Abstract
Procaspase-8, the zymogen form of the apoptosis-initiator caspase-8, undergoes phosphorylation following integrin-mediated cell attachment to an extracellular matrix substrate. Concordant with cell attachment to fibronectin, a population of procaspase-8 becomes associated with a peripheral insoluble compartment that includes focal complexes and lamellar microfilaments. Phosphorylation of procaspase-8 both impairs its maturation to the proapoptotic form and can promote cell migration. Here we show that the cytoskeletal adaptor protein CrkL promotes caspase-8 recruitment to the peripheral spreading edge of cells, and that the catalytic domain of caspase-8 directly interacts with the SH2 domain of CrkL. We show that the interaction is abolished by shRNA-mediated silencing of Src, in Src-deficient MEFs, and by pharmacologic inhibitors of the kinase. The results provide insight into how tyrosine kinases may act to coordinate the suppression caspase-8 mediated apoptosis, while promoting cell invasion.
Introduction
Tumor cell invasion of, and survival within, diverse microenvironments is a key feature of aggressive metastasis. Anoikis, or cell death triggered by lack of an appropriate extracellular matrix, opposes tumor metastasis by promoting cell death as tumor cells invade new tissues. Integrin-mediated death is a subtype of anoikis mediated by caspase-8, an apical protease in the “extrinsic” apoptosis cascade.Citation1-Citation3 Procaspase-8 typically matures to caspase-8 via recruitment to ligated death receptor complexes, undergoing auto-processing in a concentration-dependent, or “induced proximity” model.Citation4 The mechanisms by which integrins, cell adhesion receptors for the extracellular matrix, can recruit or activate procaspase-8 are unknown. Recruitment does not appear to require death receptors,Citation1 but may involve interaction with other integrin-associated components.
(Pro)caspase-8 functions in roles other than apoptosis. Mice deficient for caspase-8 are nonviable, demonstrating numerous embryonic cardiac and neural deficiencies.Citation5 Survival is mostly rescued by a secondary knockout of RIPK3, implicating caspase-8 as a suppressor of RIPK3-induced cellular necrosis during some critical step in embryo survival.Citation6,Citation7 However, additional roles have been shown by our lab and others, including influencing cellular differentiation, altering endosome trafficking, and enhancing motility and attachment to the extracellular matrix.Citation8-Citation14 Molecular toggling between apoptotic and migratory roles can switch caspase-8 from anti-metastatic to pro-metastatic function,Citation2,Citation3,Citation13 and this may account, in part, for the continued expression of caspase-8 in many carcinoma.Citation15 Several mechanistically linked processes are implicated in caspase-8 enhanced cell migration, including the enhancement of tyrosine kinase receptor signaling, the promotion of calpain and Rac1 activity, alterations to integrin trafficking, and crosstalk with the PI3 kinase pathway.Citation3,Citation9,Citation13 Proteolytic function, which is necessary for apoptosis and the suppression of RIPK3-mediated necrosis, is not required for the regulation of cell migration.Citation2,Citation13
The activity of the tyrosine kinase c-Src promotes cell invasion, and Src family kinase activity may therefore be required for procaspase-8-enhanced cellular migration. Src phosphorylates, and can directly associate with, procaspase-8 as well as other cellular “migration machinery” proteins such as integrins, focal adhesion kinase (FAK), and the cytoskeletal adaptor protein p130CAS.Citation3,Citation11 The latter was first identified as a Crk-associated substrate of Src, and both Crk and the related CrkL genes encode cytosolic adaptor proteins. Crk encodes the protein products Crk and CrkII, each bearing an Src-homology 2 domain (SH2) and one or two Src homology 3 (SH3) domains, respectively. CrkL (Crk-like) has a similar primary structure to CrkII, with an SH2 domain and two SH3 domains. Crk family proteins perform diverse cellular functions, including regulation of cell survival and migration.Citation16 Interestingly, Crk and Crkl knockouts exhibit cardiovascular and neural crest defects, resulting in embryonic lethality.Citation17,Citation18 Here, we provide evidence that caspase-8 interacts with the SH2 domain of CrkL in a Src- and adhesion-dependent manner, and that this interaction promotes cellular migration.
