Abstract
NO is an important regulator of cardiovascular remodelling and function. ADMA, an endogenous L-arginine analogue, reduces NO production by inhibiting the activity of NOS. ADMA levels in turn, are regulated by DDAH, which metabolises ADMA. High levels of ADMA and dysregulated DDAH activity are risk factors for cardiovascular disease and morbidity. To investigate this link, the DDAH I null mouse has been recently generated, and has a lethal phenotype. Studies on vascular function in the DDAH I heterozygous knockout mouse, which is viable, demonstrates a causal link between reduced DDAH I activity, increased ADMA levels and reduced NO signalling and vascular dysfunction. In another study, detailed in vitro analyses reveal that the DDAH/ADMA pathway critically regulates endothelial cell motility and angiogenesis, and establishes some of the molecular mechanisms involved. These studies highlight the importance of DDAH and ADMA in regulating NO dependent vascular homeostasis.
NO is generated from L-arginine by NOS; a process which is competitively inhibited by the arginine analogues ADMA and L-NMMA. These endogenous factors are products of proteolytic degradation of methylated proteins. ADMA and L-NMMA are metabolised by DDAH I and II, thereby enhancing NO generation. Of relevance to vascular biology, dysfunctional DDAH activity and ADMA accumulation are risk factors for cardiovascular disorders, including hypertension, artherosclerosis, diabetes, insulin resistance, hypercholesterolemia and homocysteinemia (reviewed in ref. Citation1).
The DDAH I null mouse was generated recently by Leiper et al.Citation2 to facilitate investigation of the role of the DDAH/ADMA pathway in the pathology of cardiovascular disorders. While the absence of DDAH I causes a lethal phenotype, heterozygotes (HT) did not display any obvious abnormalities. However, ADMA levels were raised in tissues and plasma, in association with raised blood pressure and systemic vascular resistance, and reduced cardiac output and heart rate. Synthetic DDAH I inhibitors were designed by the authors and were shown by crystallography to bind to the active site of the enzyme and induce local distortions at this region. Confirming that loss of DDAH I was responsible for ADMA accumulation, these inhibitors enhanced ADMA levels in wildtype mice, and resulted in cardiovascular changes similar to those seen in the HT background. Inhibitor treatment also promoted ADMA release from wildtype blood vessels maintained ex vivo, indicating that the DDAH/ADMA pathway is directly responsible for maintaining cardiovascular function in this model.
Evidence was also presented for a causal link between ADMA metabolism and reduced NO levels. In an ex vivo model, aortic rings from HT mice displayed enhanced phenylephrine-induced contraction and reduced acetylcholine-induced relaxation, while DDAH I inhibitors induced similar responses in aortic rings from wildtype mice; indicative of reduced levels of endothelial-derived NO. Further demonstrating an ADMA/NO-dependent mechanism, exogenous L-arginine restored a normal response to these vasomodulators in the HT model (by competing with ADMA for interaction with NOS). Similarly, cultured endothelial cells from HT vessels produced more ADMA and less NO than cells from wildtype vessels, and DDAH I inhibitors induced a similar phenotype in wildtype endothelial cells. The significance of DDAH I/ADMA and NO in vascular disease was tested in a disease model. Endotoxic shock was induced in rats by intravenous infusion of LPS, which induces excess NO production, resulting in systemic hypotension. After blood pressure had fallen by 20%, infusion of a DDAH I inhibitor was able to rapidly stabilise blood pressure, in accordance with inhibition of NO production through reduced ADMA metabolism. Thus, when DDAH I is reduced, ADMA is increased and endogenous NO inhibited, resulting in altered vascular function.
