CD44 positive or CD44+/CD24- served as an excellent criterium with regard to the identification of TICs from breast,Citation9 colorectalCitation10 or pancreatic carcinomas.Citation11
Besides its frequent overexpression on TICs of various entities,Citation16 EpCAM was identified in 2008 as a marker for human embryonic stem cells in an antibody based screening assay.Citation17 Several publications of the last years confirmed EpCAM as a suitable marker for pluripotency and proliferation in murine and human embryonic stem cells.Citation18,Citation19
In this review, we will focus on the functional aspects of EpCAM in stem cells and TICs, and on possible therapeutic approaches using this molecule as a target (see also Box 1).
General Facts on EpCAM
EpCAM was first described in 1979 as an antigen, which induced the production of specific antibodies after immunization of mice with human colorectal carcinoma cells and subsequent fusion of splenocytes with myeloma cells to create hybridomas.Citation20 Biochemical approaches to define the actual antigen recognized by the Co-17-1A antibody were conducted several years later and identified EpCAM as a glycosylated protein with an apparent molecular weight of 33–40 kDa.Citation21,Citation22 Early on, anti-EpCAM antibodies were assessed for the capacity to serve as diagnostic and therapeutic toolsCitation23-Citation27 including early phase I clinical trials.Citation28,Citation29 The mid 1990s brought about large clinical phase studies utilizing the monoclonal anti-EpCAM antibody EdrecolomabCitation30,Citation31 and which resulted in FDA approval and market introduction of Panorex for the treatment of metastasized colon cancer. In parallel to this development the group of S. Litvinov concentrated on molecular and functional aspects of EpCAM to reveal his role as a homophilic cell adhesion molecule and a correlation to proliferation.Citation32-Citation34 In 1999 the same group compiled the first comprehensive review on EpCAM expression pattern and function.Citation35 It must be noted that Panorex was retrieved from the German market in 2000 and the actual benefit of this particular anti-EpCAM antibody was eventually refuted in a large cohort of patients suffering from stage III colon cancer.Citation36 Besides intense discussions and arguments on reasons for such a clinical failure even after FDA approval,Citation37,Citation38 it appears advisable to heed the following important issues when considering EpCAM as a target for therapy:
• Expression levels of EpCAM matter with respect to potential benefit of EpCAM-specific antibody therapy.Citation39
• Effects of anti-EpCAM antibody on EpCAM signaling must be paid attention to.Citation40
• Inhibition of EpCAM signaling with specific inhibitors of regulated intramembrane proteolysis is a novel option to be considered.
In the great majority of cancer entities, EpCAM overexpression strongly correlates with worse overall survival and bad prognosis,Citation41,Citation42 and distinguishes patients at high risk for recurrence.Citation43 However, in some entities such as pancreatic and gastrointestinal cancers EpCAM overexpression rather correlates with better prognosis.Citation44 The molecular basis for this discrepancy is as yet unknown.
EpCAM and Metastases
Early work by Jojovic et al. reported on a substantial decrease of EpCAM expression in micrometastases as compared with primary carcinomas and established metastases in the mouse model.Citation45 Already at this time, it was postulated that EpCAM was subject to “regulatory processes in epithelial-mesenchymal transitions during metastases.” This is in line with a function of EpCAM as a cell adhesion molecule and as a promoter of cell proliferation. Initial steps of metastatic spread, which are associated with migration and a decreased proliferative rate during circulation and spreading to distant organs, would rather be expected to be associated with a loss of EpCAM expression. However, induction of epithelial-mesenchymal transition (EMT) upon the expression of regulatory molecules such as FoxM1 correlated with increase of vimentin and EpCAM expression.Citation46 Thus, at present the actual role of EpCAM in the process of metastatic spread is not properly understood. We can speculate on a potential requirement for the loss of EpCAM during initial phases of metastatic spread, which could facilitate detachment of tumor cells from the bulk and favor a dormant state of circulating cells. Upon invasion and formation of micrometastases, a second phase of EpCAM expression might be necessary to foster proliferation and formation of multicellular, large metastases.
Structure of EpCAM
Epithelial cell adhesion molecule EpCAM (murine CD326) is a type I transmembrane glycoprotein consisting of a large extracellular (EpEX), a single transmembrane and a short intracellular (EpICD) domain. Three independent glycosylation sites in the EpEX part dictate the stability of the whole protein at the cell surface. N-glycosylation of the most membrane-proximal asparagine198 in tumor cells resulted in a 3-fold increase of the half-life of EpCAM at the membrane and might thus impact on the functionality of EpCAM.Citation47 This is of special interest as EpCAM was shown to be hyperglycosylated in the great majority of head and neck carcinomas as compared with autologous normal mucosa.Citation48 EpCAM was shown to mediate cell-cell adhesion via intra- and intercellular homophilic interactions, which are provided by an EGF-like domain and a thyroglobulin domain in the extracellular part.Citation49 Additionally, EpCAM is connected to the cytoskeleton upon interactions of EpICD with α-actinin, and thus EpICD is required for proper intercellular adhesions.Citation50 Interestingly, the short intracellular domain comprising only 26 amino acids is crucial for EpCAM-dependent signal transduction from the plasma membrane to the nucleus,Citation51 as will be described in more detail in the next section.
