Abstract
Adhesion between cells and the extracellular matrix is mediated by different types of transmembraneous proteins. Their associations to specific partners lead to the assembly of contacts such as focal adhesions and hemidesmosomes. The spatial overlap between both contacts within cells has however limited the study of each type of contact. Here we show that with “stampcils” focal contacts and hemidesmosomes can be spatially separated: cells are plated within the cavities of a stencil and the grids of the stencil serve as stamps for grafting an extracellular matrix protein—fibronectin. Cells engage new contacts on stamped zones leading to the segregation of adhesions and their associated cytoskeletons, i.e., actin and intermediate filaments of keratins. This new method should provide new insights into cell contacts compositions and dynamics.
Introduction
Separation of phases in soft matter physics is often studied for characterizing systems.Citation1-Citation3 It involves a variety of approaches, ranging from lipid chemistry to statistical mechanics. Beyond the understanding of the dynamics of polymers and lipids at the mesoscopic scale, these studies allow to isolate zones within vesicles.Citation4-Citation6
Mammalian cells can be viewed in a similar manner: they exhibit structures which are entangled, and this can lead to limitations in understanding the nucleation and growth of the associated structures. Here we applied these ideas on cells and we successfully separated two adhesion structures: “focal adhesions” and “hemidesmosomes,” and their associated cytoskeletons, which are naturally strongly entangled. For that purpose, we promoted cell motility and forced cells to engage motion on specific adhesions in the explored zone. The scope of this study is to show the separation of entangled structures while opening a new way for characterizing their molecular components and signaling pathways.
We now outline briefly basic elements of cell adhesion to present the rationale for the separation. Cells engage adhesion with their environment by assembling contacts.Citation7 Their areas are typically about few micrometers squared, they nucleate, grow and disassemble rapidly within minutes. They consist schematically of three layers, (1) the extracellular matrix protein layer, (2) a specific adhesion complex of transmembraneous proteins and associated cytoplasm proteic partners and (3) a specific cytoskeleton. For example, focal contacts are composed of (1) fibronectin, (2) integrins associated with vinculin, paxillin, FAK, etc and (3) the actin stress fibers.Citation8,Citation9 Likewise, hemidesmosomes are structured of (1) laminin, (2) integrin associated with plectin and BPAG1e and (3) the intermediate filaments.Citation10 Generally, it is the specific binding of an extracellular matrix protein to a transmembraneous adhesion complex which dictates the type of the adhesion contacts.
These distinct types of contacts do often co-localize. This spatial feature hinders their thorough characterizations, which makes it difficult to distinguish their respective components. In addition, this overlap most likely generates some interplay between proteins of the contacts and the associated cytoskeletons; signaling pathways can be also entangled for the same reasons. It seems therefore important to design an assay for disentangling both contacts spatially in order to be able to dissect their specificity.
For this purpose, it is needed to trigger cell motility on a new zone of the surface potentially allowing one type of contact to be assembled, i.e., focal contacts—and not hemidesmosomes—for promoting motion. Imposing a cell to go in a given direction was indeed required to characterize new features of cell motion.Citation11 The so-called wound healing assay was designed to trigger such motion.Citation12-Citation15 A pipette or a syringe “scratches” a cell monolayer thus removing some rows of cells. Cells on both sides of the wound get polarized, they start to migrate to close the space devoid of cells. This approach has allowed to unravel key features of cell motility.Citation16
The scratch has however some limitations. Cells are detached in a non-controlled manner which potentially generates differences in results from experiments to experiments and between laboratories. In addition, the first row of migrating cells is damaged during the procedure. Also, the instrument can affect the surface in several ways: it can remove the extracellular matrix protein layer deposited by the cells or by the experimentalists on the scratched region; it can also generate some trenches within the surface by engraving the plastic or glass coverslip; this can also affect the migration.
An alternative method to wound healing was designed to remove cells in a controlled manner: the stencil consists in placing cells in holes surrounded by microfabricated structures.Citation17-Citation19 By removing the structure, cells can migrate, thus mimicking the traditional wound healing method. However, cells migrate on a surface depleted from a specific coating; it is then probably covered non-specifically by proteins of the serum after removal of the structure.
Microcontact printing (μCP) allows the controlled patterning of biomolecules on which cells can be deposited.Citation20-Citation22 We propose to combine the stencil approach together with the stamping method. Cells are placed into the holes of the stencil while the lower side of the stencil itself stamps proteins of the extracellular matrix, here fibronectin, in order to promote focal contacts formation and thereby cell motility. Because of its dual role, we name stampcil the microfabricated tool. When the stampcil is removed, cells start migrating on a surface coated with fibronectin. We are able to show that focal contacts are then separated from hemidesmosomes, and their associated cytoskeletons are also segregated.
