Abstract
Cell adhesion and migration are important determinants of homing and development of hematopoietic stem and progenitor cells (HSPCs) in bone marrow (BM) niches. The extracellular matrix protein transforming growth factor-β (TGF-β) inducible gene H3 (BIGH3) is involved in adhesion and migration, although the effect of BIGH3 is highly cell type-dependent. BIGH3 is abundantly expressed by mesenchymal stromal cells, while its expression in HSPCs is relatively low unless induced by certain BM stressors. Here, we set out to determine how BIGH3 modulates HSPC adhesion and migration. We show that primary HSPCs adhere to BIGH3-coated substrates, which is, in part, integrin-dependent. Overexpression of BIGH3 in HSPCs and HL60 cells reduced the adhesion to the substrate fibronectin in adhesion assays, which was even more profound in electrical cell-substrate impedance sensing (ECIS) assays. Accordingly, the CXCL12 induced migration over fibronectin-coated surface was reduced in BIGH3-expressing HSPCs. The integrin expression profile of HSPCs was not altered upon BIGH3 expression. Although expression of BIGH3 did not alter actin polymerization in response to CXCL12, it inhibited the PMA-induced activation of the small GTPase RAC1 as well as the phosphorylation and activation of extracellular-regulated kinases (ERKs). Reduced activation of ERK and RAC1 may be responsible for the inhibition of cell adhesion and migration by BIGH3 in HSPCs. Induced BIGH3 expression upon BM stress may contribute to the regulation of BM homeostasis.
Introduction
Cell adhesion and migration are important in many biological processes, including development, immune responses, and wound healing. Similarly, the translocation of hematopoietic stem and progenitor cells (HSPCs) between distinct bone marrow (BM) niches and the homing of HSPCs to the BM following transplantation are dependent on proper regulation of adhesion and migration.Citation1 Within the BM microenvironment, the interactions of HSPCs with extracellular matrix (ECM) components and adhesion molecules on neighboring cells are critical for the maintenance of the HSPC population. Cells of the BM niche, including mesenchymal stromal cells (MSCs), osteoblasts, and fibroblasts, produce soluble and ECM- or cell-associated proteins that modulate the adhesion and homing of HSPCs.Citation2-Citation5
Directional cell migration is induced by asymmetrically presented chemokines, whereas random cell migration, also termed cell motility, occurs when the direction of the movement is not defined. The main chemokine that induces HSPC migration is CXCL12 that signals through its G-coupled receptor CXCR4, which upon ligand-binding promotes the exchange of GDP for GTP on the G-α subunit.Citation6 Subsequently, CXCR4 associates with endolyn and the integrins VLA4 and VLA5 at the leading edge of the cell.Citation7 This induces phosphoinositol-3-kinase (PI3K) and phospholipase-C-β (PLC) activity, which respectively activate AKT and PKC-zeta, whereas PKC subsequently induces RAC1 and extracellular regulated kinase (ERK) signaling.Citation8-Citation10 These signaling pathways activate a cascade of events, including polarized signaling, integrin-mediated adhesion, and cytoskeleton remodeling, ultimately leading to migration. Whereas activated RAC1 induces polarization by its polarized localization at the cell’s front, activated ERK induces FAK activation and myosin-light-chain phosphorylation in the cytosol, thereby regulating the formation of focal adhesions and actomyosin contractility.Citation11
BM mobilizing agents, such as granulocyte-colony-stimulating-factor (G-CSF), release HSPCs into peripheral blood, by remodeling the BM-niche. Comparative gene-expression profiling of purified murine HSPCs in homeostatic and regenerative conditions identified transforming growth factor-β (TGF-β) inducible gene H3 (BIGH3) as a gene that is upregulated in regenerative conditions, 1 d post-G-CSF and/or -cyclophosphamide treatment and 6 d post-5FU treatment.Citation12,Citation13
Little is known about the function of the extracellular matrix protein BIGH3 in the hematopoietic BM microenvironment. Expression of BIGH3 is highest in MSCs, whereas expression is low but detectable in human HSPCs,Citation14 (Klamer et al. unpublished results). In vitro, BIGH3 expression is increased in HSPCs that adhere to MSCs compared with hematopoietic stem cells (HSCs) adherent to fibronectin, or to non-adherent HSPCs.Citation15
BIGH3 has been identified as a major TGF-β responsive gene in the lung adenocarcinoma cell line A549Citation16,Citation17 and is also upregulated by lyso-phosphatidic acid (LPA) in MSCs and fibroblasts.Citation18,Citation19 BIGH3 is a secreted protein detected in various human cell types.Citation16,Citation20 It is a 68-kDa protein that consists of four fasciclin-1 domains and a C-terminal arginyl-glycyl-aspartic acid (RGD) sequence, both of which can bind integrins, including αVβ3, αVβ5, α5β1, α3β1, α1β1, αMβ2, and α6β4, as reviewed.Citation21 Being one of the ECM components, BIGH3 interacts with other ECM molecules, including collagens, fibronectin, laminins, and glycosaminoglycans,Citation17 and BIGH3 can function as a linker protein in the interaction between ECM and integrins.Citation22 However, there is limited knowledge on the impact of BIGH3 on intracellular signaling during integrin-mediated adhesion and migration.
We studied the role of BIGH3 in adhesion and migration of human HSPCs and specifically on CXCL12-induced signaling. Overexpression of BIGH3 decreased adhesion and migration of hematopoietic cells. Because CXCL12-induced signaling is critically dependent on PKC activity, we used the PKC activator phorbol ester phorbol-12-myristate-13-acetate (PMA) to induce adhesion.Citation23-Citation25 Our analysis of PMA-induced signaling pathways showed decreased activation of the small GTPase RAC1 and the ERK MAP-kinases upon BIGH3 overexpression. Therefore, regulation of BIGH3 expression in HSPCs may control BM homeostasis at the level of homing and mobilization of HSPCs.
Results
Surface-coated BIGH3 supports HSPC adhesion and is partly mediate by β1- and β2-integrins
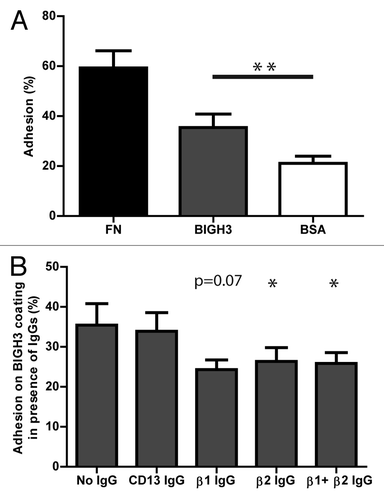
BIGH3 is expressed on the plasma membrane of stromal cells in the BM environment.Citation14,Citation26 These cells bind HSPCs and support their maintenance, but the role of BIGH3 in these processes is unknown. The adhesive capacity of human HSPCs to BIGH3 was analyzed by means of a static adhesion assay in which fibronectin and bovine serum albumin (BSA) coatings were used as positive and negative controls, respectively. On average, 35% (± 5.4%) of HSPCs adhered to the BIGH3 coating, whereas 21% (± 2.9%) adhered to BSA, and 59% (± 6.8%) to fibronectin (), which indicated that significantly more HSPCs adhered to BIGH3 as compared with BSA (P = 0.004). Pre-incubation of HSPCs with blocking antibodies directed at β1- or β2-integrins resulted in 24% (± 2.4%) and 26% (± 3.5%) adhesion, respectively. This indicates an inhibition of the adhesion by 27% and 23% and, almost reduced the adhesion to the level of aspecific binding to BSA (). Pre-incubation of HSPCs with an irrelevant antibody (anti-CD13) did not affect BIGH3-mediated adhesion. Engagement of β1- or β2-integrins appeared to be redundant, as simultaneously blocking of β1- and β2-integrins did not further increase inhibition of HSPC adhesion. These data indicate that BIGH3 supports HSPC adhesion, which is, at least in part, mediated by β1- and β2-integrins.
Figure 1. HSPC adhere to BIGH3 in static adhesion assays, dependent on β1- and β2-integrins. (A) The percentage static adhesion of MPB-derived HSPCs of six mobilized donors on plastic coated by BSA (2%), BIGH3 (10 μg/mL), or fibronectin (FN, 20 μg/mL). The percentage adhesion was calculated by the absorbance of a well relative to the absorbance of a 100% input control. (B) The percentage static adhesion of MPB-derived HSPCs on a BIGH3 (10 μg/mL) coating, in the presence of functional blocking antibodies against the indicated integrin subunits. Statistics were performed based on the percentage adhesion with the indicated antibody relative to that without antibodies. Blocking antibodies against β1- and β2-integrins inhibit the static adhesion of HSPCs to BIGH3. Shown are means ± SEM (n = 6) and each samples was performed in duplicates. * P < 0.05, ** P < 0.01.