Another related study investigated a mechanistic understanding of the role of ADMA/DDAH/NO in angiogenesis.Citation3 The authors demonstrated that ADMA regulates endothelial cell motility and phenotype by inhibiting NO-dependent changes in activity of Rho-GTPases; key mediators of cytoskeletal dynamics and motility. Treatment of pulmonary artery endothelial cells with ADMA enhanced stress fibres and focal adhesion formation in conjunction with increased activity of RhoA in pull-down assays. In accordance with these observations, motility, tracked by time-lapse microscopy, was inhibited by ADMA treatment, and ADMA effects were reversed by a Rho kinase inhibitor (Y-27632) or by adenoviral-mediated gene transfer of a dominant negative RhoA mutant. RhoA activity is mediated by PKG, which mediates RhoA-Ser188 phosphorylation, preventing RhoA localization to the membrane and inhibiting its activity.Citation4 In further support of a RhoA-dependent mechanism, ADMA reduced phosphorylation at RhoA-Ser188, while a PKG activator was also able to revert ADMA effects on motility. Further, a non-phosphorylatable mutant of RhoA, Ala188RhoA, or a specific PKG inhibitor, each inhibited cell motility to a similar level as ADMA treatment alone. Inhibition of NO production and endothelial cell motility by ADMA was also reversed by a NO donor, SNAP, or by DDAH I or II overexpression via adenovirus-mediated gene transfer. Thus, reduction of NO/PKG levels by ADMA reduces RhoA phosphorylation at Ser188 resulting in enhancement of RhoA activity and inhibition of cell motility.
The significance of these molecular mechanisms to angiogenesis was demonstrated using endothelial cells and aortic ring explants from HT DDAH I and wildtype mice. HT endothelial cells, which secrete more ADMA and produce less NO than their wildtype counterparts, exhibit enhanced RhoA activity and stress fibre formation in conjunction with reduced motility. Reduced sprouting from ex vivo aortic rings was also observed in the HT model, which was mimicked by addition of exogenous ADMA in the wildtype background. These data demonstrate that in vivo, DDAH/ADMA levels are likely to play a key role in control of endothelial cell motility and angiogenesis by regulating NO production.
Summary
DDAH and ADMA are known risk factors for cardiovascular disease (see ). These two studies indicate that the DDAH/ADMA pathway is critical in maintaining vascular homeostasis, and highlight some of the molecular mechanisms responsible. ADMA inhibits endothelial cell motility and angiogenesis by suppressing NO-mediated inhibition of RhoA, thereby inducing RhoA-dependent stress fibre formation and enhancing cell-substratum adhesion. Regulated control of ADMA metabolism by DDAH is crucial in control of vascular homeostasis. DDAH overexpression restores normal endothelial cell responses in vitro and in vivo, and clearance of ADMA by DDAH is responsible for maintaining normal vascular remodelling and cardiovascular responses. Modulation of ADMA metabolism by DDAH may therefore prove important in future clinical applications.
Abbreviations
| NO | = | nitric oxide |
| NOS | = | nitric oxide synthase |
| ADMA | = | asymmetric dimethylarginine |
| DDAH | = | dimethylarginine dimethylaminohydrolase |
| HT | = | heterozygotes |
| L-NMMA | = | monomethylarginine |
| LPS | = | lipopolysaccharide |
| PKG | = | protein kinase G |
Figures and Tables
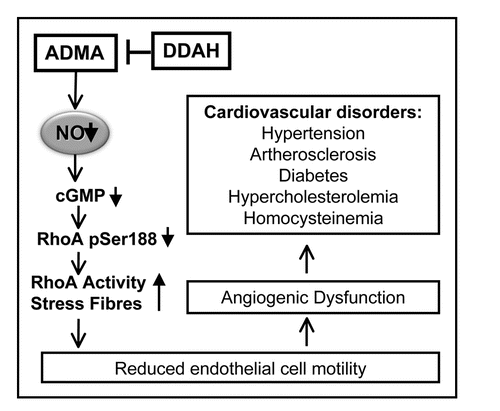
Figure 1 Summary of the role of DDAH/ADMA in vascular function and homeostasis. Increased ADMA levels are associated with cardiovascular disorders, which are related to dysfunctional endothelial cell responses and may be reversed by modulating DDAH activity. ADMA inhibits NO production, resulting in activation of RhoA and stress fibres though inhibition of PKG activity and reduction of RhoA phosphorylation at Ser188. This pathway results in reduced endothelial cell motility and angiogenesis, which is reversed by DDAH activation.
References
- Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol 2007; 293:3227 - 3245
- Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med 2007; 13:198 - 203
- Wojciak-Stothard B, Torondel B, Tsang LY, Fleming I, Fisslthaler B, Leiper JM, Vallance P. The ADMA/DDAH pathway is a critical regulator of endothelial cell motility. J Cell Sci 2007; 120:929 - 942
- Ellerbroeck SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 2003; 278:19023 - 19031