Results
Caspase-8 interacts with CrkL SH2 domain
We noted the de novo phosphorylation of several proteins, selectively in caspase-8 expressing cells, following cell adhesion to fibronectin substrates. These included a phosphoprotein at ~37 kDa (). To determine whether the phosphoprotein might be part of a complex associated with the caspase, we performed caspase-8 immunoprecipitations, resolved the proteins and probed the precipitates with a panel of cytoskeletal and apoptotic antibodies, including one that recognized Crk family proteins. A Crk-reactive protein was seen to interact with procaspase-8, and this interaction required cellular attachment to a fibronectin substrate (), but did not occur in cells maintained in suspension (first lane). Other phosphoproteins co-precipitated in the procaspase-8 containing complex, including the Crk-interacting protein p130CASCitation16 (). Immunoprecipitation using the monoclonal antibody to Crk family proteins reciprocally revealed the presence of p130CAS and procaspase-8 in the precipitate. The results confirmed the presence of these proteins in a common complex ().
Figure 1. Identification of an interaction between Caspase-8 and Crk proteins. (A) Serum starved NB7 (Casp8−/−) and NB7C8 (Casp8 reconstituted) cells were trypsinized and held in suspension (Susp) or allowed to attach to fibronectin for 10, 30, or 60 min and lysates probed with indicated antibodies. (B) NB7C8 cells were subjected to immunoprecipitation with antisera to caspase-8 after spreading on a fibronectin substrate for 30 min (FN), or after being held in suspension (Susp) and probed for indicated proteins. (C) Similarly treated NB7C8 cells were subjected to Crk protein-immunoprecipitation and immunoblotting for the proteins indicated. NB7 cells with genetic deletion of caspase-8 were used as controls to clearly differentiate the procaspase-8 band from precipitating Ig. (D) Immobilized recombinant SH2 domains of Crk-I/II, CrkL or Syk were probed with recombinant caspase-8 catalytic domain treated with recombinant Src kinase (phosphocasp-8) or not (casp-8) (mean ± SE, *P < 0.01). (E) Recombinant SH2 domains of Crk-II and CrkL were used in pulldown assays, and the precipitants resolved and probed for indicated proteins. (F) Lysates prepared as in (A) expressing control shRNA or shRNA against CrkL. (G) NB7 or NB7C8 cells expressing CrkL shRNA or control shRNA were analyzed for their ability to migrate in a wound assay for four hours (n = 40/group, mean ± SE, *P < 0.01).
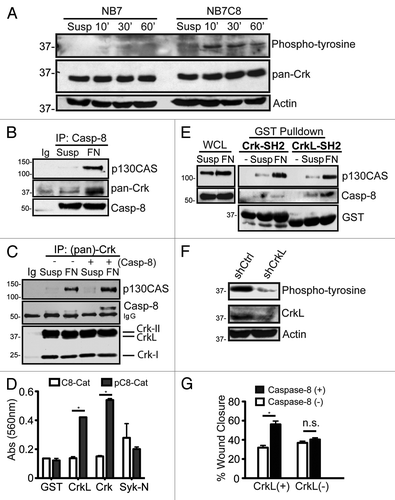
It remained unclear which specific Crk protein(s) associated with procaspase-8. Crk proteins are comprised of one or two SH3 domains and an SH2 domain.Citation16 Notably, some SH2 domains have been shown to interact with the catalytic domain of procaspase-8Citation9,Citation11,Citation13 suggesting that the observed interaction might be direct. To test this, we generated GST-fused SH2 domains of CrkL and Crk-I/II, and evaluated their interaction with recombinant caspase-8 catalytic domain using a modified ELISA assay. Tyrosine phosphorylation of caspase-8 has been previously reported to be required for SH2 binding,Citation9,Citation19,Citation20 therefore, both phosphorylated and untreated recombinant forms of the caspase-8 catalytic domain were evaluated for interaction with the Crk fusion proteins. As shown, the SH2 domains of both Crk and CrkL interacted selectively with phosphocaspase-8, but not with GST alone or a non-Crk SH2 domain (the N-terminal SH2 domain of Syk) ().
Next, we tested whether the SH2 domain of Crk or CrkL interacted with cellular forms of procaspase-8. Pull-down assays were performed using the GST-SH2 domains of Crk and CrkL. Both SH2 domains were capable of co-precipitating a complex containing p130CAS, selectively following cellular adhesion to fibronectin (), a result that was consistent with the “adhesion-dependent” formation of the “p130CAS-Crk” complex.Citation21 In contrast to the biochemical interactions observed, however, procaspase-8 was co-precipitated selectively when CrkL, but not Crk, was used for pulldowns (). The data suggest that the SH2 domain of CrkL, but not Crk, preferentially interacts with procaspase-8 during cell adhesion. Since both Crk and CrkL form complexes with p130CAS, it further suggested that caspase-8 does not interact indiscriminately with complexes containing p130CAS following cell attachment to fibronectin. Suppression of CrkL expression reduced the abundance of the phosphoprotein at 37kDa, suggesting that in live cells, CrkL selectively interacted with a procaspase-8 containing complex in response to signaling events associated with cell adhesion ().