BIGH3 is expressed and secreted by hematopoietic cells upon overexpression
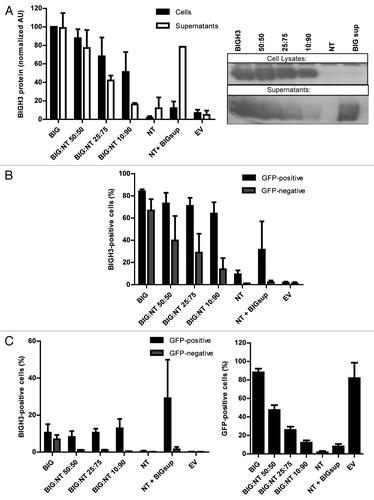
BIGH3 is highly expressed by stromal cells, whereas its expression is relatively low in HSPCs,Citation14 (Klamer et al., manuscript in preparation). Certain environmental conditions that cause BM-stress, such as chemotherapy, increase BIGH3 expression in HSPCs.Citation12 To examine the function of BIGH3 in HSPCs, we increased the expression of BIGH3 in immature hematopoietic cells. First, we used HL60 cells in which endogenous BIGH3 expression is nearly undetectable (, NT). HL60 cells were transduced with a lentiviral expression vector containing the BIGH3-IRES-GFP or a GFP control sequence, and sorted for GFP expression. BIGH3 protein expression in cell lysates and supernatants were determined by western blot (, first lane) and BIGH3 was detected in both fractions, indicating that BIGH3 is excreted. The surface and intracellular expression of BIGH3 was analyzed by flow cytometry in non-transduced (NT), BIGH3-expressing cells (BIG), and transduced control cells (EV) (). Endogenous expression of BIGH3 on HL60 cells was undetectable, whereas 82% of the transduced and sorted cells stained positive for intracellular BIGH3. Surface expression was detected on 19% of the transduced cells (). These data show that cells transduced by BIGH3-GFP effectively express BIGH3 that is partly secreted, while the cellular BIGH3 is mainly present intracellular, although a small fraction of BIGH3 is detected at the cell surface.
Figure 2. BIGH3 is secreted by cells with BIGH3 overexpression and internalized by wild-type cells. L60 cells with BIGH3 overexpression (BIG) were mixed with non-transduced (NT) cells in various ratios (50:50, 25:75, and 10:90) and co-cultured during 5 d. A condition with NT cells cultured during 5 d in conditioned medium from cells with BIGH3 overexpression (NT + BIGsup) was taken along. (A) Western blot with cell lysates and supernatants of the mixtures of HL60 cells, stained for BIGH3 (right panel, representative experiment, n = 3). An equal number of input cells were used for the cell lysates in each lane. The proteins in the medium (Supernatants) were precipitated and an amount corresponding to 2 × 105 cells was used in each lane. The lanes on Western Blot were quantified by use of ImageJ software (left panel). With decreasing fractions of cells with BIGH3 overexpression, the amount of BIGH3 in the cell lysates is disproportionably stable, while the amount of BIGH3 in the supernatants is disproportionably fast decreasing. Shown are means ± SEM (n = 3). (B and C) Flow cytometry for BIGH3 expression in the mixtures, depicted as the percentage BIGH3-positive cells in the GFP-positive fraction (cells with BIGH3 overexpression; GFP-positive) and the GFP-negative fraction (wild-type cells; GFP-negative), by intercellular staining, indicating the internalization (B) and by surface staining, indicating the surface binding (C). The cells were considered BIGH3-positive if the BIGH3 signal exceeded the background signal in empty-vector transduced control cells (EV) and NT cells, stained for BIGH3. Shown are means ± SEM (n = 3). (D) The percentage of transduced cells in the mixtures was verified by flow cytometry for GFP, and reflected the input ratios. Shown are means ± SEM (n = 3).
BIGH3 is secreted upon overexpression and internalized by wild-type cells
Cells transduced with BIGH3 express and secrete BIGH3 protein, which might be bound by non-transduced cells in co-culture. To explore this, the surface and intracellular expression of BIGH3 was analyzed in HL60 cell populations consisting of defined ratios of NT and BIGH3-expressing (GFP-positive sorted) cells. In addition, we added BIGH3-conditioned medium to NT cells. The percentage of GFP-positive cells faithfully represented the ratio of BIGH3-transduced cells as shown by flowcytometry (, first bars). Furthermore, the NT cells, identified as GFP-negative cells, were able to recruit soluble BIGH3 from the (conditioned) medium. Up to 39.8% of the NT-cells showed intercellular BIGH3 staining in a culture containing 1:1 NT and transduced cells (). Even in the presence of only 10% of BIGH3-expressing cells, 13% of the NT cells recruited BIGH3 intracellularly (). Because membrane staining for BIGH3 was not observed in NT cells (), the BIGH3 is probably internalized. However, NT HL60 cells did not recruit BIGH3 from conditioned medium of BIGH3-expressing cells (, “NT + BIGsup”), although sufficient BIGH3 was present (, last bar), which suggested that additional (cellular) signals are required to trigger the uptake of BIGH3.
From the same type of co-culture with decreasing fractions of BIGH3-expressing cells, we measured cell-bound BIGH3 (i.e., in cell lysates) and BIGH3 in supernatants (). Whereas the presence of BIGH3 in the supernatant decreased with decreasing numbers of BIGH3-transduced cells, the cell-bound fraction of BIGH3 decreased to a far lesser extend (when the BIG:NT ratio was 10:90, the cellular BIGH3 decreased by 48%, whereas the soluble BIGH3 decreased by 84% compared the levels detected in BIGH3-overexpressing cells). This again suggests that NT cells recruit BIGH3 from the supernatant, thereby depleting it from the supernatant. Together, these data suggest that HL60 cells with BIGH3-overexpression secrete BIGH3 and that wild-type HL60 cells can recruit BIGH3 but only when in contact with BIGH3-expressing cells.
Overexpression of BIGH3 in hematopoietic cells inhibits adhesion
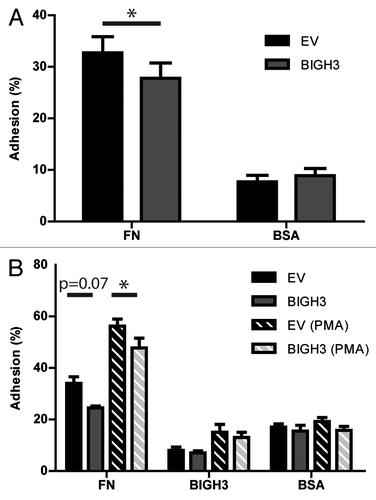
Presentation of BIGH3 at the cell surface of HSPCs may influence the cell–cell or cell–matrix interactions in the niche. We therefore analyzed whether hematopoietic cells with increased BIGH3 expression have altered adhesive capacities compared with control cells. HL60 cells were transduced with the BIGH3-GFP vector or a control GFP vector. The cells were incubated on fibronectin or BSA coatings and the fraction of adherent cells was quantified. Adhesion to the BSA coating was similar in BIGH3-expressing and control cells (). Adhesion to a fibronectin coating, however, was 19% (27.8% ± 3.0% vs 32.7% ± 3.1%) reduced in BIGH3-expressing HL60 cells compared with control cells (). In addition, we analyzed the adhesion of primary HSPCs transduced with the BIGH3-GFP or a control vector (). We observed a trend toward a smaller percentage of BIGH3-transduced primary HSPCs that adhered to fibronectin (24.5 ± 1.5%) compared with control transduced HSPCs (34.0 ± 5.1%; P = 0.07). Next, we stimulated adhesion of HSPCs by exposing the cells to the PMA. PMA-induced adhesion bypasses the integrins in the initiation of the signaling by its direct activation of PKC, but integrins are still required for the execution of adhesion, by the formation of adhesive complexes. PMA treatment induced the adhesion of control HSPCs on fibronectin by 2-fold. Furthermore, expression of BIGH3 significantly reduced adhesion in PMA-stimulated HSPCs (), in line with the results in HSPCs without PMA. However, no significant differences in adhesion to BSA or BIGH3 were observed, regardless of stimulation with PMA.
Figure 3. Overexpression of BIGH3 inhibits the static adhesion of HL60 cells and HSPCs. The percentage adhesion of HL60 cells (A) or HSPCs (B) with BIGH3 overexpression (BIGH3), with empty-vector (EV) and non-transduced cells (NT) on a fibronectin (20 μg/mL, FN), BSA (2%), or BIGH3 (10 μg/mL) coating. The HSPC were incubated in the presence (PMA) or absence of PMA during the adhesion period. The percentage adhesion was calculated by the absorbance of a well relative to the absorbance of the 100% input control of the corresponding cell type. Shown are means ± SEM (n = 7 in A, n = 4 in B) and each samples was performed in duplicates. * P < 0.05, ns, not significant.
Quantitative analysis of cell spreading and adhesion can be measured as changes in electrical resistance of the cell layer using electrical cell–substrate impedance sensing (ECIS). In this assay, control of BIGH3 transduced HL60 cells were seeded on fibronectin-coated gold electrodes, and in the presence of PMA the relative resistance of the cell layer was determined as a measure for cell spreading and adhesion. The relative resistance peaked at ~50 min post PMA treatment, and was 1.39-fold increased compared with t = 0 in control cells. In BIGH3-expressing cells, however, the relative resistance was only 1.14-fold increased compared with t = 0 (). These differences were statistically significant over a time period of 9–71 min post-PMA. BIGH3-expressing cells showed lower resistance compared with control cells, meaning less efficient spreading and/or adhesion.
Figure 4. Overexpression of BIGH3 decreases transcellular resistance after PMA induction. (A and B) The transcellular resistance of HL60 cells with BIGH3 overexpression and control cells (EV) on a fibronectin (20 μg/mL) coating (A) or poly-l-lysine coating (B) was analyzed by Electrical Cell-substrate Impedence Sensing (ECIS). PMA was added at time point zero. The resulted resistance-values were normalized to basal levels. Shown are means ± SEM (n = 6 in [A], n = 4 in [B]) and each samples was performed in duplicates. (C) The transcellular resistance of PB-derived HSPC with BIGH3 overexpression and control cells on fibronectin (20 μg/mL) upon PMA induction was analyzed by ECIS. Shown are means ± SEM (n = 3) and each samples was performed in duplicates. (D) Cell spreading of HL60 cells with BIGH3 overexpression (BIGH3) and control cells (EV) was analyzed by live-imaging microscopy. The surfaces of 10 randomly selected cells per condition were quantified during time. The data was normalized per cell to basal surface size. Shown are means ± SEM (n = 6 in BIGH3, n = 5 in control). * P < 0.05.