EpCAM Signaling
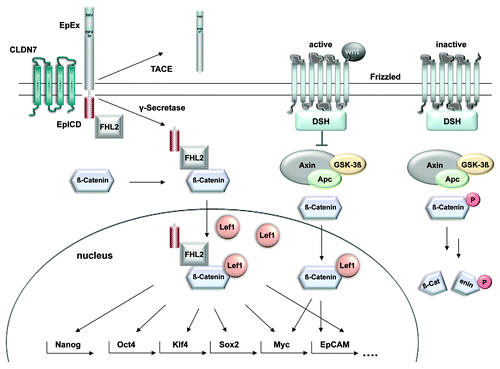
Some TIC markers such as EpCAM, CD44, and CD133 are shared in a number of entities and represent the most frequently used markers for the enrichment of tumor-initiating cells from primary human cancer samples.Citation14 Reasons for the high frequency and high-level expression of CD44 in TICs have been reviewed recently by ZöllerCitation12 and clearly relate to long-known cell communication and signaling properties of CD44. Knowledge on the functions of CD133 is rather scarce and so are the assumptions on a particular functional role of CD133 in the generation and/or maintenance of the TIC phenotype.Citation52 When concentrating on EpCAM as a marker of TICs, implications of the molecule in signaling events were for long time no matter of discussion, since EpCAM was viewed as cell adhesion molecule only. However, as for the case of many cell adhesion molecules, EpCAM has dual properties in that it can mediate cell-to-cell contact but also transmit signals from the plasma membrane into the nucleus in order to regulate gene transcription (see left part of for schematic view of EpCAM signaling).Citation53 In addition, EpCAM is not solely expressed in epithelial cells but likewise strongly expressed in various tissue stem cells, precursors, and in embryonic stem (ES) cells of murine and human origin.Citation53,Citation54 In ES cells, EpCAM is mandatory for the maintenance of self-renewal and the pluripotent phenotype.Citation18,Citation19 Its mode of signaling proceeds via regulated intramembrane proteolysis (see left part of ). After being cleaved off by the tumor necrosis factor α converting enzyme (TACE, ADAM17), the extracellular part of the protein, termed EpEX, can act as a shed ligand to induce further cleavage of intact EpCAM molecules.Citation51 The second cleavage is mediated by γ-secretase complexes, which contain presenilin 2 and results in the release of the intracellular domain EpICD in the cytosol of the cell (). Soluble EpICD constitutes the signaling active intracellular compound, which is found in a large nuclear complex together with FHL2, β-catenin and Lef-1. FHL2 is composed of four-and-a-half LIM domains, which represent protein-protein interaction moieties.Citation55 Known interaction partners of FHL2 are among many others TACE,Citation56 presenilin 2Citation57 and β-catenin.Citation58 Hence, FHL2 might act as a scaffold protein, which could be essential for the initial formation of the EpCAM signalosome. Although additional solid evidence is required, first hints that indeed FHL2 is of paramount importance for EpCAM signaling stem from siRNA-mediated knock down experiments. Knock-down of FHL2 inhibited EpCAM-mediated proliferation of HEK293-EpCAM cellsCitation51 and carcinoma cells (O.G., unpublished data). Importantly, FHL2 links EpCAM to the Wnt pathway via interactions with the major components of this central pathway, i.e., β-catenin and Lef-1 (), as will be discussed in more detail in the following.
Figure 1. Proposed cross-talk between EpCAM signaling and the Wnt pathway. Activation of the frizzled receptor by members of the Wnt family of ligands induces the inhibition of GSK3β and the subsequent stabilization of β-catenin. Upon nuclear translocation, β-catenin controls Lef-1 dependent transcription. EpICD interacts with the very same components to form a nuclear complex comprised of β-catenin, FHL2 and Lef-1. This nuclear complex is licensed to bind promoters of genes involved in cell cycle regulation and stemness. Further research would be necessary to describe in-depth the mechanism of transcriptional regulation of Nanog, Oct4, Klf4 and Sox2 by EpCAM. The actual composition of EpICD nuclear complexes recruited to the promoters of stemness genes are unexplored until now.
EpCAM and Central Signaling Pathways in TICs
TICs most probably require a multitude of signals in order to maintain a phenotype characterized by self-renewal and pluripotency. These signals include the Wnt/β-catenin pathway,Citation59,Citation60 the Sonic Hedgehog and the Notch pathways,Citation61 which play a decisive role in the regulation and maintenance of stemness, and in tumor formation. Hence, it is not surprising that all these pathways are known to be major regulators of TICs.Citation7,Citation11,Citation62 Uncontrolled activation of these and other pathways are presumed to play essential roles in the initial formation of TICs and therefore in tumorigenesis in general.Citation14,Citation63 As these pathways are frequently involved in the regulation of the phenotype of various stem cells, it is further tempting to speculate that gain-of-function mutations of members of those pathways are instrumental in the formation of TICs.
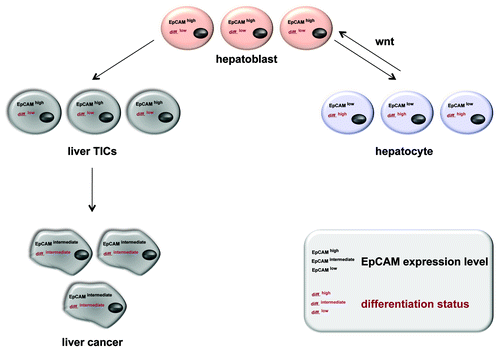
Briefly, binding of Wnt ligands to the frizzled receptor facilitates the phosphorylation of the disheveled protein (DSH), which inactivates glucogen synthase kinase 3β (GSK3-β). Thereby, the phosphorylation and subsequent degradation of β-catenin by the proteasome are inhibited. The resulting accumulation of β-catenin in the cytoplasm allows for its transfer into the nucleus, where it interacts with transcription factors of the Lef1 family (). This functional complex induces the transcription of prominent targets like CD44,Citation64 cyclin D1Citation65,Citation66 and cmyc,Citation67 which is also a major target of EpCAM signaling.Citation68 Moreover cmyc can trigger the induction of a stem-like transcriptional profile in normal and cancer cells and represents the central switch from adult to embryonic stem cells.Citation69 So far, it remains unknown at which point in the signaling cascades of EpCAM and Wnt/Frizzled cross-talk occurs (). However, EpICD does not only interact with β-catenin and Lef-1, it also binds to Lef-1 consensus sites in the promoter of Wnt target genes such as cyclin D1.Citation51,Citation66 Interestingly, EpICD appeared to be essential for the formation of one of the two major nuclear protein/DNA complexes formed at Lef-1 consensus sites in EpCAM-positive carcinoma cells,Citation51 suggesting that EpICD can provide additional levels of regulation to Wnt target genes, which are central in cell cycle regulation and thus could play important roles in self-renewal. Since Wnt signaling is reportedly instrumental in TICsCitation7,Citation70 and because TICs rely on Wnt pathway inducing signals from their microenvironment for the maintenance of their phenotype,Citation71 it is tempting to speculate that EpCAM overexpression and signaling are instrumental in this respect, too. For example, in addition to cmyc other key factors such as nanog, klf4, sox2 and oct4, which are central to the conversion of somatic cells into induced pluripotent stem cells (iPS),Citation72,Citation73 were also described as direct targets of EpCAM in human embryonic stem cells (hESC).Citation74 Thus, a loss of regulation of EpCAM expression and signaling would equip cells with some of the characteristic traits of TICs such as proliferative potential and multipotency.Citation14 Evidence in support of a central role of EpCAM in the reprogramming of iPS came from studies involving mouse embryonic fibroblast transduced with expression constructs for Oct4, Sox2, Klf4 and cmyc (OSKM). Induction of expression and synergistic effects of Claudin-7 (Cldn7) and EpCAM during efficient generation of iPS indicated that Cldn7 