Caspase-8 enhanced migration requires CrkL
Caspase-8 can promote cell migration 20–40% following recruitment to the leading edge of migrating cells. We next tested whether CrkL was involved in regulating caspase-8-influenced cell motility. Cells were allowed to attach to fibronectin substrates as confluent monolayers, the monolayers wounded, allowing cells to migrate into the empty space. The migration rate of the cells was determined, revealing that CrkL appeared to be required for caspase-8 to enhance cell migration (). Suppression of CrkL did not dramatically impact cell proliferation in long-term culture (Fig. S1), suggesting that the effects on wound closure (over 4 h) resulted directly by impacting cell migration.
CrkL mediates procaspase-8 recruitment to the cell periphery and to focal adhesions
Procaspase-8 recruitment to the cellular periphery is required for caspase-8 enhancement of cell migration and invasiveness.Citation3,Citation10,Citation11 We therefore considered that CrkL localization to the periphery of cells migratingCitation21 or spreading on a fibronectin substrate, might similarly be critical to this event. CrkL was found to colocalize with procaspase-8 (Fig. S2A), possibly reflecting the biochemical complex identified (). To evaluate whether CrkL and procaspase-8 recruitment to the cell periphery were associated, we next characterized procaspase-8 recruitment during cell spreading in cells expressing, or deficient, in CrkL. Suppression of CrkL expression attenuated procaspase-8 recruitment to the cell periphery (). By contrast, lack of caspase-8 did not suppress CrkL recruitment to the periphery (Fig. S2B). Suppression of the related protein, Crk, did not impact caspase-8 localization in the periphery (Fig. S2C). Together, the results support the notion that CrkL, but not Crk, interacts with cellular procaspase-8 and aids in its recruitment, while procaspase-8 is not required for recruitment (or stabilization) of CrkL in the cell periphery.
Figure 2. CrkL promotes recruitment of caspase-8 to the periphery. (A) NB7C8 cells expressing CrkL shRNA or control shRNA were allowed to spread on fibronectin coated coverslips, then fixed cells were stained for procaspase-8 and with phalloidin to identify actin-rich adhesion ruffles, then imaged by confocal microscopy. Representative images are shown. Scale bars indicate 10 μm. (B) Recruitment of caspase-8 to adhesion ruffles was evaluated (n = 20/group, mean ± SE, *P < 0.01). (C) NB7C8 cells were allowed to attach to fibronectin-coated plates for 30 min before sequential isolation of cytosolic or focal adhesion complex containing fractions and immunoblot analysis. Bars indicate ratio of caspase-8 to FAK (C8:FAK) as normalized to “control” shRNA, as determined by densitometry via ImageJ software from NIH (n = 2/group, mean ± SE, *P < 0.05).
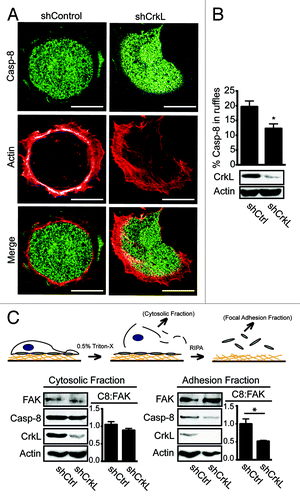
A fraction of total cellular procaspase-8 associates with an insoluble focal complex formed during cell spreading and migration.Citation3,Citation13 To evaluate the effect of CrkL suppression on the ability of caspase-8 to localize to the insoluble compartment, cellular fractions enriched in cytosolic, or focal adhesion proteins, were isolated from cells expressing, or deficient, in CrkL. The presence of procaspase-8 in each fraction was assessed. Suppression of CrkL expression did not compromise the expression of FAK or its recruitment to the FA/cytoskeletal fraction. Rather, there was a trend of increased FAK content in the FA/cytoskeletal fraction relative to control cells. The abundance of actin, however, appeared somewhat decreased in the CrkL-deficient cells, consistent with reported roles of Crk proteins in Rac activation and actin remodeling.Citation16 The abundance of procaspase-8 was similarly decreased in the FA/cytoskeletal enriched fractions deficient in CrkL, although procaspase-8 was not detectably decreased in the cytosolic fractions (). The results were consistent with the notion that CrkL promotes caspase-8 recruitment to the peripheral cytoskeleton, and/or to focal adhesions, following integrin-medated attachment to a fibronectin substrate.