![Figure 4. Overexpression of BIGH3 decreases transcellular resistance after PMA induction. (A and B) The transcellular resistance of HL60 cells with BIGH3 overexpression and control cells (EV) on a fibronectin (20 μg/mL) coating (A) or poly-l-lysine coating (B) was analyzed by Electrical Cell-substrate Impedence Sensing (ECIS). PMA was added at time point zero. The resulted resistance-values were normalized to basal levels. Shown are means ± SEM (n = 6 in [A], n = 4 in [B]) and each samples was performed in duplicates. (C) The transcellular resistance of PB-derived HSPC with BIGH3 overexpression and control cells on fibronectin (20 μg/mL) upon PMA induction was analyzed by ECIS. Shown are means ± SEM (n = 3) and each samples was performed in duplicates. (D) Cell spreading of HL60 cells with BIGH3 overexpression (BIGH3) and control cells (EV) was analyzed by live-imaging microscopy. The surfaces of 10 randomly selected cells per condition were quantified during time. The data was normalized per cell to basal surface size. Shown are means ± SEM (n = 6 in BIGH3, n = 5 in control). * P < 0.05.](/cms/asset/d08f2acf-ba6f-4c34-a235-56e641cfe9e1/kcam_a_10926596_f0004.gif)
The HL60 cells that express BIGH3 also showed impaired induction of electrical resistance on a poly-l-lysine-coated surface, compared with control cells (), although the overall induction of adhesion on poly-l-lysine was much lower compared with the fibronectin-coated condition. Finally, the electrical resistance of BIGH3-expressing HSPCs (1.50 ± 0.16) was significantly reduced compared with control vector transduced HSPCs (1.84 ± 0.18; ).
The reduced resistance in BIGH3-expressing cells may imply reduced adhesion and/or reduced spreading of these cells. We therefore analyzed the spreading of HL60 cells after PMA treatment by live-cell microscopy. Spreading was quantified by measuring the average cellular surface during time. Surprisingly, cell spreading was similar with or without BIGH3 expression (). Together, these data indicate that overexpression of BIGH3 does not influence cell spreading, but impairs adhesion of cells, both on fibronectin substrates that bind cells dependent on integrin expression, and on poly-l-lysine substrates that bind cells independent of integrins.Citation27
Overexpression of BIGH3 in hematopoietic cells inhibits migration
Because adhesion and cell–matrix interactions are critical for efficient migration, the migratory capacity of BIGH3-expressing cells was examined. HL60 cells efficiently migrate over fibronectin-coated Transwell filters, dependent on the concentration of the chemoattractant CXCL12, with the highest migration at 100 ng/mL CXCL12.Citation28 In the presence of 30, 100, and 300 ng/mL CXCL12, approximately half of the control cells migrated across the fibronectin-coated filter, which was reduced by 23%, 22%, and 27% in BIGH3-expressing cells (41 ± 9% vs 54 ± 12%, P = 0.03; 48 ± 8% vs 61 ± 12%, P = 0.03; 39 ± 8% vs 54 ± 11%, P = 0.03) (). In addition, the spontaneous migration was also significantly reduced with 48% (0.6 ± 0.2% vs 1.7 ± 0.3%, P = 0.04) by BIGH3 expression (). These data show that BIGH3 overexpression impaired both the directional migration toward CXCL12 as well as the spontaneous migration.
Figure 5. Overexpression of BIGH3 inhibits the migration of HL60 cells toward CXCL12. Dose-dependent Transwell-based migration of HL60 cells with BIGH3 overexpression and control cells (EV). The cells in the upper compartment were allowed for 4 h to migrate through 5 μm pores toward CXCL12 in the lower compartment. The transmigrated cells were collected and quantified by flow cytometry and quantification beads. (A) The percentage migration was calculated by the ratio of cells and beads relative to this ratio in the 100% input control of the corresponding cell type. (B) Spontaneous migration of HL60 cells with BIGH3 overexpression and control cells (EV). (C) Horizontal TaxiScan-based migration of HL60 cells with BIGH3 overexpression and control cells toward a CXCL12-gradient, equivalent to 100 ng/mL in Transwell-based migration. The percentage migrating cells was quantified by the fraction of cells that migrated at least 50 pixels. (D) Considering the migrating cells, the total migration distance per cell was quantified by depiction of the migration-path per cell. In addition, the directed migration distance of the migrating cells was quantified in the direction of the CXCL12-gradient. Shown are means ± SEM (n = 4; P = 0.03 at t = 30, t = 100 and t = 300; P = 0.04 at t = 0) and samples were performed in duplicates. * P < 0.05.
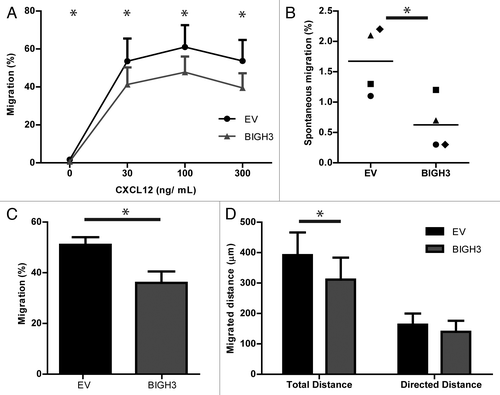
In TAXIScan-based migration experiments, cells migrate on a 2D-horizontal-plane trough 260-μm wide and 6-μm thick silicon chips on fibronectin-coated glass plates. We analyzed the CXCL12-induced migration capacity of BIGH3-expressing HL60 cells (). The fraction of cells that migrated was significantly smaller in the BIGH3-expressing cells compared with control cells (37 ± 4% vs 49 ± 3%, n = 4, P < 0.05; ). From the cells that migrated, the migration path was analyzed in real-time. The migrated distance of BIGH3-expressing cells was reduced as compared with control cells, i.e., 319 ± 69 vs 409 ± 63 µm, corresponding to 0.625 micron per pixel (, right bars). The migration component in the direction of the chemoattractant showed no significant reduction (, left bars). Together, these data indicate that sensing of CXCL12 and subsequent polarization toward CXCL12 is not affected by BIGH3, but that overall motility of the cells is reduced by BIGH3 overexpression. This conclusion is in line with the observed reduction in spontaneous migration in Transwells that also indicated impaired general motility of BIGH3-expressing cells.
BIGH3 overexpression does not affect actin polymerization and integrin expression, but modulates adhesive morphology
Cell migration and adhesion are coordinated by receptor activation and fast intracellular responses of e.g., the actin cytoskeleton. Therefore, the actin polymerization responses and receptor expression levels were analyzed in cells with BIGH3 overexpression. HL60 cells show rapid polymerized actin (F-actin) formation upon CXCL12 exposure.Citation29 In cells that overexpress BIGH3 and in control cells, the F-actin signal peaked at 15 s, followed by a rapid decline to basal levels after 60–120 s. No significant differences were found in the intensity or the duration of the F-actin signal (). This indicated that BIGH3 does not alter CXCL12-induced actin polymerization.
Figure 6. Overexpression of BIGH3 does not influence F-actin formation in HL60 cells upon CXCL12 treatment, but modifies the adhesive cell morphology. (A) The time course of CXCL12-induced F-actin formation in HL60 cells with BIGH3 overexpression and control cells was analyzed by flow cytometry. The mean fluorescence intensity (MFI) was normalized relative to basal MFI levels. Shown are means ± SEM (n = 3) and samples were performed in duplicates. (B and C) Confocal imaging of HL60 cells with BIGH3 overexpression (B) and control vector expression (C) 60 min after PMA stimulation. The adherent cells were fixed, permealized, and stained for F-actin (green), β1-integrin (red), and Hoechst (blue). Projections of Z-stacks are shown in upper panels of (B and C). Depth-analyses were performed based on the β1-integrin signal, shown in the lower panels of (B and C). The adhesive structures at the cell boundaries appear deeper (more yellow) in control cells compared with cells with BIGH3 overexpression. Four representative cells are shown for each condition, out of two independent experiments. Scaling is indicated by a white bar in the lower left corner, representing 5 μm. (D) The maximal length of cell and nucleus (red lines in projections) and the maximal height (white lines in cross sections) were determined (representative example). (E) The maximal length and height of cell and nucleus from HL60 cells with BIGH3 overexpression and control cells. The cells with BIGH3 overexpression appear flatter and have a slightly bigger nuclear diameter, although the maximal cellular diameter is not changed. This indicates BIGH3 induces structural changes in cell morphology. Shown are means ± SEM (n = 20). * P < 0.05, ** P < 0.01, ns, not significant.