Results and Discussion
We illustrate the preparation of the stampcil on . Briefly a master is prepared to generate a grid of typical dimensions with cavities of 900 × 900 × 200 μm3 in volume separated by walls of 100 μm in width. This particular width was selected because it matches the width of a pipette-induced wound on a confluent cell monolayer. Liquid PDMS is poured on the structure and is allowed to polymerize for 4 h at 65°C while being pressed (see Materials and Methods). The stampcil is then removed and its lower surface is inked with 10 μg ml−1 fibronectin solution (; Fig. S1). The stampcil is then brought in contact with the coverslip and simultaneously allows stamping fibronectin while generating chambers where cells are deposited (). illustrates the cell monolayer embedded inside a squared chamber, with its surrounding fibronectin layer.
Figure 1. Schematic of the stampcil fabrication procedure: (A) SU-8 mold; (B) pouring of PDMS onto the template and pressing; (C) curing and peeling-off of the stampcil; (D) inking with 10 μg ml−1 fibronectin (see Fig. S1); (E) stamping onto the surface; (F) incubating with cells; (G) releasing of the stampcil.
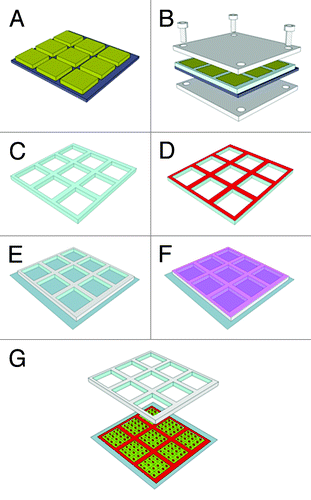
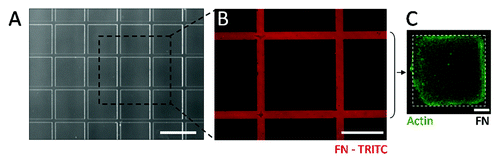
Figure 2. (A) Optical image of the PDMS stampcil (scale bar: 1 mm). (B) Fibronectin network (in red) patterned with the stampcil. Scale bar: 500 μm. (C) Fluorescent image after releasing the stampcil of 1 cavity filled with cells and stained for actin (in green). Scale bar: 200 μm.
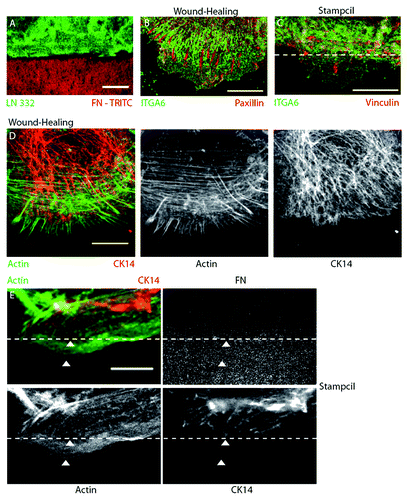
Then, the stampcil is removed which generates a straight border of cells allowing them to migrate (). Strikingly, focal contacts appear first on the migrating fronts (; Fig. S2B), and hemidesmosomes are lagging behind. This feature is in sharp contrast with the classical observation of mixed localization of focal contacts and hemidesmosomes in wound healing (; Fig. S2A). The respective cytoskeletons are also separated; while actin and intermediate filaments are mixed in cells in standard assays (; Fig. S2C), they are separated when cells are migrating on the stamped fibronectin (see , arrowheads; Fig. S2D). Altogether, our approach shows that contacts can be segregated by using stampcils. We are currently using this setup to fully characterize the signaling proteins involved in phase separation.
Figure 3. The stampcil segregates the focal adhesions/hemidesmosomes adhesions and the actin/intermediate filaments cytoskeletons. (A) The stampcil and the HaCaT cells were removed. Laminin (green) was stained directly on the coverslip stamped with fibronectine-rhodamine (red). Scale bar: 20 µm. (B) HaCaT cells were left to migrate for 4 h after wounding the monolayer and then fixed and stained for hemidesmosomes (ITGA6, green) and focal adhesions (paxillin, red). Scale bar: 10 µm. (C) After removing the stampcil, cells were left to migrate for 1 h and then fixed and stained for hemidesmosomes (ITGA6, green) and focal adhesions (vinculin, red). Scale bar: 10 µm. The dashed line indicates the FN border. (D) After wounding the monolayer, cells were left to migrate for 4 h and then fixed and stained for actin (phalloidin, green) and intermediate filaments (keratin 14, red). Scale bar: 10 µm. (E) After removing the stampcil, cells were left to migrate for one hour fixed and stained for actin (phalloidin, green) and intermediate filaments (keratin 14, red). The dashed line indicates the fibronectin border. White arrowheads indicate actin (lower) and intermediate filaments (upper) segregation. Scale bar: 10 µm.