Src activity is critical for CrkL SH2–procaspase-8 interaction
The Src kinases phosphorylate several focal adhesion complex proteins, including FAK, CrkL, p130CASCitation16,Citation21,Citation22 and procaspase-8.Citation19 Src kinase activity is enhanced following fibronectin attachment. Nano-molar concentrations of the Abl and Src-family tyrosine kinase inhibitor dasatinib block this activity (). By contrast, dasatinib displayed no effect on fibronectin-induced FAK activation in these studies (). Dasatinib treatment also prevented co-precipitation of procaspase-8 by the SH2 domain of CrkL (). The data are consistent with a role for Src family kinases, rather than FAK, to act as critical signaling elements promoting the association of CrkL with procaspase-8. The result was somewhat unexpected, given our prior implication of FAK in promoting procaspase-8 recruitment to the cell periphery.Citation3
Figure 3. Role of Src kinase in the caspase-8-CrkL interaction. (A) Serum-starved NB7C8 cells were treated with increasing concentrations of dasatinib and allowed to attach to a fibronectin substrate for 30 min. (B) Lysates of adherent NB7C8 cells were prepared as in (A) with or without 30 nM dasatinib treatment and assessed for changes in activity and expression of Src and FAK. (C) Lysates of adherent NB7C8 cells were prepared as in (A) with or without 30 nM dasatinib treatment and probed with recombinant CrkL SH2 domain to assess conditional ability to bind caspase-8. (D) Murine embryonic fibroblasts (MEF) with indicated genetic deletions were allowed to attach to a fibronectin substrate and whole cell lysate (WCL) probed for indicated proteins, or with recombinant CrkL SH2 domain, which was then assessed for ability to interact with indicated proteins. +, FAK-GFP reconstitution. (E) NB7C8 cells expressing c-Src shRNA or control shRNA were allowed to spread on fibronectin-coated coverslips, and lysates probed with recombinant CrkL SH2 domain to assess interaction with indicated proteins.
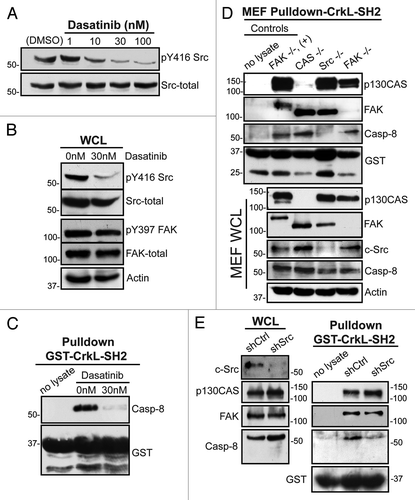
Since pharmacologic inhibition may exert pleiotropic or off-target effects, we further tested a role for Src using murine embryo fibroblasts (MEF) genetically deficient for c-Src, FAK or p130CAS. We again allowed cells to attach to fibronectin, created cell lysate, and performed pull-down assays to evaluate respective roles in procaspase-8 association with CrkL. Both FAK and p130CAS were found to be co-precipitated in complex with CrkL–SH2. Although decreased procaspase-8 may co-precipitate in FAK-deficient MEFs, neither FAK nor p130CAS appeared essential for co-precipitation of procaspase-8 by CrkL–SH2. By contrast, Src-deficient cells lacked association of caspase-8 with the CrkL–SH2, suggesting a key role for Src ().
To complement these genetic and pharmacologic approaches, we also performed coprecipitaion studies following shRNA-mediated suppression of c-Src in the neuroblastoma cells. Consistent with the prior observations, silencing of Src expression compromised association of procaspase-8 with CrkL, further implicating fibronectin-induced Src activation in CrkL-procaspase-8 association ().