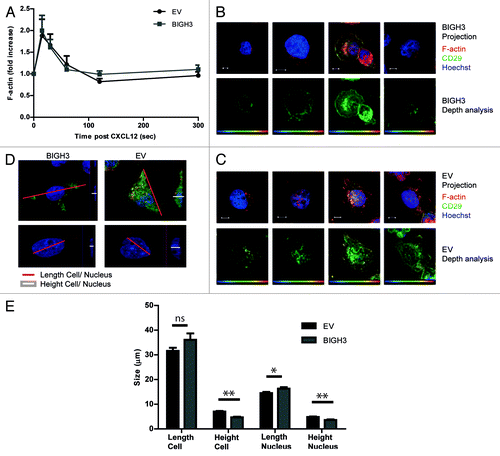
The expression levels of the integrins α4β1 (VLA4) and α5β1 (VLA5) and the CXCL12 receptor CXCR4, as measured by flowcytometry, were not altered in cells with BIGH3 expression compared with controls (data not shown). The CXCL12-induced receptor internalization of CXCR4 showed similar kinetics during a timeframe of 1 h in BIGH3 cells and in control cells (data not shown). Although the signal intensity of F-actin and the integrins was not changed by BIGH3 expression, we analyzed the spatial distribution of β1-integrin and F-actin in more detail (). Cells were seeded on a fibronectin-coated surface and exposed to PMA. To examine cell morphology, the β1-integrin signal of these cells was subjected to more detailed analyses. By computerized depth-analysis, based on confocal Z-scanning, the adhesive structures at the cell boundaries of control cells appear at a lower confocal plane (depicted as more yellow) compared with cells with BIGH3 expression. This means that BIGH3 alters the morphology of adhesive structures, suggestive for reduced adhesion to fibronectin.
In addition, in similar HL60 cells that adhered to fibronectin after PMA-treatment, geometric parameters were quantified. The maximum height of the nucleus (3.6 ± 0.2 vs 4.8 ± 0.2 μm) and the cell (4.7 ± 0.2 vs 7.0 ± 0.3) were significantly reduced in cells with BIGH3-expression compared with control cells (). This is in line with increased nuclear height observed in endothelial cells treated with blebbistatin, which inhibits myosin-II activity and induces membrane extensions, or in endothelial cells with a disrupted actomyosin-nuclear connection.Citation30 And these two described conditions are also associated with increased substrate-adhesion. Together, our data indicate that BIGH3 disturbs normal cell morphology in adhesive conditions. The reduced nuclear height of BIGH3-expressing cells confirms that the substrate-adhesion of these cells is weakened, because the opposite is known to happen when myosin-based contractility is inhibited and substrate-adhesion is enhanced.
PMA-induced RAC1 activation is restrained in cells with BIGH3 overexpression
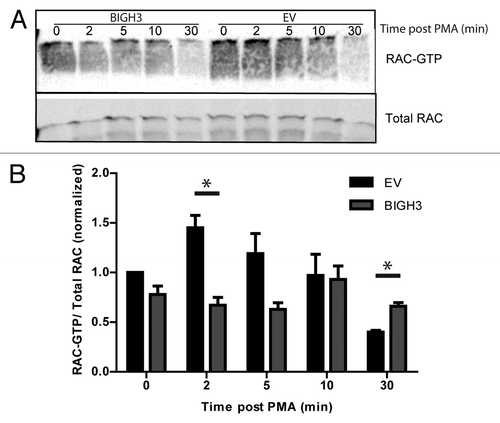
Activation of the small GTPase RAC1 is essential for protrusion of the leading edge of a migrating cell. Activated RAC1 stimulates protrusion at the leading edge and stabilizes the polarized migratory phenotype.Citation31 We investigated whether BIGH3 expression alters PMA-induced RAC1 activation, using a pull-down assay for active RAC1 by a Cdc42/Rac1-binding (CRIB) bait peptide.Citation32 In resting cells, RAC1-GTP levels were similar in BIGH3-expressing and control cells (). Two minutes after PMA administration, RAC-GTP levels were upregulated in control conditions (1.3-fold), but not in BIGH3-expressing cells, resulting in significant lower RAC-GTP levels compared with control cells (P = 0.01). RAC-GTP levels in BIGH3-expressing cells hardly responded to PMA, although the maximum activation was reached 10 min post-PMA. After 30 min, the initial induction was followed by an inhibitory response, especially in control cells. This pattern caused significant lower overall RAC-GTP levels in control cells as compared with BIGH3-expressing cells (P = 0.03). These data indicate that RAC1 activation is inhibited in cells with BIGH3 overexpression. The impaired RAC1 activation could well explain the impaired migration and adhesion of BIGH3-expressing cells.
Figure 7. Overexpression of BIGH3 impairs the RAC1 activation by PMA. To study the activation of RAC1, a CRIB-peptide pool-down for endogenous RAC1-GTP was performed in HL60 cells with BIGH3 overexpression and control cells (EV). (A) Activated RAC1 was detected by immunoblotting in total cell lysates (total RAC1) and in CRIB-peptide pool-down lysates (RAC1-GTP). A representative experiment is shown. (B) Quantification of activated RAC1 normalized for the total RAC1 content. Shown are means ± SEM (n = 4). * P < 0.05
PMA-induced ERK phosphorylation is decreased in BIGH3-expressing cells
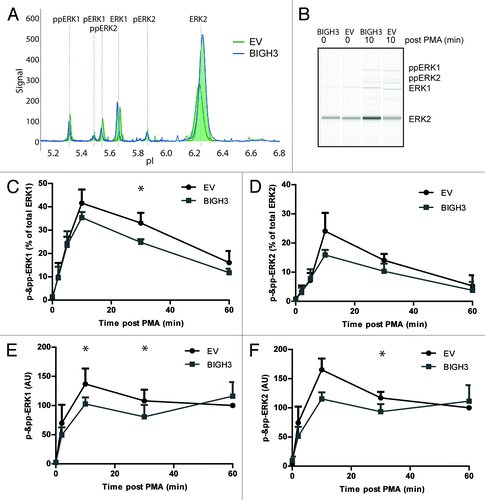
Differential engagement of ERK signaling was found to explain differences in CXCL12-induced migration of murine HSPCs.Citation33 In addition, inhibition of ERK activity resulted in decreased cell migration on ECM.Citation34 In specific situations, activated RAC1 was required for full activation of the ERKs, e.g., in EGF- and PAK-induced ERK activation.Citation35 We therefore compared ERK activation of BIGH3-overexpressing cells and control cells using the NanoPro technology.Citation36,Citation37 This assay separates proteins by capillary iso-electric focusing followed by immunodetection (). The phosphorylated and unphosphorylated forms of ERK1 and ERK2 can be quantified within the same sample, using one anti-ERK1/2 antibody. In steady-state cells, the majority of ERK1/2 was in its unphosphorylated state. Upon PMA activation, both ERK1 and ERK2 shifted partly to the phosphorylated states. However, in BIGH3-expressing HL60 cells () and HSPCs (), the PMA-induced phosphorylation of ERK1 and ERK2 was reduced compared with cells expressing a control vector. As RAC1 is upstream of ERK1/2, the reduced phosphorylation of ERK1/2 may be caused by lower RAC1 activation and may explain the decreased adhesive and migratory responses of BIGH3-expressing cells.
Figure 8. Overexpression of BIGH3 reduces ERK1 and ERK2 protein phosphorylation. The NanoPro Assay was used to quantify ERK expression and phosphorylation. (A) Peaks represent phosphorylated or unphosphorylated ERK1 or ERK2 protein levels, as indicated, in HL60 cells (A–C) or HSPCs (D). Shown are the samples at t = 10 min from a representative experiment. (B) The generated peaks are transformed in a western-blot-like representation of the data. The band intensity represents (phospho-)ERK protein levels. (C) The phosphorylated ERK1 (left panel) and ERK2 (right panel) protein levels as percentage of total ERK1 or ERK2 expression, respectively, in HL60 cells with BIGH3 overexpression or control cells (EV) were depicted. In resting state, the unphosphorylated isoforms are dominant. Shown are means ± SEM (n = 3). (D) The phosphorylated ERK1 (left panel) and ERK2 (right panel) protein levels were analyzed in HSPCs with BIGH3 overexpression or control cells (EV). Data in each experiment was normalized to the signal in control cells 60 min after PMA treatment. Shown are means ± SEM (n = 3). * P < 0.05
Discussion
We studied the function of BIGH3 in adhesion and migration of hematopoietic cells. BIGH3 is part of the BM extracellular matrix. It is expressed at high levels by MSCs but also detected in HSPCs. BIGH3 expression is increased in HSPCs during BM-regeneration. To investigate its function, we increased BIGH3 expression in hematopoietic cells. BIGH3 was secreted and subsequently bound by non-transduced cells. BIGH3-expressing cells showed reduced adhesion to fibronectin, as observed in adhesion- and ECIS-assays, resulting in decreased migration across fibronectin-coated filters. BIGH3 expression did not change actin polymerization upon stimulation of migration, but reduced PMA-induced RAC1 activation and ERK phosphorylation. We identified BIGH3 as a modulator of HSPC adhesion and migration, associated with inhibited RAC1- and ERK-signaling pathways.
Expression of BIGH3 impairs cell adhesion to fibronectin
The effect of BIGH3 on adhesion differs between cell types, possibly because it depends on the expression of BIGH3 or other adhesion proteins by the cells. Coating the cell culture surface with BIGH3 results in increased adhesion for most cells;Citation18,Citation38-Citation43 however, decreased adhesion of melanoma and endothelial cells to BIGH3-coated surfaces is seen, possibly due to their high expression of endogenous BIGH3.Citation21,Citation44 In our study, primary HSPCs showed adhesion to BIGH3-coated surfaces, at least in part via interaction with integrins on these cells. Concordantly, addition of BIGH3 in cell-suspension inhibited cell adhesion to fibronectin, which supports the notion that BIGH3 binds and blocks adhesion molecules.Citation45-Citation47
However, in cultured HSPCs, no significant increased adhesion of control cells (EV) on BIGH3-substrate was observed as compared with adhesion on BSA-substrate (), in contrast to adhesion of primary non-cultured HSPCs on BIGH3-substrates (). This may indicate that the BIGH3 binding capacity is changed upon culturing of the HSPCs. Interestingly, we observed that endogenous BIGH3 RNA expression was induced upon culturing (unpublished data), which may explain the reduced BIGH3 binding capacity of cultured HSPCs, as endogenous BIGH3 may block integrins and integrin-mediated adhesion.