This forced separation provides a simple mechanism for explaining the formation of a cellular adhesion structure. Protrusions are sent by the cell, and when the front reaches the surface, a transmembraneous protein is captured by the grafted ligand. Such a simple lock-and-key interaction suggests that cells may be probing their environments through this search and capture being filtered out by the recognition of ligands. The cytoskeleton and the other adhesive structures seem to follow then this first binding event. Even though such a mechanism is expected, our study demonstrates that the phenomenon is at play in migrating cells. It is worth noting that this approach is distinct from experiments where adhesions such as focal contacts and adherens junctions are separated:Citation23,Citation24 our contacts are naturally physically mixed and we triggered their separations through a new approach combining induced motility and specific adhesions. In addition, other approaches would allow the separation of entangled contacts by suppressing contacts targeting integrins through siRNA or by blocking contact formation through antibodies. However, such approaches would not promote a local control provided by our stampcil approach, which allows normal entanglement within cells.
In addition, this approach could be extended to any combinations of contact types, because it relies on the proven formation of biological organelles upon stamping of a variety of proteins.Citation25 The key feature is just to ink the stampcil with a specific protein of the extracellular matrix and to match the cell type so that the expected contacts can be formed. This could also work for studying adherens junctions by inking cadherins on the stampcils: it would allow us to study the interplay between focal contacts and cell-cell contacts on planar surfaces. Other developments such as imposing a topography on the stamped regions of the stampcil could generate also constraints on the migration with potential applications. Further studies will demonstrate the extent of general applicability of our method. Finally, because the mechanism of segregation is based on a specific ligand-receptor recognition, the separation of adhesions and cytoskeletons should be observed on any cell lines.
If phase separation in vesicles is often associated with interaction energies between lipids, segregation of adhesion structures relies on other rules. The binding of the ligands to the associated receptors dictates the nature of the future cellular structure. Our study illustrates this rule: cells migrate and the exclusive binding of integrin to fibronectin triggers only one type of contacts to be assembled. The induced migration is needed since these cells are not migrating without free space being generated (either by scratch or by the stampcil removal), and the saturation of the new zone under exploration allows unique assembly of the cellular structure. We anticipate that such simple rules for phase separation in cells should have broad applications in a variety of situations in vivo and in vitro.
Materials and Methods
Chemicals and reagents
Poly(dimethylsiloxane) (PDMS) (Sylgard 184) was obtained from Dow Corning; SU-8 2025 and SU-8 developer were purchased from Microchem Corp.
HaCaT cells (obtained from the German Cancer Research center, DKFZ) were cultivated in DMEM 10% fetal calf serum supplemented with gentamicin (40 µg ml−1). The following reagents were used: anti-integrin α6 (clone GoH3, BD Bioscience PharMingen), anti-vinculin (clone VIN-11-5, Sigma-Aldrich), anti-paxillin (clone Z035, Zymed), anti-keratin14 (Convance), phalloïdin Alexa Fluor 488 (Molecular Probes), anti-laminin 332 (Epiligrin, clone P3H9, Chemicon) and fibronectin-rhodamine (Cytoskeleton). All secondary antibodies were obtained from Molecular Probes.
Fabrication of the stampcil
The stampcil consisted of an array of 10 × 10 square-shaped wells of 900 × 900 μm2 in internal area with a separation between wells of 100 μm. It was fabricated using PDMS and standard soft-lithography techniques. First, a photolithography mask was designed with a CAD software (AutoCAD, Autodesk, Inc.) and purchased from Selba SA. A mold of SU-8 photoresist (MicroChem Corp.), 200 μm in height, was fabricated by first spin-coating SU-8 photoresist onto a silicon wafer (Siltronix) and patterned by UV exposure (MJB3 contact mask aligner; SUSS MicroTec). Finally, the mold was developed by using SU-8 developer (MicroChem Corp.) (). Then, a 1:10 (w/w) mixture of cross-linker and Sylgard 184 silicone elastomer (Dow Corning Corp) was degassed under vacuum and a small drop poured onto the SU-8 template. A thin film (125 μm) of polystyrene (Goodfellow) was placed onto the PDMS drop and carefully pressed against it in a custom-made four-screw metal press (). Finally, the whole set-up was cured at 65°C for 4 h. The stampcil was then carefully peeled off ( and ) and UV-sterilized prior to use.