Caspase-8 expression and CrkL depletion independently promote apoptosis
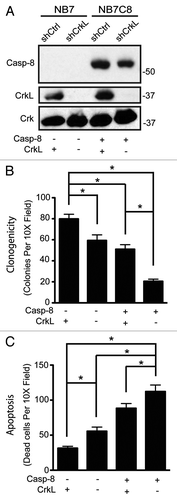
Successful metastasis is enhanced by cellular abilities to adhere, migrate and survive. Since caspase-8, FAK, p130CAS, and Crk proteins influence cell survival,Citation3,Citation4,Citation9,Citation11,Citation13,Citation23 it was tempting to speculate that CrkL may be a master regulator of caspase-8. To test whether caspase-8 influenced cell survival in a CrkL-dependent manner, we evaluated the clonogenicity of tumor cells expressing CrkL shRNA (or control shRNA) as a function of caspase-8 expression (). Suspended in soft agar, cells which expressed caspase-8 and CrkL together yielded an intermediate level of colony formation, with ~50 colonies observed per low power field. Those expressing caspase-8 but lacking CrkL yielded only ~20 colonies per field (), supporting the concept that CrkL was a key pro-survival element. However, the impact of CrkL expression extended beyond caspase-8, as cells deficient in caspase-8, but expressing CrkL, were most advantaged and formed 70–80 colonies. The results support a model in which the effects of caspase-8 and CrkL on anchorage-independent survival are not co-dependent, but additive with each other, with CrkL as a positive element and caspase-8 as a deleterious element. To evaluate whether this was, in fact, due to differences in cell survival, rather than proliferation (Fig. S1), cells were seeded in a three-dimensional collagen matrix to trigger anoikis via integrin signaling.Citation1,Citation2 Viability was assessed after 72 h; as shown, the expression of caspase-8 was associated with increasing rates of dead cells, which corresponded to decreasing colony formation in the clonogenic assay (). Altogether, our results reveal CrkL as both a mediator of caspase-8 enhanced cell migration and as a promoter of survival in caspase-8 expressing cells.
Figure 4. CrkL and caspase-8 independently affect survival of cells in suspension conditions. (A) Whole cell lysates from NB7 or NB7C8 cells expressing CrkL shRNA or control shRNA were probed with antibodies against indicated proteins. Cells plated in 0.3% soft agar and grown for 14 d. Clonogenic ability assessed (n = 12/group, mean ± SE, *P < 0.01). (B) Cells from (A) were suspended in collagen droplets and apoptotic cells counted 72 h later (n = 12/group, mean ± SE, *P < 0.01).
Discussion
Caspase-8 triggers cell death in a context-dependent manner. In response to death receptor ligands, metabolic miscues, or an incorrect ECM microenvironment, the recruitment of procaspase-8 to key initiator sites in the cell results in maturation and induction of the apoptotic cascade.Citation2-Citation4,Citation9 However, the contextual clues that direct caspase-8 to its different roles remain poorly understood. In this study, we implicate the cytoskeletal “adaptor” protein CrkL in caspase-8 localization to peripheral lamella.
Src phosphorylation of the caspase-8 catalytic domain significantly enhanced interaction with the CrkL SH2 domain in vitro. The SH2 domain of the related protein Crk similarly interacted with phosphocaspase-8 in vitro, yet was unable to precipitate procaspase-8 from cell lysates following adhesion to fibronectin substrates. It is not yet clear why, but the differences may reflect changes in the accessibility of the binding site on procaspase-8 in living cells. Alternatively, the caspase-8 catalytic domain may be phosphorylated on additional “non-physiologic” tyrosine residues in vitro, which may provide additional binding sites not induced by fibronectin adhesion in live cells.
CrkL and caspase-8 influenced cell survival in opposing manners in “anoikis” type apoptosis assays, but were additive in cell migration. The results are consistent with context dependent roles for caspase-8, since phosphorylation of caspase-8 both inhibits apoptosisCitation19,Citation24 and enhances cell invasion.Citation11 At, or near, the focal adhesion, caspase-8 may activate calpain 2 as well as the small GTPases Rab5 and Rac1.Citation3,Citation13 Indeed, Rab5 activity appears to enhance focal adhesion turnover (in review). Our current study supports a model of modulated procaspase-8 interaction with CrkL that mediates recruitment to, or simply retention within, the periphery, that is regulated by tyrosine kinase (Src) signaling.