Overexpression of BIGH3 most likely affects cell adhesion similar to the addition of soluble BIGH3, because BIGH3 is present in the cell-suspension after secretion. However, the effect of BIGH3 overexpression has hardly been studied, and depends on the cell type, with decreased adhesion in neuroblastoma and colon cancer cells, but increased adhesion in adenocarcinoma cells.Citation18,Citation48,Citation49 We observed decreased adhesion of HSPCs with BIGH3 overexpression on a fibronectin-surface in adhesion assays and electric resistance-based assays. This ECIS assays revealed more significant differences between BIGH3-overexpressing cells than classic adhesion assays did, which may be due to the higher sensitivity of ECIS assays. In addition, classic adhesion assays measure the on-off phenomena of a cell being adherent or not, whereas ECIS assays sense the nature of the adhesive interaction of adherent cells. This may indicate that BIGH3 modulates the cell–matrix interaction, whereas cell spreading was not affected.
In addition, we found that hematopoietic cells could bind and internalize BIGH3 from their environment, at least in co-cultures of BIGH3-overexpressing-cells and control cells. Thus, excreted BIGH3 most likely binds to adhesion complexes, such as integrins. As a result, these complexes are not available to bind to, for instance, fibronectin, which may explain the anti-adhesive effects of BIGH3 overexpression.Citation50,Citation51 As these complexes are not involved in the limited cell adhesion to BSA and poly-l-lysine, BIGH3 overexpression does not affect adhesion to BSA and poly-l-lysine.
We observed that BIGH3 from conditioned medium could not be internalized, whereas secreted BIGH3 could be internalized in direct co-cultures. This suggests that other factors are required to support the BIGH3 internalization, although the identity of these factors is yet unclear. Possibly, soluble BIGH3 is rapidly modified by soluble or cellular factors, although it stays detectable by antibodies.
BIGH3 expression decreases fibronectin-assisted cell migration
In line with our results on adhesion, we found that the motility of hematopoietic cells is decreased upon BIGH3 overexpression as measured by decreased spontaneous and CXCL12-induced migration across fibronectin-coated Transwells. This could be due to reduced CXCL12 signaling, or to a decreased overall motility. The decreased spontaneous migration suggests that the overall motility of these cells is decreased. Cells that have a lower overall motility are less prone to find a micropore to migrate through, resulting also in lower migration rates toward the CXCL12 in Transwell systems. We showed in the 2D-migration experiments that polarization and CXCL12-directed migration was not influenced by BIGH3-expression, although the total migrated distance, associated with overall motility, was reduced by BIGH3. We also observed a decreased percentage of cells that responded to CXCL12 in the 2D-migration experiments, which may also be caused by a decreased overall motility, as more cells stay below the cut-off level for the migrated distance and are counted as non-migrating cell. The conclusion that BIGH3 inhibits migration is in line with its inhibitory effect on adhesion, as HSPC cell migration requires adhesion, which was provided by the fibronectin-coated substrate in our experiments. Reduced adhesion on fibronectin-substrates of BIGH3-expressing HSPCs is expected to result in decreased fibronectin-based cell migration.
Concordantly, BIGH3 expression is known to be implicated in a number of migration-related cellular disease processes, including tumorigenesis, angiogenesis, and metastasis.Citation17,Citation48,Citation52,Citation53 BIGH3 enhances metastasis of glioma, melanoma, and colon cancer cells,Citation21,Citation49,Citation54 but its expression is decreased in bronchial epithelial tumorigenic cells, primary lung carcinomas, and neuroblastomas and BIGH3 overexpression reduced tumorigenicy of Chinese hamster ovary cells.Citation45,Citation55-Citation58 Although tumorigenesis is determined by more factors than migration capacity and invasiveness only, our results of reduced migration upon BIGH3-expression in HSPCs are in line with e.g., the reduced BIGH3 expression in more tumorigenic bronchial cells.
Despite the inhibited migration of BIGH3-expressing cells, the actin polymerization upon CXCL12 was not affected by BIGH3 expression. Actin polymerization is essential for proper migration,Citation59 but we observed that other signaling components, independent of F-actin formation, are required as well, as described for MSC migration.Citation60 CXCL12-induced phosphorylation of AKT and p38 was not altered by BIGH3 expression, which is in accordance with the unchanged basal expression of CXCR4 (data not shown).
BIGH3 impairs Rac1- and ERK1-signaling
Migration requires the concerted activation of integrin and chemokine receptors such as CXCR4, the CXCL12 receptor. Their engagement results in the activation of multiple signaling pathways. CXCL12 is a G-protein coupled receptors that signal through Protein Kinase C (PKC). Hence, adhesion and migration-related processes can be activated by PMA, a potent direct activator of PKC and its downstream targets, RAC1 and the ERKs.
Complete RAC1-activation is required for a complete adhesive phenotype, including lamellipodia formation. Inhibition of RAC1 in carcinomasarcoma cells results in low-adherent blebs instead of strong-adherent lamellipodia.Citation61 We observed that PMA-induced adhesion and RAC1 activation were reduced in BIGH3-expressing cells. This was accompanied by the morphological differences that we observed by confocal imaging, which suggested low-adherent protrusions in BIGH3-expressing cells. The more rounded cell morphology of BIGH3-expressing cells and, in addition, the reduced nuclear height, suggest more actinomyosin contractility in BIGH3-expressing cells.Citation30 Our results are in line with results obtained by Rac1/Rac2-deficient mice that showed reduced adhesion and migration of HSPCs and massive HSPC egress.Citation62,Citation63
In most cells, RAC1 activation is tightly balanced with RhoA activation by reciprocal inhibitory regulation.Citation64 The impaired adhesion and migration of BIGH3-expressing cells might be explained by a switch in the GTPase balance, from a RAC1-induced protrusions toward RhoA-induced actomyosin contractility.Citation65 We measured RhoA activation levels using RhoA G-lisa assays and found a trend toward increased RhoA activation in BIGH3-expressing cells, although the results were variable (basal active RhoA is induced in BIGH3-expressing cells by a fold-change of 1.76 ± 0.8, n = 4, P = 0.16; in addition, 10 min after CXCL12-treatment, active RhoA-levels were normalized to levels of EV at t = 0, and revealed active RhoA expression of 1.16 ± 0.07 in EV vs. 2.13 ± 0.99 in BIGH3, n = 4, P = 0.15).
In addition, we found that BIGH3 overexpression decreased ERK activation in HSPCs. This is in line with signaling in endothelial cells, where a BIGH3-peptide also decreased ERK phosphorylation.Citation47 In addition, ERK was required for BIGH3-mediated migration in vascular smooth muscle cells, as migration over a BIGH3-substrate was dependent on ERK phosphorylation.Citation43 Various other studies established the regulatory role of ERKs in chemokine-induced migration, as ERKs, besides their function in the nucleus, also induce FAK activation and myosin-light-chain phosphorylation in the cytosol, thereby regulating the formation of focal adhesions and actomyosin contractility.Citation11,Citation66-Citation72 This indicates that decreased ERK phosphorylation in BIGH3-expressing cells might contribute to the decreased overall motility in these cells. We conclude that BIGH3 inhibits activation of RAC1 and ERK, which coordinately control the adhesive and migratory response of HSPCs ().
Figure 9. Overexpression of BIGH3 reduces adhesion and migration via RAC1 and ERK responses. We propose that induced BIGH3 expression inhibits RAC1 and ERK activation. As RAC1 and RhoA are mutual repressive, the phenotype of cells with high BIGH3 expression switch toward more actomyosin contraction and reduced protrusion formation. The reduced ERK activity may results in reduced FAK activity and a lower stability of adhesive junctions. Ultimately, high BIGH3 expression results in reduced cell adhesion and migration. Bold arrows represent activation or induction; dashed lines indicate indirect relationships; small arrows behind proteins indicate increased or decreased activation or expression levels in BIGH3-overexpressing cells.
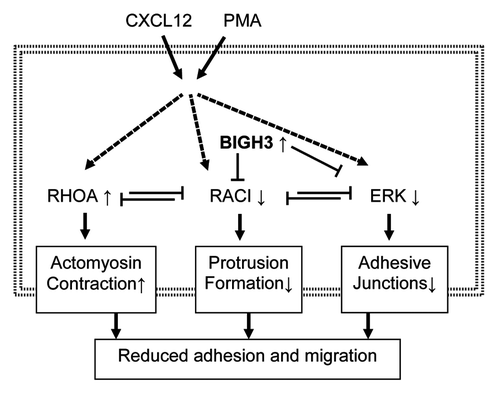
Migration and adhesion and the adhesive cell-morphology are very well interdependent, as migration is the result of a balance between transmitted forces of cell-substrate-adhesion, actinomyosin-contraction, and actin-polymer-network expansion in protrusions.Citation73 Although migration of neutrophils, dendritic cells, and lymphocytes may be integrin-independent,Citation73,Citation74 HSPC migration involves the formation and degradation of integrin-mediated adhesion complexes.Citation75-Citation80 Therefore, we propose that inhibited RAC1 and ERK activity alter the cytoskeletal-mediated forces and the dynamics of adhesion complexes, thereby modulating HSPC adhesion and migration ().