Scratch assay
Cells were seeded on a 12 mm coverslip. The confluent monolayer was scratched using a blunt glass microneedle. Cells were left to migrate for 2 to 4 h and fixed.
Stampcil assay
The stampcil was placed face-up onto a parafilm sheet and a PDMS frame matching its external dimensions was deposited on top of it thereby forming a cavity (see Fig. S1). The stamping side of the stampcil was then incubated for 1 h with fibronectin-rhodamine (10 µg ml−1) and dried for 30 min (). Fibronectin was stamped on a 18 × 18 mm2 coverslip placed into a Petri dish by applying pressure for 10 min ().
Cells were then seeded into the cavities of the stampcil and incubated for 2 h at 37°C in 5% CO2 (). Medium was then added into the dish holding the coverslip and cells were left to grow for 2–3 d. Then, the stampcil was removed and the medium was replaced with fresh medium supplemented with 1% BSA. Cells were left to migrate for 1 h and fixed (). Note that migration time was lower compared with the scratching assay, because in the latter case the first row of cells needed more time to recover from the wound and migrate through the layer of dead cells whereas cells started to migrate immediately after the stampcil release. No major difference was observed between 1 h and 4 h when using both assays.
Immunofluorescence
Cells were fixed in paraformaldehyde 4% in PBS for 10 min, permeabilized with 0.2% Triton in PBS for 10 min and blocked with sodium borohydride (0.5 mg ml−1) in PBS for 10 min. Treated cells on coverslips were then incubated for 1 h with primary antibodies, washed in PBS and incubated for 30 min with secondary antibodies. Finally, coverslips were washed and mounted in Mowiol-DABCO buffer. All images were acquired on a Leica TCS SP2 confocal microscope using a 63× NA 1.4 Plan Apochromat HCX oil objective controlled by the LCS software (Leica Microsystems).
Conclusions
Using the stampcil method, we showed that a segregation of different types of contacts and the corresponding cytoskeletons can be induced. The grids of a stencil were used as stamps for grafting fibronectin on the surface. Keratinocytes were plated within its cavities and allowed to migrate on the fibronectin surface. A separation of distinct adhesion complexes (focal contacts and hemidesmosomes) and associated cytoskeleton (actin and intermediate filaments of keratins) was observed, in contrast with conventional cell migration methods, such as scratching-assay where no separation was observed. This micro-stencil printing method should provide new insights into cell-contact composition and dynamics. Future studies will be instrumental in assessing the benefit of using this controlled assay which trigger and control cell motility on controlled ligands.
| Abbreviations: | ||
| BPAG1e | = | bullous pemphigoid antigen 1e |
| CAD | = | computer-aided design |
| DABCO | = | 1,4-diazabicyclo[2.2.2]octane |
| DMEM | = | Dulbecco’s modified Eagle medium |
| FAK | = | focal adhesion kinase |
| μCP | = | microcontact printing |
| PBS | = | phosphate buffer saline |
| PDMS | = | poly(dimethylsiloxane) |
| UV | = | ultra-violet |
Additional material
Download Zip (2 MB)Acknowledgments
This work was supported by funds from the CNRS (ATIP and PIR grants), the University of Strasbourg and the ci-FRC of Strasbourg. N.O, E.G.-L. and M.L. also thank the ANR for financial support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Chaikin PM, Lubensky TC. Principles of Condensed Matter Physics. Cambridge: Cambridge University Press, 1995.
- de Gennes P-G. Scaling Concepts in Polymer Physics. Ithaca: Cornell University Press, 1979.
- Safran SA. Statistical Thermodynamics of Surfaces, Interfaces, and Membranes. Reading: Westview Press, 2003.