The capacity for caspase-8 to promote migration may explain why many epithelial tumors maintain expression of this potentially harmful protein.Citation15 It is also possible that procaspase-8 expression is retained to suppress necrosis, through its capacity to cleave RIPK3. The dis-regulated activity of Src or other tyrosine kinases commonly observed in tumors may be sufficient to suppress caspase-8 proapoptotic functions. While only a fraction of total caspase-8 is phosphorylated, it is evident that key, small compartmentalized pools of caspase-8 are sufficient for killing.Citation25
In this regard, the inhibition of Src may enhance apoptosis induced by death receptors. Dasatinib, an FDA-approved Abl tyrosine kinase inhibitor, is also an excellent Src kinase inhibitor. It is possible that the use of this class of drug may ameliorate the pro-invasive actions of Src, while enhancing the apoptotic capacity of caspase-8 in response to local inflammatory signals or chemotherapies such as TRAIL. An increased understanding of these molecular interactions may be particularly relevant to the clinic: as one considers the coming impact of genomics and transcriptomics to personalized medicine, the capacity to interpret roles for cellular effectors such as caspase-8 and CrkL will be of increasing value.
Materials and Methods
Immunoprecipitation and immunoblot analysis
Performed as described previously.Citation11
Cell culture
Human neuroblastoma NB7 (CASP8−/−), NB7C8 (NB7 with CASP8 stably re-introduced), human lung carcinoma A549 cell lines, and murine embryonic fibroblasts (MEF) were described and cultured as previously.Citation2,Citation3 SF-9 cells were cultured in BacVector Insect Medium from Novagen. Knockdown of caspase-8 or CrkL used lentivirus (Open Biosystems) and confirmed by immunoblot with specific antibodies below. Control shRNA cells were infected with a lentivirus encoding a nonspecific shRNA sequence (plasmid 1864; Addgene). Knockown was maintained with puromycin (1 μg/ml).
Materials
Polyclonal anti-caspase-8 (559932) monoclonal anti-caspase-8 (551242) monoclonal anti-p130CAS (610272) monoclonal anti-FAK (610082) and monoclonal anti-Crk (610036) and monoclonal anti-Crk-L (551242) antibodies were obtained from BD Bioscience. The monoclonal anti-phosphotyrosine (clone 4G10, 05-321) was obtained from Millipore. The monoclonal anti-Src (2102) polyclonal anti-GST (2622S) and rabbit polyclonal anti-p-Src (P-Y416, V2101) were from Cell Signaling. Rabbit polyclonal anti-p-FAK (p-Tyr 397; 44-625G) from BioSource International Antibody. Anti-actin (clone AC-15, 5441) from Sigma. Goat anti-rabbit and goat anti-mouse antibodies coupled to horseradish peroxidase were from Bio-Rad. Alexa Fluor® 488- and Alexa Fluor® 555-labeled secondary antibodies, PMSF, Complete “mini” protease inhibitor from Roche Diagnostics. The c-Src inhibitor dasatinib was obtained from Selleck Chemicals (BMS-354825). Geneticin® (G418 sulfate, 11811) was from Invitrogen. Puromycin (P8833) and fibronectin from bovine plasma (F1141) are from Sigma. Glutathione-sepharose™ 4B was from GE Healthcare. Chemiluminescent substrate (34080) and protein A/G beads were from Pierce.
Plasmids
CrkL and Crk-II plasmid were a kind gift from Dr. Richard Klemke. The SH2 domains of CrkL or Crk-II were subcloned into pGEX-6P-1 vector (GE Healthcare) by PCR using the primers 5′-CCCGGATCCA TGGCGGGCAA CTTCGACTCG-3′ and 5′-CGGTCGACTC ATGATCTGGA AACTGGTTCT AT-3′ for and 5′-CCCGGATCCA TGTCCTCCGC CAGGTTCGAC-3′ and 5′-CGGTCGACTC AATACCTGGG CGCAGGCTCG AT-3′ for Crk-II and CrkL respectively containing the restriction sites BamHI/SalI. Tagless caspase-8 catalytic domain was purified as described previously.Citation24
Bead-bound SH2-Crk-L or SH2-Crk-II pulldown assay
Serum starved cells were placed in suspension or plated into fibronectin coated plates (2 μg/ml) before being lysed in radioimmuno-precipitation assay (RIPA) buffer as described previously.Citation11 One thousand milligrams of extracts were probed with 25 μl GSH beads were pre-coated with 20 μg of GST-SH2-CrkL or GST-SH2-Crk-II for 2 h at 4 °C. Pulldowns were incubated overnight in a rotating shaker at 4 °C before washing and analysis by immunoblot.