regulated the functional activity of EpCAM.Citation75 Accordingly, conventional stem cell markers such as SSEA-1 and alkaline phosphatase were observed during reprogramming even in incompletely reprogrammed cells, which did not turn into iPS, whereas EpCAM and E-cadherin could only be detected in successfully reprogrammed iPS.Citation76 With these findings in mind and the knowledge of EpCAM signaling capacities, we suggest a role for EpCAM in the induction and/or maintenance of the phenotype of tissue precursors, stem cells, iPS cells, cancer cells, and TICs. This function most probably relates primarily to proliferation and the maintenance of an undifferentiation state. This hypothesis is so far best exemplified in the liver, where EpCAM expression and Wnt signaling both are associated with a tissue stem cell phenotype and regenerative capacity of cells.Citation77-Citation80 It is important to note that EpCAM expression was only detected in regenerating cells like hepatobiliary stem and progenitor cells, while it was lost in mature hepatocytesCitation77,Citation81 and thus a potential role for EpCAM in the regulation of stemness of liver progenitors was discussed recently.Citation81,Citation82 An interrelation of EpCAM and Wnt in hepatocellular carcinomas was further substantiated upon the finding that the epcam gene becomes transcriptionally activated by Tcf-4, a member of the Lef family of transcription factors.Citation83 Accordingly, EpCAM is a marker for TICs with a stem/progenitor phenotype in hepatocellular carcinomasCitation84 (). Taken together these facts on EpCAM expression patterns and signaling capacities present a solid rationale for the frequent overexpression of EpCAM in TICs of various tumor entities.Citation16 It is conceivable that EpCAM becomes upregulated or is per se strongly expressed in tissue progenitors and in cells which regenerate damaged or injured organs, as for the case of liver and kidney. Under non-pathological conditions, mature cells of these organs will cease to express EpCAM and thereby silence self-renewal and pluripotency traits mediated by EpCAM. Upon mutations and/or abnormal activation of the Wnt pathway this regulative decrease of EpCAM expression might be obviated and result in cells, which are primed for further transformation.
Figure 2. EpCAM expression in normal and regenerating liver, mature hepatocytes, liver carcinomas and TICs thereof. During liver morphogenesis and regeneration upon injury or chronic inflammation, EpCAM expression correlates with a proliferative and low differentiation. Liver precursors (hepatoblasts) are characterized by EpCAMhigh and downregulate this EpCAM expression upon differentiation into mature hepatocytes (differentiationlow). This process is partly regulated by the Wnt signaling pathway, which also dictates EpCAM expression. For the case of malignant transformation, EpCAM remains highly expressed in liver TICs and show an intermediate phenotype in hepatocellular carcinomas (differentiationintermediate).
It must however be noted that controversy exists, as it was depicted that the regulation of pluripotency genes except for cmyc were not or just marginally affected by the knockdown of EpCAM in hESC,Citation19 which contradicts work by Lu et al.Citation74 Furthermore, there is still no consensus on the mechanism of EpCAM regulation during differentiation in human ES cells. On the one hand it was reported that EpCAM is controlled by epigenetic modifications, which correlated with a reduced transcription,Citation74 while on the other hand a posttranscriptional regulation was proposed.Citation19 Hence, further research would be necessary to solve this controversial discussion. Also, EpCAM is expressed in normal tissue with a differentiated phenotype and lacking both proliferation and pluripotency. So far nothing was reported on the actual function of EpCAM in these tissues. A comparison of EpICD localization in normal colonic mucosa and colon carcinomas revealed striking differences. EpICD translocated into the nucleus, where it deploys its function,Citation85 in malignant but not in normal colon cells.Citation51 Molecular mechanisms underlying these observations are still unknown.
Besides its expression, a second level of regulation of EpCAM in stem cells and TICs remains unexplored, namely the induction of EpCAM signaling. Data available on the induction of EpCAM cleavage originate from human cancer cells but not from stem cells or mature somatic cells. In principle, evidence for a regulated intramembrane proteolysis of EpCAM and nuclear translocation of EpICD is indirect. Lu et al. reported on chromatin immunoprecipitation of EpICD at promoters of stemness genes,Citation74 strongly suggesting EpCAM cleavage in stem cells too. However, a formal proof of this notion is still pending. Assuming that EpCAM is cleaved in stem cells and TICs, and knowing that cleavage in carcinoma cells is initially induced by cell-to-cell contact,Citation85 we can speculate on the repercussions on stem cells and TICs. Upon strong overexpression of EpCAM, stem cells and TICs might become primed for EpCAM signaling pathways such that signaling can be induced upon cell-to-cell contacts of stem cells or TICs among themselves, or with EpCAM-positive cells of the microenvironment. In line with this hypothesis was the interesting finding that aldehyde dehydrogenase 1- and EpCAM-positive cells from patients suffering from chronic colitis were the cells of origin during transformation to colorectal cancers.Citation86 This implies that the expression of ALDH1 and EpCAM marks cells with pre-malignant traits, which are prone to tumor formation in vivo. Such a mechanism would allow the setting of a threshold for minimal numbers of TICs required for efficient amplification in a given microenvironment and promote a self-perpetuating amplification of EpCAM signals and, as a result, of TICs. In turn, active shut-down of EpCAM expression and signals in TICs could participate in the reported aberrant differentiation of TICs into bulk tumor cells, as was observed in normal embryonic stem cells.Citation18,Citation19
EpCAM as a Target in Cancer Therapy
Conventional therapies for the treatment of cancer such as chemotherapy or radiation target predominantly the tumor bulk. Unfortunately, TICs are not or only marginally affected for several reasons: at the time point of treatment it is assumable that TICs are low proliferating and almost quiescent cells with an infrequent cell cycle transition. In other words, transient amplification of TICs to generate the tumor bulk already had happened at the time point of diagnosis. Hence, TICs will be sub-optimally affected by conventional approachesCitation87,Citation88 or even be rather refractory to standard chemotherapy and increase proportionally in numbers upon treatment.Citation89-Citation91 The preference for hypoxic niches makes these cells to inappropriate targets also for angiogenesis inhibitors. Furthermore, the overexpression of detoxifying enzymes or multidrug resistant pumps makes it challenging to target TICs with classical treatments. Therefore, a combination of differentiated treatment strategies against the tumor mass and TICs are required. In this context, EpCAM might be a promising target because it is highly expressed on the bulk of cancer cells as well as on TICs. Since EpCAM is also expressed endogenously on healthy epithelial tissue