- Li Y, Lipowsky R, Dimova R. Membrane nanotubes induced by aqueous phase separation and stabilized by spontaneous curvature. Proc Natl Acad Sci U S A 2011; 108:4731 - 6; http://dx.doi.org/10.1073/pnas.1015892108; PMID: 21383120
- Muddana HS, Chiang HH, Butler PJ. Tuning membrane phase separation using nonlipid amphiphiles. Biophys J 2012; 102:489 - 97; http://dx.doi.org/10.1016/j.bpj.2011.12.033; PMID: 22325271
- Tsafrir I, Caspi Y, Guedeau-Boudeville M-A, Arzi T, Stavans J. Budding and tubulation in highly oblate vesicles by anchored amphiphilic molecules. Phys Rev Lett 2003; 91:138102; http://dx.doi.org/10.1103/PhysRevLett.91.138102; PMID: 14525338
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol 2001; 13:584 - 92; http://dx.doi.org/10.1016/S0955-0674(00)00255-6; PMID: 11544027
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, et al. Nanoscale architecture of integrin-based cell adhesions. Nature 2010; 468:580 - 4; http://dx.doi.org/10.1038/nature09621; PMID: 21107430
- Wolfenson H, Henis YI, Geiger B, Bershadsky AD. The heel and toe of the cell’s foot: a multifaceted approach for understanding the structure and dynamics of focal adhesions. Cell Motil Cytoskeleton 2009; 66:1017 - 29; http://dx.doi.org/10.1002/cm.20410; PMID: 19598236
- Zhang H, Labouesse M. The making of hemidesmosome structures in vivo. Dev Dyn 2010; 239:1465 - 76; PMID: 20205195
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science 2003; 302:1704 - 9; http://dx.doi.org/10.1126/science.1092053; PMID: 14657486
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008; 453:314 - 21; http://dx.doi.org/10.1038/nature07039; PMID: 18480812
- Loeb L. Wound healing in experimental (Ceilfibrin) tissue. Science 1919; 50:502 - 4; http://dx.doi.org/10.1126/science.50.1300.502; PMID: 17806318
- Osmani N, Peglion F, Chavrier P, Etienne-Manneville S. Cdc42 localization and cell polarity depend on membrane traffic. J Cell Biol 2010; 191:1261 - 9; http://dx.doi.org/10.1083/jcb.201003091; PMID: 21173111
- Rodriguez LG, Wu X, Guan JL. Wound-healing assay. Methods Mol Biol 2005; 294:23 - 9; PMID: 15576902
- Osmani N, Vitale N, Borg J-P, Etienne-Manneville S. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr Biol 2006; 16:2395 - 405; http://dx.doi.org/10.1016/j.cub.2006.10.026; PMID: 17081755
- Folch A, Jo B-H, Hurtado O, Beebe DJ, Toner M. Microfabricated elastomeric stencils for micropatterning cell cultures. J Biomed Mater Res 2000; 52:346 - 53; http://dx.doi.org/10.1002/1097-4636(200011)52:2<346::AID-JBM14>3.0.CO;2-H; PMID: 10951374
- Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, et al. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci U S A 2007; 104:15988 - 93; http://dx.doi.org/10.1073/pnas.0705062104; PMID: 17905871
- Weibel DB, Diluzio WR, Whitesides GM. Microfabrication meets microbiology. Nat Rev Microbiol 2007; 5:209 - 18; http://dx.doi.org/10.1038/nrmicro1616; PMID: 17304250
- Caballero D, Samitier J, Bausells J, Errachid A. Direct patterning of anti-human serum albumin antibodies on aldehyde-terminated silicon nitride surfaces for HSA protein detection. Small 2009; 5:1531 - 4; http://dx.doi.org/10.1002/smll.200801735; PMID: 19296562
- Kumar A, Whitesides GM. Features of gold having micrometer to centimeter dimensions can be formed through a combination of stamping with an elastomeric stamp and an alkanethiol “ink” followed by chemical etching. Appl Phys Lett 1993; 63:2002 - 4; http://dx.doi.org/10.1063/1.110628
- Théry M, Pépin A, Dressaire E, Chen Y, Bornens M. Cell distribution of stress fibres in response to the geometry of the adhesive environment. Cell Motil Cytoskeleton 2006; 63:341 - 55; http://dx.doi.org/10.1002/cm.20126; PMID: 16550544
- Borghi N, Lowndes M, Maruthamuthu V, Gardel ML, Nelson WJ. Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proc Natl Acad Sci U S A 2010; 107:13324 - 9; http://dx.doi.org/10.1073/pnas.1002662107; PMID: 20566866
- Tsai J, Kam L. Rigidity-dependent cross talk between integrin and cadherin signaling. Biophys J 2009; 96:L39 - 41; http://dx.doi.org/10.1016/j.bpj.2009.01.005; PMID: 19289031
- Reymann A-C, Boujemaa-Paterski R, Martiel J-L, Guérin C, Cao W, Chin HF, et al. Actin network architecture can determine myosin motor activity. Science 2012; 336:1310 - 4; http://dx.doi.org/10.1126/science.1221708; PMID: 22679097