Immunofluorescence studies
Cells were plated at sub-confluence on 2 μg/ml fibronectin pre-coated glass coverslips and allowed to spread for 30 min. After rinsing with phosphate buffered saline (PBS), cells were fixed in PBS plus 4% paraformaldehyde for 10 min, permeabilized in PBS plus 0.1% Nonidet-P-40 for 2 min and blocked at room temperature for 90 min in 3% bovine serum albumin (BSA) in PBS. Cells were stained with polyclonal antibody to procaspase-8 (BD Biosciences) (1:100) or mouse anti-CrkL antibody (BD Biosciences) (1:100) for 2 h. After multiple PBS/BSA washes, cells were exposed to respective Alexa Fluor conjugated secondary antibodies (1:250). In some cases, cells were co-incubated with Alexa Fluor Phalloidin to stain fibrillar actin (1:70). Samples were then washed and mounted onto slides with the Vectashield Hard-Set mounting media (Vector Laboratories) and visualized on a Nikon Eclipse C1 confocal microscope with a 1.4 NA 60× oil immersion lens using minimum pinhole (30 μm). Images were captured using Nikon EZ-C1 3.50 imaging software and analyzed using NIH ImageJ software by subtracting areas of caspase-8 channel associated with actin-rich ruffles.
In vitro kinase assays
Recombinant caspase-8 catalytic domain was purifiedCitation26 and phosphorylated as described previouslyCitation24 by recombinant Src kinase in Src Reaction Buffer (20 mM PIPES pH 7.0, 10 mM MnCl2, 1 mM DTT, 1 mM ATP) for 30 min at 32 °C. Phosphorylation was detected by immunoblot analysis with anti-phosphotyrosine 4G10 antibody concurrently with anti-GST antibody to detect phosphorylation and recombinant GST-caspase-8 catalytic domain on Li-Cor Odyssey and quantified using Li-Cor software.
ELISA assays
Recombinant SH2 domains of Crk-I/II, CrkL and SYK-N (100 ul of 1 μM) were allowed to bind to 96-well ELISA plates for 2 h at room temperature. Following 2 h 37 °C blocking in 5% BSA in PBS, 100 ul of 0.1 μM of recombinant caspase-8 catalytic domain was allowed to bind for 1 h after phosphorylation by recombinant Src kinase, as described above, or control mock reaction.
Enzymes
Recombinant full-length Src kinase was induced by baculovirus expression in SF-9 cells. Cultures were harvested once viability dropped to 70% and purified using Ni-affinity chromatography as previously described.Citation27
Adhesion-dependent signaling
Cells were serum starved for 6 h, placed in suspension for 30 min at room temperature (representing the 0 time point), or plated into 10 cm non-tissue culture-treated dishes (1 × 106 per dish) precoated with fibronectin substrate (2 µg/ml). Cells were allowed to attach for 30 min and directly lysed in RIPA buffer as described previously.Citation11 Where indicated, cells were incubated in the presence of dasanitib before (10 min) and during cell adhesion. DMSO vehicle served as a control. Cell lysates were analyzed by immunoblot.
Focal adhesion enrichment and isolation
Fractionation was performed as previously described.Citation3 Cells were plated onto fibronectin-coated dishes and at 30 min adherent cells were treated with 0.5% Triton X-100 lysis buffer, yielding the cytosolic fraction. The focal adhesion enriched fraction was prepared from the remaining adhesion complexes bound on the plates, which was lysed in RIPA buffer and scraped off the dish. Lysates from the two different fractions were analyzed by immunoblotting as described previously.Citation11
Clonogenic survival assays
Cells in single cell suspension were embedded in media solidified with 0.3% agar before 14-d incubation in normal tissue culture conditions described above. Colony formation was quantified using contrast and counting algorithms from NIH ImageJ software. Collagen survival assay was performed as described previously.Citation2
Cell migration assays
Wound assay was performed as described previously.Citation3 Cells were allowed to migrate in complete media for four hours and imaged at zero and four hour time points with a Nikon Eclipse Ti bright field microscope using 10× objective. Wound closure was quantified using NIH ImageJ software.
Statistical analysis
P values of means calculated using the two-sided Student t tests. Error bars reflect standard error.