and stem cells the question arises if these cells are also affected by EpCAM-based strategies or whether differences in expression levels allow for an appropriate therapeutic window. In normal tissue, EpCAM is arranged in a complex with CD9, CD44 and Claudin-7, and is localized to basolateral membranes.Citation35,Citation92 Thus, the accessibility for EpCAM-binding antibodies is lower in normal cells than for cancer cells. In these cells EpCAM is strongly overexpressed and therefore might be partly unbound and better accessible for targeting antibodies. In 2001, McLaughlin et al. generated a transgenic mouse model expressing human EpCAM under the control of the endogenous human promoter. In this animal model, the authors observed a favored binding of the monoclonal antibody MOC31 to xenotransplanted tumor cells in comparison to healthy tissue expressing human EpCAM, indicating a higher accessibility of EpCAM in tumor cells.Citation93 During the last decades several efforts were made to generate therapeutic antibodies. The first EpCAM targeting monoclonal IgG2a murine antibody tested in patients was termed Edrecolomab (Panorex). First clinical studies enrolling patients treated with Edrecolomab revealed a reduced tumor recurrence and death rate, whereas these effects could not be reproduced in larger clinical trials.Citation30,Citation31,Citation36 This failure of clinical response might be due to the short serum half-life of the murine antibody and to a lack of randomization of patients according to their actual EpCAM status on tumor cells. Clearly, it is expected that only patients with high-level EpCAM expression on tumor cells would benefit from antibody treatment and therefore randomization appears mandatory. Nonetheless, severe doubts on the efficiency and feasibility of EpCAM-specific antibodies were raisedCitation37 and partly rebutted.Citation38 As a matter of fact, knowledge on the actual function of a target will help to develop more effective therapeutics with lower side-effect, as has been beautifully shown for the case of Her and Trastuzumab. By the time EpCAM-specific antibodies have been developed, knowledge on EpCAM's capacities to signal and induce proliferation was not available. Accordingly, differences in the influence of EpCAM-specific antibodies on the proliferation of EpCAM-positive cells have recently been demonstratedCitation40 and must be taken into consideration for future developments. Several chimeric (chimeric Edrecolomab), humanized (3622W94), human-engineered (ING-1), and fully human (Adecatumumab) anti-EpCAM antibodies with different target affinities were designed. Interestingly, antibodies with highest affinities such as 3622W94 and ING-1 induced acute pancreatitis even at low concentrations (1 mg/kg body weight).Citation94,Citation95 High affinity obviously increases the binding of EpCAM-specific antibodies to healthy tissue such as pancreas or the respiratory tract. In contrast, the human antibody Adecatumumab (MT201), which is characterized by an intermediate affinity, showed only minor side effects like nausea, chill, fatigue and diarrhea using higher doses (2–6 mg/kg body weight).Citation39 In this particular clinical phase II, randomization between high and low EpCAM expression in metastatic breast cancer revealed that a high EpCAM level is associated with a good prognosis in terms of overall survival after treatment with Adecatumumab. In 2009, the first antibody targeting EpCAM, called Catumaxomab (Removab), obtained approval for the European market. This trifunctional antibody has the ability to bind EpCAM expressing cancer cells as well as cytotoxic T-cells via the CD3 receptor. The Fcγ receptor allows the molecule to further bind and activate accessory cells such as macrophages, NK cells and DCs.Citation96 Clinical trials revealed humoral responses against this antibody after treatment. This might be due to the chimeric structure consisting of mouse IgG2a and rat IgG2b. Surprisingly, this kind of response against Catumaxomab correlated with the clinical outcome in a positive manner and treatment of patients with malignant ascites with Catumaxomab caused a prolonged overall survival.Citation97
So far, all clinical trials reported herein did not specifically deal with the eradication of TICs in particular, but rather with the treatment of EpCAM-positive cancers. Recently, the bispecific antibody MT110 was tested for its ability to target TICs derived from colorectal cancers. This antibody has binding affinities for EpCAM and CD3, which allow it to initiate the formation of a cytolytic synapse between T-cells and TICs. A combination of the antibody and PBMCs (Peripheral Blood Mononuclear Cells) led to decreased or absent colony formation in soft agar assays. Additionally, the treatment with MT110 prevented tumor formation in a xenograft model, where mice were inoculated with TICs.Citation98
Despite considerable improvements in the field of therapeutic antibodies, there are still challenges to manage. Variations in carbohydrate compositions, which are related to cytotoxicity can occur in the production of therapeutic antibody and thereby exhibit a safety risk during treatment. Additionally, for several antibodies the short serum half-life is a remaining problem. Therefore, RNA aptamers represent an attractive alternative. The advantages of these molecules are a cheap production, higher stability and chemical synthesis providing better reproducibility. Shigdar et al. generated a specific nuclease resistant aptamer targeting EpCAM, known as EpDT3. This molecule consisting of only 19 nucleotides offers a similar moderate binding affinity as for instance Catumaxomab or Adecatumumab antibodies. Importantly, EpDT3 is internalized into the cell via endocytosis after binding to EpCAM on the surface. Hence, this mechanism can be used to channel diverse substances such as chemotherapeutic drugs, toxins and therapeutic radioisotopes. The high accessibility due to the small molecular size is considered to be an advantage compared with antibodies. For the case of EpCAM it could however be a disadvantage since EpCAM expressing healthy tissue might be targeted as well.Citation48
Based on the novel understanding of the functions of EpCAM, another interesting approach relies on the interference with the signaling cascade of EpCAM. The knowledge of proteases involved in the activating proteolytic cleavage of EpCAM allows for the systematic testing of combinations of inhibitors of these proteases, i.e. TACE and presenilin 2, therapeutic antibodies, and conventional treatments involving chemotherapeutics. Even more interesting, inhibition of the EpICD-FHL2 interaction by small molecules generated upon structure-based rational design and bioinformatics is a promising and highly innovative strategy to specifically target EpCAM and its signaling. In vitro experiments support inhibitory effects of TACE and gamma-secretase inhibitors on EpCAM-dependent proliferation and target gene induction. Likewise, knockdown of FHL2 resulted in an inhibition of EpCAM-specific effects.Citation51 Last but not least, the differential cleavage and localization of EpICD in normal and malignant colon epithelium represents a solid in vivo rationale for a therapeutic intervention at the level of EpCAM cleavage and signaling. Future work will concentrate on the pre-clinical and clinical translation of these therapeutic approaches.