Function of BIGH3 in hematopoiesis
BIGH3 expression is upregulated in murine BM HSPCs after G-CSF/ CY5 or 5FU treatment.Citation12,Citation13 These treatments induce mobilization and egress of HSPCs from the BM, by massive remodeling of the extracellular matrix. Upregulated BIGH3 secretion may prepare the HSPCs for BM egress, by decreased adhesion via blockage of integrins and other adhesion molecules. In line with this, overexpression of BIGH3 in colon cancer cells decreases VE-cadherin-mediated endothelial junctions, resulting in more aggressive metastasis.Citation49 BIGH3 may weaken endothelial junctions in HSPC egress as well. The HSPC pool in the BM after G-CSF or 5FU treatment differs from the original pool of HSPCs in the BM: it contains more mature cells that show more mitotic activityCitation13,Citation81 and have lower affinity for the BM niche. This means that our in vitro observation that BIGH3-decreased adhesion of HSPCs may have physiologic implications for the HSPC adhesion in the BM.
On the other hand, we found decreased overall motility of BIGH3-expressing cells, but these cells could still polarize and their migration in the direction of CXCL12 was not influenced. The upregulation of BIGH3 may help loosen the adhesive contacts of the hematopoietic cells with their originating niche and may make them susceptible for polarization and subsequent egress.
In this paper we established that BIGH3 overexpression in HSPCs decreased adhesion and migration and downstream RAC1 and ERK activation. Therefore, upregulation of BIGH3 in regenerative BM might contribute to HSPC homeostasis, through decreased adhesion and an altered GTPase balance of HSPCs, which may regulate homing and mobilization of HSPCs.
Materials and Methods
HSPC purification
All human material was obtained with approval of the medical ethical committee of the Academical Medical Centre (AMC) and after informed consent. Mobilized peripheral blood (MPB) progenitor cells were enriched from leukapheresis material following treatment with chemotherapy and/or G-CSF (5–10 mg/kg/day) to induce stem cell mobilization. Cord blood (CB) was collected according to the guidelines of Eurocord Nederland. Mononuclear cells were enriched by density gradient centrifugation over Ficoll-Paque PLUS (1.077 g/mL; GE Healthcare Life Sciences; 17-1440-03). Mononuclear cells were resuspended in MACS buffer, consisting of phosphate buffered saline (PBS), 0.5% bovine serum albumin (BSA; fraction V; Invitrogen; 15260-037), and 5 mM EDTA (Sigma-Aldrich; E5134). CD34-positive cells were isolated by magnetic cell sorting (MACS; Miltenyi Biotec), using the human CD34 Microbead Kit (Miltenyi; 130-046-703) according to the manufacturer instructions and within 48 h after initial sample harvest. This resulted in a purity of more than 90% CD34+ cells, as determined by flow cytometry.
Cell lines
HL60 and 293T HEK cells were obtained from the ATCC. 293T HEK cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Lonza; BE12-604F), HL60 cells were cultured in Iscove's modified Dulbecco's media (IMDM; Lonza; BE12-722F), and both media were supplemented with L-glutamine, penicillin 100 U/mL, streptomycin 100 mg/mL, and 10% v/v FCS (Life Technologies, 16140071). Twenty-four hours before each migration experiment, the HL60 cells were reseeded at 300 000 cells/mL.
Lentivirus production
Lentiviral vector pSIN-SFFV-BIGH3-IRES-GFP (BIG) was generated by replacing the EGFP sequence of pSIN-SFFV-GFP (empty vector, EV, kindly provided by Prof Dr G Wagemaker from Erasmus MC, Rotterdam) by the BIGH3-IRES-GFP sequence. Lentiviral particles were produced by transient co-transfection of the vectors with pVSV-G, pRSV.Rev, and pMDL.RRE into 293T HEK cells by calcium phosphate transfection methods. 293T cells were cultured in poly-l-lysine (Sigma-Aldrich; P8920) coated T80 flasks (Thermo Scientific; 153732). The medium of the cultures was refreshed 12 h after transfection and cell culture supernatants were collected at 36 and 60 h after transfection, filtered through a 0.45-μm pore size cellulose-acetate filter (Whatman, 10404006) and concentrated by high-speed centrifugation (Beckman Coulter) at 20 000 G for 2 h.
Lentiviral transductions
CD34-positive cells were cultured 1–2 d in serum-free medium CellGro (CellGenix; 20802-0500) supplemented with a cytokine maintenance cocktail of 20 ng/mL thrombopoietin (TPO; Miltenyi Biotec; M9515520), 100 ng/mL stem cell factor (SCF; R&D Systems; 255-SC), 100 ng/mL Fms-like tyrosine kinase 3 ligand (FLT3L; Miltenyi Biotec; 130-093-854), and 20 ng/mL IL-6 (Miltenyi Biotec; 130-093-931) in Costar 24-well plates (Corning; 3527). CD34-positive cells, 30 000–100 000 cells in a total of 100 μl CellGro supplemented by the maintenance cytokine cocktail, were infected at an equal multiplicities of infection (MOI) of 500 infectious units per cell. The cells were centrifuged 20 min at 2000 rpm, incubated 20 min at 37 °C in the incubator, centrifuged again 20 min at 2000 rpm, and incubated for 24 h at 37 °C and 5% CO2. The cells were washed and resuspended in Cellgro, supplemented by the maintenance cytokine cocktail, and kept in culture until used for migration assays.
For transduction of HL60 cells, virus was added to 100 000 cells at an equal MOI in all the conditions of about 100 infectious units per cell. The cells were centrifuged for 20 min at 2000 rpm, incubated for 24 h at 37 °C and 5% CO2, then washed and kept in culture until used for experiments. Each set of replicate experiments was performed with at least two independent cell batches retrieved by independent transductions.
Adhesion assays
A MaxiSorp 96-wells immuno-plate (Thermo Scientific/NUNC; 439454) was coated with 20 μg/mL Fibronectin (Sigma-Aldrich, F1141), 20 μg/ml recombinant BIGH3 (R&D Systems; 3409-BG-050), or 2% w/v BSA for 1 h at 37 °C, and blocked by 2% w/v BSA for 20 min at 37 °C. In parallel, the HL60 cells with overexpression of BIGH3 and control cells were incubated for 30 min in assay medium (IMDM + 0.25% w/v BSA) at 37 °C. For experiments with HSPCs, 400 000 cells per well were used, while for HL60 cells 250 000 cells per well were sufficient to coffer the complete surface. For inhibition experiments, the cells were incubated for 20 min with 20 μg/mL blocking antibodies: anti-CD13, anti-CD18, and/or anti-CD29, respectively clones Q20 (Sanquin Pharmaceutical Services; M1387), P4C10 (EMD Millipore; MAB1987Z), and IB4 (Abnova; MAB6945). The cells were stained with Calcein AM at 6.7 μg/mL (Invitrogen; C3100MP), added to the coated plate, and allowed to adhere for 30 min at 37 °C. When PMA (Promega; V1171) treatment was used, PMA (0.5 μg/mL) was present during this adhesion period. Subsequently, the non-adherent cells were carefully removed by washing the cells two times with PBS. Lysisbuffer (PBS + 0.1% v/v Triton; Sigma-Aldrich; T9284) was used to lyse the adherent cells and to release the Calcein AM. The intensity of the fluorescent signal was measured at 485 nm excitation and 535 nm emission on a TECAN GENios Plus platereader (Tecan Group). As an input control, the total number of input cells was added to an empty well and lysed. The percentage adhesion was calculated by a ratio of the fluorescent signal of each sample relative to the fluorescent signal of the input control.
Transwell migration assays
Transwell migration assays were performed in Corning Transwell plates (Corning; 3421), where the upper and lower compartment of each well is separated by a membrane filter, with pore sizes of 5 μm. The membranes were coated with 20 μg/mL fibronectin for 1 h at 37 °C, and subsequently washed twice gently with assay medium (IMDM + 0.25% BSA). In parallel, the HL60 cells were incubated in assay medium for 30 min at 37 °C. Subsequently, 100 000 cells in 100 μL assay medium were seeded in each upper compartment and 600 μl assay medium with the indicated concentrations of CXCL12 (ranging from 0–300 ng/mL; Peprotech; 300-28B) was added to the lower compartments. The upper compartments were placed in the lower compartments, and the cells were allowed to migrate at 37 °C for 4 h. As an input control, 500 μL assay medium was taken together with 100 μL assay medium containing 100 000 cells. The cells that did migrate to the lower compartment were harvested and quantified by flow cytometry. For this quantification, twice 200 μL from each well, including the input control, was transferred to a 96-wells plate and a fixed amount of CountBright fluorescent counting beads (Invitrogen/Life Technologies; C36950) was added. Flow cytometry was performed by use of an FACS LSR II HTS (BD Biosciences). The percentage migration was calculated by dividing the ratio of the number of cells vs. the number of beads in the experimental conditions by the ratio of the number of cells vs. the number of beads in the input control.
TAXIScan migration assays
Real-time horizontal chemotaxis was examined of HL60 cells and CB-derived HSPCs with overexpression of BIGH3 and control cells, using a TAXIScan device (ECI), comprising two compartments connected by a bridge of 260 μm length and 6–8 μm height for each of the six channels. The glass slide used as migration surface was coated by 20 μg/mL fibronectin for 1 h at 37 °C and blocked by 2% w/v BSA for 20 min at 37 °C. The cells were injected in the first compartment and flow was induced by drawing some medium from the second compartment to align the cells in front of the connecting bridge. Two μl CXCL12 (10 μg/mL) was added to the second compartment, equivalent to a final concentration gradient of ~100 ng/mL. The cells were allowed to migrate through the connecting bridge for 8 h at 37 °C, while taking pictures each 2 min. Migration pathways were subsequently analyzed by use of ImagePro software, by clicking on the cells though the sequential frames. Directed migration distance was calculated as the net migrated distance in the direction of the concentration gradient of the chemoattractant. The total migration distance was calculated as the total distance of the migrated pathway. The velocity of the cell migration is expressed in μm/s, considering all time points between the first and last time points where the speed is above 10 μm/s.