Additional material
Download Zip (236.6 KB)Acknowledgments
The authors thank Guy Salvesen and Richard Klemke for the gift of reagents. This work was supported by NIH/NCI grant CA107263 to D. Stupack and CA102310 to D. Schlaepfer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol 2001; 155:459 - 70; http://dx.doi.org/10.1083/jcb.200106070; PMID: 11684710
- Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 2006; 439:95 - 9; http://dx.doi.org/10.1038/nature04323; PMID: 16397500
- Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D, et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res 2009; 69:3755 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-08-3937; PMID: 19383910
- Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS. RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell 2011; 44:9 - 16; http://dx.doi.org/10.1016/j.molcel.2011.09.003; PMID: 21981915
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998; 9:267 - 76; http://dx.doi.org/10.1016/S1074-7613(00)80609-3; PMID: 9729047
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011; 471:368 - 72; http://dx.doi.org/10.1038/nature09857; PMID: 21368762
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471:363 - 7; http://dx.doi.org/10.1038/nature09852; PMID: 21368763
- Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Bong Kang T, et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res 2006; 66:4273 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-4183; PMID: 16618751
- Senft J, Helfer B, Frisch SM. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res 2007; 67:11505 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-5755; PMID: 18089778
- Finlay D, Vuori K. Novel noncatalytic role for caspase-8 in promoting SRC-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res 2007; 67:11704 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-07-1906; PMID: 18089800
- Barbero S, Barilà D, Mielgo A, Stagni V, Clair K, Stupack D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J Biol Chem 2008; 283:13031 - 4; http://dx.doi.org/10.1074/jbc.M800549200; PMID: 18216014
- Torres VA, Mielgo A, Barilà D, Anderson DH, Stupack D. Caspase 8 promotes peripheral localization and activation of Rab5. J Biol Chem 2008; 283:36280 - 9; http://dx.doi.org/10.1074/jbc.M805878200; PMID: 18974049
- Torres VA, Mielgo A, Barbero S, Hsiao R, Wilkins JA, Stupack DG. Rab5 mediates caspase-8-promoted cell motility and metastasis. Mol Biol Cell 2010; 21:369 - 76; http://dx.doi.org/10.1091/mbc.E09-09-0769; PMID: 19923319
- Scharner D, Rössig L, Carmona G, Chavakis E, Urbich C, Fischer A, et al. Caspase-8 is involved in neovascularization-promoting progenitor cell functions. Arterioscler Thromb Vasc Biol 2009; 29:571 - 8; http://dx.doi.org/10.1161/ATVBAHA.108.182006; PMID: 19122169
- Stupack DG. Caspase-8 as a therapeutic target in cancer. Cancer Lett 2013; 332:133 - 40; http://dx.doi.org/10.1016/j.canlet.2010.07.022; PMID: 20817393
- Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal 2009; 7:13; http://dx.doi.org/10.1186/1478-811X-7-13; PMID: 19426560
- Park TJ, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol 2006; 26:6272 - 82; http://dx.doi.org/10.1128/MCB.00472-06; PMID: 16880535
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet 2001; 27:293 - 8; http://dx.doi.org/10.1038/85855; PMID: 11242111
- Cursi S, Rufini A, Stagni V, Condò I, Matafora V, Bachi A, et al. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J 2006; 25:1895 - 905; http://dx.doi.org/10.1038/sj.emboj.7601085; PMID: 16619028
- Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem 2008; 283:5402 - 13; http://dx.doi.org/10.1074/jbc.M706462200; PMID: 18086677
- Li L, Guris DL, Okura M, Imamoto A. Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol Cell Biol 2003; 23:2883 - 92; http://dx.doi.org/10.1128/MCB.23.8.2883-2892.2003; PMID: 12665586
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest 2009; 119:252 - 66; PMID: 19147981
- Cho SY, Klemke RL. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol 2000; 149:223 - 36; http://dx.doi.org/10.1083/jcb.149.1.223; PMID: 10747099
- Keller N, Grütter MG, Zerbe O. Studies of the molecular mechanism of caspase-8 activation by solution NMR. Cell Death Differ 2010; 17:710 - 8; http://dx.doi.org/10.1038/cdd.2009.155; PMID: 19851329
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012; 287:12455 - 68; http://dx.doi.org/10.1074/jbc.M111.309104; PMID: 22362782
- Keller N, Mares J, Zerbe O, Grütter MG. Structural and biochemical studies on procaspase-8: new insights on initiator caspase activation. Structure 2009; 17:438 - 48; http://dx.doi.org/10.1016/j.str.2008.12.019; PMID: 19278658
- Wu L, Bernard-Trifilo JA, Lim Y, Lim ST, Mitra SK, Uryu S, et al. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene 2008; 27:1439 - 48; http://dx.doi.org/10.1038/sj.onc.1210770; PMID: 17828307