Conclusions
Epithelial cell adhesion molecule EpCAM has long been neglected as a central molecule in cancer development and was rather perceived as a cell adhesion molecule, whose overexpression in cancer was rather concomitant than causal. With the emerging understanding of EpCAM being a signaling active receptor involved in the transformation of malignant cells, in the regulation of self-renewal and pluripotency in stem and progenitor cells, and possibly with a strong impact on the activity of TICs, novel therapeutic approaches were conceived. Specific inhibition of the activating cleavage of EpCAM is a major challenge of future developments and might provide powerful agents to fight TICs (see also Box 1). Along with these advances, further understanding of the role of EpCAM in the regulation of proliferation and pluripotency in any given cell type is in great demand.
Box 1. At a glance.
• Tumor-initiating cells (TICs) share a common phenotype with stem cells including self-renewal, multipotency and increased proliferative capacity.
• EpCAM is a well-established marker for TICs, cancer cells and stem cells.
• EpCAM belongs to the type I transmembrane glycoproteins.
• EpCAM signaling is initiated by regulated intramembrane proteolysis. After being cleaved off, the intracellular domain EpICD translocates into the nucleus and regulates transcription in a complex with FHL2, β-catenin and Lef1.
• As TICs are not effectively targeted by conventional cancer treatments like chemotherapy or radiation, therapeutic strategies have to be adapted. Recently, an EpCAM/CD3-bispecific antibody analogon, which engages T-cells was generated. This BiTE antibody was effective in the prevention of tumor formation by TICs in a mouse model.
References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- Bhaijee F, Pepper DJ, Pitman KT, Bell D. Cancer stem cells in head and neck squamous cell carcinoma: A review of current knowledge and future applications. Head Neck 2011; In press http://dx.doi.org/10.1002/hed.21801; PMID: 21850700
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755 - 68; http://dx.doi.org/10.1038/nrc2499; PMID: 18784658
- Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest 1994; 70:6 - 22; PMID: 8302019
- Sawyers CL, Denny CT, Witte ON. Leukemia and the disruption of normal hematopoiesis. Cell 1991; 64:337 - 50; http://dx.doi.org/10.1016/0092-8674(91)90643-D; PMID: 1988151
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105 - 11; http://dx.doi.org/10.1038/35102167; PMID: 11689955
- Espada J, Calvo MB, Diaz-Prado S, Medina V. Wnt signalling and cancer stem cells. Clin Transl Oncol 2009; 11:411 - 27; http://dx.doi.org/10.1007/s12094-009-0380-4; PMID: 19574199
- Chumsri S, Matsui W, Burger AM. Therapeutic implications of leukemic stem cell pathways. Clin Cancer Res 2007; 13:6549 - 54; http://dx.doi.org/10.1158/1078-0432.CCR-07-1088; PMID: 18006753
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434:843 - 50; http://dx.doi.org/10.1038/nature03319; PMID: 15829953
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003; 423:409 - 14; http://dx.doi.org/10.1038/nature01593; PMID: 12717450
- Bolos V, Blanco M, Medina V, Aparicio G, Diaz-Prado S, Grande E. Notch signalling in cancer stem cells. Clin Transl Oncol 2009; 11:11 - 9; http://dx.doi.org/10.1007/s12094-009-0305-2; PMID: 19155199
- Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule?. Nat Rev Cancer 2011; 11:254 - 67; http://dx.doi.org/10.1038/nrc3023; PMID: 21390059
- Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res 2008; 68:6932 - 41; http://dx.doi.org/10.1158/0008-5472.CAN-07-5779; PMID: 18757407
- Gires O. Lessons from common markers of tumor-initiating cells in solid cancers. Cell Mol Life Sci 2011; 68:4009 - 22; http://dx.doi.org/10.1007/s00018-011-0772-9; PMID: 21786143
- Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol 2007; 23:675 - 99; http://dx.doi.org/10.1146/annurev.cellbio.22.010305.104154; PMID: 17645413
- Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. [author reply] Nat Rev Cancer 2009; 9:143; http://dx.doi.org/10.1038/nrc2499-c1; PMID: 19132011
- Choo AB, Tan HL, Ang SN, Fong WJ, Chin A, Lo J, et al. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells 2008; 26:1454 - 63; http://dx.doi.org/10.1634/stemcells.2007-0576; PMID: 18356574
- Gonzalez B, Denzel S, Mack B, Conrad M, Gires O. EpCAM is involved in maintenance of the murine embryonic stem cell phenotype. Stem Cells 2009; 27:1782 - 91; http://dx.doi.org/10.1002/stem.97; PMID: 19544432
- Ng VY, Ang SN, Chan JX, Choo AB. Characterization of epithelial cell adhesion molecule as a surface marker on undifferentiated human embryonic stem cells. Stem Cells 2010; 28:29 - 35; http://dx.doi.org/10.1002/stem.221; PMID: 19785009
- Herlyn M, Steplewski Z, Herlyn D, Koprowski H. Colorectal carcinoma-specific antigen: detection by means of monoclonal antibodies. Proc Natl Acad Sci USA 1979; 76:1438 - 42; http://dx.doi.org/10.1073/pnas.76.3.1438; PMID: 286328
- Gottlinger H, Johnson J, Riethmuller G. Biochemical and epitope analysis of the 17-1A membrane antigen. Hybridoma 1986; 5:Suppl 1 S29 - 37; PMID: 2427436
- Gottlinger HG, Funke I, Johnson JP, Gokel JM, Riethmuller G. The epithelial cell surface antigen 17-1A, a target for antibody-mediated tumor therapy: its biochemical nature, tissue distribution and recognition by different monoclonal antibodies. Int J Cancer 1986; 38:47 - 53; http://dx.doi.org/10.1002/ijc.2910380109; PMID: 3721623
- Herlyn D, Herlyn M, Ross AH, Ernst C, Atkinson B, Koprowski H. Efficient selection of human tumor growth-inhibiting monoclonal antibodies. J Immunol Methods 1984; 73:157 - 67; http://dx.doi.org/10.1016/0022-1759(84)90041-3; PMID: 6491298
- Herlyn D, Herlyn M, Steplewski Z, Koprowski H. Monoclonal antibodies in cell-mediated cytotoxicity against human melanoma and colorectal carcinoma. Eur J Immunol 1979; 9:657 - 9; http://dx.doi.org/10.1002/eji.1830090817; PMID: 499332
- Sears HF, Atkinson B, Mattis J, Ernst C, Herlyn D, Steplewski Z, et al. Phase-I clinical trial of monoclonal antibody in treatment of gastrointestinal tumours. Lancet 1982; 319:762 - 5; http://dx.doi.org/10.1016/S0140-6736(82)91811-6; PMID: 6121224
- Sears HF, Herlyn D, Steplewski Z, Koprowski H. Effects of monoclonal antibody immunotherapy on patients with gastrointestinal adenocarcinoma. J Biol Response Mod 1984; 3:138 - 50; PMID: 6374043
- Weiner LM, Steplewski Z, Koprowski H, Sears HF, Litwin S, Comis RL. Biologic effects of gamma interferon pre-treatment followed by monoclonal antibody 17-1A administration in patients with gastrointestinal carcinoma. Hybridoma 1986; 5:Suppl 1 S65 - 77; PMID: 3091476
- Lobuglio AF, Saleh M, Peterson L, Wheeler R, Carrano R, Huster W, et al. Phase I clinical trial of CO17-1A monoclonal antibody. Hybridoma 1986; 5:Suppl 1 S117 - 23; PMID: 3488950
- Verrill H, Goldberg M, Rosenbaum R, Abbott R, Simunovic L, Steplewski Z, et al. Clinical trial of Wistar Institute 17-1A monoclonal antibody in patients with advanced gastrointestinal adenocarcinoma: a preliminary report. Hybridoma 1986; 5:Suppl 1 S175 - 83; PMID: 3527947
- Riethmuller G, Holz E, Schlimok G, Schmiegel W, Raab R, Hoffken K, et al. Monoclonal antibody therapy for resected Dukes' C colorectal cancer: seven-year outcome of a multicenter randomized trial. J Clin Oncol 1998; 16:1788 - 94; PMID: 9586892
- Riethmüller G, Schneider Gadicke E, Schlimok G, Schmiegel W, Raab R, Hoffken K, et al. Randomised trial of monoclonal antibody for adjuvant therapy of resected Dukes' C colorectal carcinoma. Lancet 1994; 343:1177 - 83; http://dx.doi.org/10.1016/S0140-6736(94)92398-1; PMID: 7909866
- Litvinov SV, Balzar M, Winter MJ, Bakker HA, Briaire-de Bruijn IH, Prins F, et al. Epithelial cell adhesion molecule (Ep-CAM) modulates cell-cell interactions mediated by classic cadherins. J Cell Biol 1997; 139:1337 - 48; http://dx.doi.org/10.1083/jcb.139.5.1337; PMID: 9382878
- Litvinov SV, van Driel W, van Rhijn CM, Bakker HA, van Krieken H, Fleuren GJ, et al. Expression of Ep-CAM in cervical squamous epithelia correlates with an increased proliferation and the disappearance of markers for terminal differentiation. Am J Pathol 1996; 148:865 - 75; PMID: 8774141
- Litvinov SV, Velders MP, Bakker HA, Fleuren GJ, Warnaar SO. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J Cell Biol 1994; 125:437 - 46; http://dx.doi.org/10.1083/jcb.125.2.437; PMID: 8163559
- Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17-1A antigen (Ep-CAM). J Mol Med 1999; 77:699 - 712; http://dx.doi.org/10.1007/s001099900038; PMID: 10606205
- Fields AL, Keller A, Schwartzberg L, Bernard S, Kardinal C, Cohen A, et al. Adjuvant therapy with the monoclonal antibody Edrecolomab plus fluorouracil-based therapy does not improve overall survival of patients with stage III colon cancer. J Clin Oncol 2009; 27:1941 - 7; http://dx.doi.org/10.1200/JCO.2008.18.5710; PMID: 19273708
- Schmoll HJ, Arnold D. When wishful thinking leads to a misty-eyed appraisal: the story of the adjuvant colon cancer trials with edrecolomab. J Clin Oncol 2009; 27:1926 - 9; http://dx.doi.org/10.1200/JCO.2008.20.6284; PMID: 19273695
- Gires O, Baeuerle PA. EpCAM as a target in cancer therapy. J Clin Oncol 2010; 28:e239 - 40; http://dx.doi.org/10.1200/JCO.2009.26.8540; PMID: 20385979
- Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol 2010; 21:275 - 82; http://dx.doi.org/10.1093/annonc/mdp314; PMID: 19633042
- Munz M, Murr A, Kvesic M, Rau D, Mangold S, Pflanz S, et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int 2010; 10:44; http://dx.doi.org/10.1186/1475-2867-10-44; PMID: 21044305
- Spizzo G, Went P, Dirnhofer S, Obrist P, Moch H, Baeuerle PA, et al. Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol 2006; 103:483 - 8; http://dx.doi.org/10.1016/j.ygyno.2006.03.035; PMID: 16678891
- Spizzo G, Went P, Dirnhofer S, Obrist P, Simon R, Spichtin H, et al. High Ep-CAM expression is associated with poor prognosis in node-positive breast cancer. Breast Cancer Res Treat 2004; 86:207 - 13; http://dx.doi.org/10.1023/B:BREA.0000036787.59816.01; PMID: 15567937
- Benko G, Spajic B, Kruslin B, Tomas D.. Impact of the EpCAM expression on biochemical recurrence-free survival in clinically localized prostate cancer. Urol Oncol 2011; In press http://dx.doi.org/10.1016/j.urolonc.2011.03.007; PMID: 21514185
- Akita H, Nagano H, Takeda Y, Eguchi H, Wada H, Kobayashi S, et al. Ep-CAM is a significant prognostic factor in pancreatic cancer patients by suppressing cell activity. Oncogene 2011; 30:3468 - 76; http://dx.doi.org/10.1038/onc.2011.59; PMID: 21399662
- Jojovic M, Adam E, Zangemeister-Wittke U, Schumacher UH. Epithelial glycoprotein-2 expression is subject to regulatory processes in epithelial-mesenchymal transitions during metastases: an investigation of human cancers transplanted into severe combined immunodeficient mice. Histochem J 1998; 30:723 - 9; http://dx.doi.org/10.1023/A:1003486630314; PMID: 9873999
- Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, et al. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem 2011; 112:2296 - 306; http://dx.doi.org/10.1002/jcb.23150; PMID: 21503965
- Munz M, Fellinger K, Hofmann T, Schmitt B, Gires O. Glycosylation is crucial for stability of tumour and cancer stem cell antigen EpCAM. Front Biosci 2008; 13:5195 - 201; http://dx.doi.org/10.2741/3075; PMID: 18508581
- Pauli C, Munz M, Kieu C, Mack B, Breinl P, Wollenberg B, et al. Tumor-specific glycosylation of the carcinoma-associated epithelial cell adhesion molecule EpCAM in head and neck carcinomas. Cancer Lett 2003; 193:25 - 32; http://dx.doi.org/10.1016/S0304-3835(03)00003-X; PMID: 12691820
- Balzar M, Briaire-de Bruijn IH, Rees-Bakker HA, Prins FA, Helfrich W, de Leij L, et al. Epidermal growth factor-like repeats mediate lateral and reciprocal interactions of Ep-CAM molecules in homophilic adhesions. Mol Cell Biol 2001; 21:2570 - 80; http://dx.doi.org/10.1128/MCB.21.7.2570-2580.2001; PMID: 11259604
- Balzar M, Bakker HA, Briaire-de-Bruijn IH, Fleuren GJ, Warnaar SO, Litvinov SV. Cytoplasmic tail regulates the intercellular adhesion function of the epithelial cell adhesion molecule. Mol Cell Biol 1998; 18:4833 - 43; PMID: 9671492
- Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol 2009; 11:162 - 71; http://dx.doi.org/10.1038/ncb1824; PMID: 19136966
- Mizrak D, Brittan M, Alison MR. CD133: molecule of the moment. J Pathol 2008; 214:3 - 9; http://dx.doi.org/10.1002/path.2283; PMID: 18067118
- Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 2009; 69:5627 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-09-0654; PMID: 19584271
- Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC. Epithelial cell adhesion molecule. More than a carcinoma marker and adhesion molecule. Am J Pathol 2007; 171:386 - 95; http://dx.doi.org/10.2353/ajpath.2007.070152; PMID: 17600130
- Johannessen M, Moller S, Hansen T, Moens U. Van Ghelue Mc-rp-p. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci 2006; 63:268 - 84; http://dx.doi.org/10.1007/s00018-005-5438-z; PMID: 16389449
- Canault M, Tellier E, Bonardo B, Mas E, Aumailley M, Juhan-Vague I, et al. FHL2 interacts with both ADAM-17 and the cytoskeleton and regulates ADAM-17 localization and activity. J Cell Physiol 2006; 208:363 - 72; http://dx.doi.org/10.1002/jcp.20671; PMID: 16619241
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, Ie L, et al. Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem 2005; 280:31537 - 47; http://dx.doi.org/10.1074/jbc.M500833200; PMID: 16014629
- Wei Y, Renard CA, Labalette C, Wu Y, Levy L, Neuveut C, et al. Identification of the LIM protein FHL2 as a coactivator of beta-catenin. J Biol Chem 2003; 278:5188 - 94; http://dx.doi.org/10.1074/jbc.M207216200; PMID: 12466281
- Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011; 8:97 - 106; http://dx.doi.org/10.1038/nrclinonc.2010.196; PMID: 21151206
- Takebe N, Warren RQ, Ivy SP. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res 2011; 13:211; http://dx.doi.org/10.1186/bcr2876; PMID: 21672282
- Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001; 411:349 - 54; http://dx.doi.org/10.1038/35077219; PMID: 11357142
- Medina V, Calvo MB, Diaz-Prado S, Espada J. Hedgehog signalling as a target in cancer stem cells. Clin Transl Oncol 2009; 11:199 - 207; http://dx.doi.org/10.1007/s12094-009-0341-y; PMID: 19380296
- Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer 2005; 5:899 - 904; http://dx.doi.org/10.1038/nrc1740; PMID: 16327766
- Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol 1999; 154:515 - 23; http://dx.doi.org/10.1016/S0002-9440(10)65297-2; PMID: 10027409
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398:422 - 6; http://dx.doi.org/10.1038/18884; PMID: 10201372
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999; 96:5522 - 7; http://dx.doi.org/10.1073/pnas.96.10.5522; PMID: 10318916
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science 1998; 281:1509 - 12; http://dx.doi.org/10.1126/science.281.5382.1509; PMID: 9727977
- Munz M, Kieu C, Mack B, Schmitt B, Zeidler R, Gires O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene 2004; 23:5748 - 58; http://dx.doi.org/10.1038/sj.onc.1207610; PMID: 15195135
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008; 2:333 - 44; http://dx.doi.org/10.1016/j.stem.2008.02.009; PMID: 18397753
- Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol 2010; 21:855 - 63; http://dx.doi.org/10.1016/j.semcdb.2010.09.004; PMID: 20837152
- Malanchi I, Huelsken J. Cancer stem cells: never Wnt away from the niche. Curr Opin Oncol 2009; 21:41 - 6; http://dx.doi.org/10.1097/CCO.0b013e32831d1faf; PMID: 19125017
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009; 462:595 - 601; http://dx.doi.org/10.1038/nature08592; PMID: 19898493
- Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 2010; 143:508 - 25; http://dx.doi.org/10.1016/j.cell.2010.10.008; PMID: 21074044
- Lu TY, Lu RM, Liao MY, Yu J, Chung CH, Kao CF, et al. Epithelial cell adhesion molecule regulation is associated with the maintenance of the undifferentiated phenotype of human embryonic stem cells. J Biol Chem 2010; 285:8719 - 32; http://dx.doi.org/10.1074/jbc.M109.077081; PMID: 20064925
- Huang HP, Chen PH, Yu CY, Chuang CY, Stone L, Hsiao WC, et al. Epithelial cell adhesion molecule (EpCAM) complex proteins promote transcription factor-mediated pluripotency Reprogramming. J Biol Chem 2011; 286:33520 - 32; http://dx.doi.org/10.1074/jbc.M111.256164; PMID: 21799003
- Chen HF, Chuang CY, Lee WC, Huang HP, Wu HC, Ho HN, et al. Surface marker epithelial cell adhesion molecule and E-cadherin facilitate the identification and selection of induced pluripotent stem cells. Stem Cell Rev 2011; 7:722 - 35; http://dx.doi.org/10.1007/s12015-011-9233-y; PMID: 21305366
- de Boer CJ, van Krieken JH, Janssen-van Rhijn CM, Litvinov SV. Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J Pathol 1999; 188:201 - 6; http://dx.doi.org/10.1002/(SICI)1096-9896(199906)188:2<201::AID-PATH339>3.0.CO;2-8; PMID: 10398165
- Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol 2011; 21:44 - 58; http://dx.doi.org/10.1016/j.semcancer.2010.12.010; PMID: 21182948
- Monga SP. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol 2011; 43:1021 - 9; http://dx.doi.org/10.1016/j.biocel.2009.09.001; PMID: 19747566
- Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, Yao HL, et al. Human hepatic stem cells from fetal and postnatal donors. J Exp Med 2007; 204:1973 - 87; http://dx.doi.org/10.1084/jem.20061603; PMID: 17664288
- Yoon SM, Gerasimidou D, Kuwahara R, Hytiroglou P, Yoo JE, Park YN, et al. Epithelial cell adhesion molecule (EpCAM) marks hepatocytes newly derived from stem/progenitor cells in humans. Hepatology 2011; 53:964 - 73; http://dx.doi.org/10.1002/hep.24122; PMID: 20967756
- Gires O. TACSTD1 (tumor-associated calcium signal transducer 1). Atlas Genet Cytogenet Oncol Haematol 2008 [Access at: http://AtlasGeneticsOncology.org/Genes/TACSTD1ID42459ch2p21.html]
- Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res 2007; 67:10831 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-0908; PMID: 18006828
- Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009; 136:1012 - 24; http://dx.doi.org/10.1053/j.gastro.2008.12.004; PMID: 19150350
- Denzel S, Maetzel D, Mack B, Eggert C, Barr G, Gires O. Initial activation of EpCAM cleavage via cell-to-cell contact. BMC Cancer 2009; 9:402; http://dx.doi.org/10.1186/1471-2407-9-402; PMID: 19925656
- Carpentino JE, Hynes MJ, Appelman HD, Zheng T, Steindler DA, Scott EW, et al. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res 2009; 69:8208 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-09-1132; PMID: 19808966
- Mueller MT, Hermann PC, Heeschen C. Cancer stem cells as new therapeutic target to prevent tumour progression and metastasis. Front Biosci (Elite Ed) 2010; 2:602 - 13; http://dx.doi.org/10.2741/e117; PMID: 20036905
- Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Cancer stem cells in solid tumors. Semin Cancer Biol 2010; 20:77 - 84; http://dx.doi.org/10.1016/j.semcancer.2010.03.004; PMID: 20371287
- Nicolini A, Ferrari P, Fini M, Borsari V, Fallahi P, Antonelli A, et al. Stem cells: their role in breast cancer development and resistance to treatment. Curr Pharm Biotechnol 2011; 12:196 - 205; http://dx.doi.org/10.2174/138920111794295657; PMID: 21044007
- Frame FM, Maitland NJ. Cancer stem cells, models of study and implications of therapy resistance mechanisms. Adv Exp Med Biol 2011; 720:105 - 18; http://dx.doi.org/10.1007/978-1-4614-0254-1_9; PMID: 21901622
- Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 2008; 3:e2428; http://dx.doi.org/10.1371/journal.pone.0002428; PMID: 18560594
- Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer 2007; 96:417 - 23; http://dx.doi.org/10.1038/sj.bjc.6603494; PMID: 17211480
- McLaughlin PM, Harmsen MC, Dokter WH, Kroesen BJ, van der Molen H, Brinker MG, et al. The epithelial glycoprotein 2 (EGP-2) promoter-driven epithelial-specific expression of EGP-2 in transgenic mice: a new model to study carcinoma-directed immunotherapy. Cancer Res 2001; 61:4105 - 11; PMID: 11358833
- LoBuglio A, Saleh M, Braddock J, Lampkin T, Khor S, Wissel P, et al. A phase I trial of the humanized anti-EGP40 monoclonal antibody 3622W94. Proc Am Soc Clin Oncol 1997; 16:436
- Goel S, Bauer RJ, Desai K, Bulgaru A, Iqbal T, Strachan BK, et al. Pharmacokinetic and safety study of subcutaneously administered weekly ING-1, a human engineere monoclonal antibody targeting human EpCAM, in patients with advanced solid tumors. Ann Oncol 2007; 18:1704 - 7; http://dx.doi.org/10.1093/annonc/mdm280; PMID: 17693421
- Seimetz D, Lindhofer H, Bokemeyer C.. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAMxanti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev 2010; 36:458 - 67; http://dx.doi.org/10.1016/j.ctrv.2010.03.001; PMID: 20347527
- Ott MG, Marme F, Moldenhauer G, Lindhofer H, Hennig M, Spannagl R, et al. Humoral response to catumaxomab correlates with clinical outcome: Results of the pivotal phase II/III study in patients with malignant ascites. Int J Cancer 2012; 130:2195 - 203; http://dx.doi.org/10.1002/ijc.26258; PMID: 21702044
- Herrmann I, Baeuerle PA, Friedrich M, Murr A, Filusch S, Ruttinger D, et al. Highly efficient elimination of colorectal tumor-initiating cells by an EpCAM/CD3-bispecific antibody engaging human T cells. PLoS ONE 2010; 5:e13474; http://dx.doi.org/10.1371/journal.pone.0013474; PMID: 20976159