Actin polymerization assays
HL60 cells with overexpression of BIGH3 and control cells were washed three times in assay medium and incubated at 37 °C for 30 min at a concentration of 1 million cells/mL. While the cells and CXCL12 was kept at 37 °C, the CXCL12 was added to the cells at a final concentration of 100 ng/mL. At the indicated time points, 100 μL cells were transferred to precooled fixation solution, 3.7% v/v formaldehyde (Merck; 1.03999) in PBS, and incubated 20 min at 4 °C. Subsequently, the cells were centrifuged and the pallet was resuspended in permeabilization reagent (Intraprep Permeabilization Reagent, Beckman Coulter; PN IM2389) and incubated for 20 min. To stain F-actin, the cells were incubated in 20 μL Palloidin-Alexa488 (Molecular Probes/Invitrogen; A12379) for 30 min at a concentration of 1 U/mL. Finally, the cells were washed in PBS and resuspended in appropriate volumes of PBS for flow cytometric analysis, which was performed by means of the FACS LSR II HTS.
Electric resistance assays
For resistance-based adhesion experiments, golden ECIS electrodes (IBIDI, Applied BioPhysics; 8W10E) were activated with l-cysteine (Sigma-Aldrich; 168149) for 20 min at 37 °C and next coated with fibronectin for 1 h at 37 °C. HL60 cells with overexpression of BIGH3 and control cells were washed three times in assay medium, incubated at 37 °C for 30 min and seeded with 350 000 cells per well in 400 μL medium. Electric cell–substrate impedance was monitored in a multivariable way for up to 12 h with the ECIS equipment (Applied BioPhysics), performing measurements of all wells each 2 min. PMA was added to the indicated wells at the indicated time points, to initiate the adhesion process. The obtained data was normalized t = 0 (addition of PMA) and further processed by using the ECIS-software.
RAC1 activity assay
HL60 with overexpression of BIGH3 and control cells were washed three times in assay medium and incubated at 37 °C for 30 min at a concentration of 1 million cells/mL. A tube was prepared with 10 million cells for each experimental condition. At the appropriate time points, PMA (0.5 μg/mL) was added to the concordant tubes. At time point zero, all samples were centrifuged, washed with cold PBS, and lysed in lysis buffer: 50 mM Tris pH 7.4 (Invitrogen; 15504-020), 0.5 mM MgCl2 (Merck; 1.05833), 500 mM NaCl (Fagron; 102231), 1% (v/v) Triton-X100 (Sigma; T9284), 0.5% (w/v) deoxycholic acid (DOC; Sigma-Aldrich; D2510), and 0.1% (w/v) Sodium Dodecyl Sulfate (SDS; Invitrogen; 15525-017), supplemented with Halt protease and phosphatase inhibitors (Thermo Scientific; 1861282). Lysates were cleared by centrifugation at 14 000 × g for 5 min and 10% of the lysate was mixed with Laemmli sample buffer (Bio-Rad; 161-0737) and snap frozen as total cell lysate. From the remaining lysate, GTP-bound Rac1 was isolated with biotinylated Pak1-Crib peptideCitation82 coupled to streptavidin-agarose beads (Sigma-Aldrich; S1638), by incubating lysates and peptide-coupled-beads for 1 h in a turning mill at 4 °C. Beads were washed four times in wash buffer: 50 mM Tris, pH 7.4, 0.5 mM MgCl2, 150 mM NaCl, 1% (v/v) Triton X-100, supplemented with protease inhibitors. The bound proteins were dissolved in Laemmli sample buffer. Pool-downs and lysates were immunoblotted with monoclonal anti-Rac1 antibodies (clone 23A8; Upstate/Millipore; 05-389).
Nanoprobe protein quantification
ERK expression and phosphorylation was measured by NanoPro 1000 System (Protein Simple) according to the manufacturer’s recommendations. HL60 cells with BIGH3 overexpression or control cells were starved for 30 min and stimulated with PMA (0.5 μg/mL) while kept at 37 °C, and lysed at indicated time points in Bicine/CHAPS lysis buffer (Protein Simple; 040-764). Lysates were centrifuged and supernatants were loaded in small capillaries (Protein Simple; CBS701) together with Pharmalyte premix G2, pH5-8 (Protein Simple; 040-973) and pI standard ladder 3 (Protein Simple; 040-646). Isoelectric focusing of proteins was performed by applying 21 000 uW for 40 min. After focusing, UV light was used to cross-link proteins to the inner capillary wall. After that, the capillary was washed and immunoprobed for the indicated proteins followed by washing to remove the unbound antibodies. Finally, Luminol (Protein Simple; 040-652) and peroxide (Protein Simple; 040-653) were added to generate chemiluminescence, which was captured by a CCD camera. Results were analyzed by Compass software (Protein Simple).
Confocal microscopy
HL60 cells with BIGH3 overexpression and control cells were starved for 30 min in assay medium (IMDM + 0.25% w/v BSA). The cells were seeded onto fibronectin-coated glass coverslips in the presence of PMA (0.5 μg/mL) for 60 min at 37 °C. The coverslips were then washed and sealed on object glasses. Fluorescent imaging was performed with a confocal laser-scanning microscope (LSM510/Meta; Carl Zeiss MicroImaging, Jena, Germany) using a 63 × NA 1.40 or a 40 × NA 1.30 oil lens (Carl Zeiss MicroImaging). Image acquisition was performed with ZEN 2009 software (Carl Zeiss MicroImaging).
Statistical analysis
To statistically analyze the differences between experimental groups, the student’s t-test was performed. A P value of ≤ 0.05 was considered significant.
| Abbreviations: | ||
| BIG | = | BIGH3-transduced |
| BIGH3 | = | TGF-beta induced gene H3 |
| BM | = | bone marrow |
| BSA | = | bovine serum albumine |
| CXCL12 | = | CXC-ligand 12 |
| CXCR4 | = | CXC-receptor 4 |
| ECIS | = | electric electrical cell-substrate impedance sensing |
| ECM | = | extracellular matrix molecules |
| ERK | = | extracellular regulated kinase |
| EV | = | empty-vector-transduced |
| FACS | = | flow cytometry activated cell sorting |
| G-CSF | = | granulocyte-colony stimulating factor |
| GFP | = | green fluorescent protein |
| HSPC | = | hematopoietic stem and progenitor cells |
| MSC | = | mesenchymal stromal cells |
| NT | = | non-transduced |
| RAC1 | = | ras-related C3 botulinum toxin substrate1 |
| RGD | = | arginyl-glycyl-aspartic acid |
| TGF-beta | = | transforming growth factor-beta |
| VLA4 | = | very late antigen 4 (CD49D) |
| VLA5 | = | very late antigen 5 (CD49E) |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank PPOC for funding this research. We further thank T Reya (University of California San Diego School of Medicine) for sharing micro-array data.
References
- Kaplan RN, Psaila B, Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med 2007; 13:72 - 81; http://dx.doi.org/10.1016/j.molmed.2006.12.003; PMID: 17197241
- Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012; 481:457 - 62; http://dx.doi.org/10.1038/nature10783; PMID: 22281595
- Wagner W, Roderburg C, Wein F, Diehlmann A, Frankhauser M, Schubert R, Eckstein V, Ho AD. Molecular and secretory profiles of human mesenchymal stromal cells and their abilities to maintain primitive hematopoietic progenitors. Stem Cells 2007; 25:2638 - 47; http://dx.doi.org/10.1634/stemcells.2007-0280; PMID: 17615262
- Warr MR, Pietras EM, Passegué E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip Rev Syst Biol Med 2011; 3:681 - 701; http://dx.doi.org/10.1002/wsbm.145; PMID: 21412991
- Nakamura-Ishizu A, Suda T. Hematopoietic stem cell niche: an interplay among a repertoire of multiple functional niches. Biochim Biophys Acta 2013; 1830:2 2404 - 9; http://dx.doi.org/10.1016/j.bbagen.2012.08.023; PMID: 22967757
- Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M, Heveker N. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem 2005; 280:9895 - 903; http://dx.doi.org/10.1074/jbc.M411151200; PMID: 15632118
- Forde S, Tye BJ, Newey SE, Roubelakis M, Smythe J, McGuckin CP, Pettengell R, Watt SM. Endolyn (CD164) modulates the CXCL12-mediated migration of umbilical cord blood CD133+ cells. Blood 2007; 109:1825 - 33; http://dx.doi.org/10.1182/blood-2006-05-023028; PMID: 17077324
- Wang JF, Park IW, Groopman JE. Stromal cell-derived factor-1alpha stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide-3 kinase and protein kinase C. Blood 2000; 95:2505 - 13; PMID: 10753828
- Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol 2004; 35:233 - 45; http://dx.doi.org/10.1023/B:HIJO.0000032355.66152.b8; PMID: 15339043
- Petit I, Goichberg P, Spiegel A, Peled A, Brodie C, Seger R, Nagler A, Alon R, Lapidot T. Atypical PKC-zeta regulates SDF-1-mediated migration and development of human CD34+ progenitor cells. J Clin Invest 2005; 115:168 - 76; PMID: 15630457
- Stupack DG, Cho SY, Klemke RL. Molecular signaling mechanisms of cell migration and invasion. Immunol Res 2000; 21:83 - 8; http://dx.doi.org/10.1385/IR:21:2-3:83; PMID: 10852105
- Voermans C, Lento WE, Uqoezwa M, et al. Identification of Novel Regulators of Hematopoietic Stem Cell Mobilization. ASH Annual Meeting Abstracts 2005;106:1724.
- Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, Goodell MA. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol 2004; 2:e301; http://dx.doi.org/10.1371/journal.pbio.0020301; PMID: 15459755
- Ren J, Jin P, Sabatino M, Balakumaran A, Feng J, Kuznetsov SA, Klein HG, Robey PG, Stroncek DF. Global transcriptome analysis of human bone marrow stromal cells (BMSC) reveals proliferative, mobile and interactive cells that produce abundant extracellular matrix proteins, some of which may affect BMSC potency. Cytotherapy 2011; 13:661 - 74; http://dx.doi.org/10.3109/14653249.2010.548379; PMID: 21250865
- Wagner W, Wein F, Roderburg C, Saffrich R, Faber A, Krause U, Schubert M, Benes V, Eckstein V, Maul H, et al. Adhesion of hematopoietic progenitor cells to human mesenchymal stem cells as a model for cell-cell interaction. Exp Hematol 2007; 35:314 - 25; http://dx.doi.org/10.1016/j.exphem.2006.10.003; PMID: 17258080
- Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol 1992; 11:511 - 22; http://dx.doi.org/10.1089/dna.1992.11.511; PMID: 1388724
- Thapa N, Lee BH, Kim IS. TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol 2007; 39:2183 - 94; http://dx.doi.org/10.1016/j.biocel.2007.06.004; PMID: 17659994
- Shin SH, Kim J, Heo SC, Kwon YW, Kim YM, Kim IS, Lee TG, Kim JH. Proteomic identification of betaig-h3 as a lysophosphatidic acid-induced secreted protein of human mesenchymal stem cells: paracrine activation of A549 lung adenocarcinoma cells by betaig-h3. Mol Cell Proteomics 2012; 11:M111.012385; http://dx.doi.org/10.1074/mcp.M111.012385; PMID: 22159598
- Jeon ES, Kim JH, Ryu H, Kim EK. Lysophosphatidic acid activates TGFBIp expression in human corneal fibroblasts through a TGF-β1-dependent pathway. Cell Signal 2012; 24:1241 - 50; http://dx.doi.org/10.1016/j.cellsig.2012.02.009; PMID: 22374302
- Zhao YL, Piao CQ, Hei TK. Downregulation of Betaig-h3 gene is causally linked to tumorigenic phenotype in asbestos treated immortalized human bronchial epithelial cells. Oncogene 2002; 21:7471 - 7; http://dx.doi.org/10.1038/sj.onc.1205891; PMID: 12386809
- Nummela P, Lammi J, Soikkeli J, Saksela O, Laakkonen P, Hölttä E. Transforming growth factor beta-induced (TGFBI) is an anti-adhesive protein regulating the invasive growth of melanoma cells. Am J Pathol 2012; 180:1663 - 74; http://dx.doi.org/10.1016/j.ajpath.2011.12.035; PMID: 22326753
- Billings PC, Whitbeck JC, Adams CS, Abrams WR, Cohen AJ, Engelsberg BN, Howard PS, Rosenbloom J. The transforming growth factor-beta-inducible matrix protein (beta)ig-h3 interacts with fibronectin. J Biol Chem 2002; 277:28003 - 9; http://dx.doi.org/10.1074/jbc.M106837200; PMID: 12034705
- Vuori K, Ruoslahti E. Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J Biol Chem 1993; 268:21459 - 62; PMID: 7691809
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 1999; 18:3909 - 23; http://dx.doi.org/10.1093/emboj/18.14.3909; PMID: 10406796
- Woods A, Couchman JR. Protein kinase C involvement in focal adhesion formation. J Cell Sci 1992; 101:277 - 90; PMID: 1629245
- Huang AH, Stein A, Mauck RL. Evaluation of the complex transcriptional topography of mesenchymal stem cell chondrogenesis for cartilage tissue engineering. Tissue Eng Part A 2010; 16:2699 - 708; http://dx.doi.org/10.1089/ten.tea.2010.0042; PMID: 20367254
- Machesky LM, Hall A. Role of actin polymerization and adhesion to extracellular matrix in Rac- and Rho-induced cytoskeletal reorganization. J Cell Biol 1997; 138:913 - 26; http://dx.doi.org/10.1083/jcb.138.4.913; PMID: 9265656
- Voermans C, van Heese WP, de Jong I, Gerritsen WR, van Der Schoot CE. Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia 2002; 16:650 - 7; http://dx.doi.org/10.1038/sj.leu.2402431; PMID: 11960346
- Voermans C, Anthony EC, Mul E, van der Schoot E, Hordijk P. SDF-1-induced actin polymerization and migration in human hematopoietic progenitor cells. Exp Hematol 2001; 29:1456 - 64; http://dx.doi.org/10.1016/S0301-472X(01)00740-8; PMID: 11750105
- Chancellor TJ, Lee J, Thodeti CK, Lele T. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys J 2010; 99:115 - 23; http://dx.doi.org/10.1016/j.bpj.2010.04.011; PMID: 20655839
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009; 461:99 - 103; http://dx.doi.org/10.1038/nature08242; PMID: 19693013
- ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor beta-Pix. J Cell Biol 2006; 172:759 - 69; http://dx.doi.org/10.1083/jcb.200509096; PMID: 16492808
- Kassmer SH, Niggemann B, Punzel M, Mieck C, Zänker KS, Dittmar T. Cytokine combinations differentially influence the SDF-1alpha-dependent migratory activity of cultivated murine hematopoietic stem and progenitor cells. Biol Chem 2008; 389:863 - 72; http://dx.doi.org/10.1515/BC.2008.099; PMID: 18627320
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 1997; 137:481 - 92; http://dx.doi.org/10.1083/jcb.137.2.481; PMID: 9128257
- Bruinsma SP, Cagan RL, Baranski TJ. Chimaerin and Rac regulate cell number, adherens junctions, and ERK MAP kinase signaling in the Drosophila eye. Proc Natl Acad Sci U S A 2007; 104:7098 - 103; http://dx.doi.org/10.1073/pnas.0701686104; PMID: 17438281
- O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, Ferrante J, Gentalen E, Glazer M, Gossett J, Hacker K, et al. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc Natl Acad Sci U S A 2006; 103:16153 - 8; http://dx.doi.org/10.1073/pnas.0607973103; PMID: 17053065
- Knittle JE, Roach D, Horn PB, Voss KO. Laser-induced fluorescence detector for capillary-based isoelectric immunoblot assay. Anal Chem 2007; 79:9478 - 83; http://dx.doi.org/10.1021/ac071537z; PMID: 18020314
- Thapa N, Kang KB, Kim IS. Beta ig-h3 mediates osteoblast adhesion and inhibits differentiation. Bone 2005; 36:232 - 42; http://dx.doi.org/10.1016/j.bone.2004.08.007; PMID: 15780949
- Li Z, Miao Z, Jin G, Li X, Li H, Lv Z, Xu HM. βig-h3 supports gastric cancer cell adhesion, migration and proliferation in peritoneal carcinomatosis. Mol Med Rep 2012; 6:558 - 64; PMID: 22710407
- Bae JS, Lee SH, Kim JE, Choi JY, Park RW, Yong Park J, Park HS, Sohn YS, Lee DS, Bae Lee E, et al. Betaig-h3 supports keratinocyte adhesion, migration, and proliferation through alpha3beta1 integrin. Biochem Biophys Res Commun 2002; 294:940 - 8; http://dx.doi.org/10.1016/S0006-291X(02)00576-4; PMID: 12074567
- Irigoyen M, Ansó E, Salvo E, Dotor de las Herrerías J, Martínez-Irujo JJ, Rouzaut A. TGFbeta-induced protein mediates lymphatic endothelial cell adhesion to the extracellular matrix under low oxygen conditions. Cell Mol Life Sci 2008; 65:2244 - 55; http://dx.doi.org/10.1007/s00018-008-8071-9; PMID: 18560760
- Kim JE, Kim SJ, Lee BH, Park RW, Kim KS, Kim IS. Identification of motifs for cell adhesion within the repeated domains of transforming growth factor-beta-induced gene, betaig-h3. J Biol Chem 2000; 275:30907 - 15; http://dx.doi.org/10.1074/jbc.M002752200; PMID: 10906123
- Lee BH, Bae JS, Park RW, Kim JE, Park JY, Kim IS. betaig-h3 triggers signaling pathways mediating adhesion and migration of vascular smooth muscle cells through alphavbeta5 integrin. Exp Mol Med 2006; 38:153 - 61; http://dx.doi.org/10.1038/emm.2006.19; PMID: 16672769
- Nam JO, Son HN, Jun E, Cha K, Lee BH, Park RW, Kim IS. FAS1 domain protein inhibits VEGF165-induced angiogenesis by targeting the interaction between VEGFR-2 and αvβ3 integrin. Mol Cancer Res 2012; 10:1010 - 20; http://dx.doi.org/10.1158/1541-7786.MCR-11-0600; PMID: 22710795
- Skonier J, Bennett K, Rothwell V, Kosowski S, Plowman G, Wallace P, Edelhoff S, Disteche C, Neubauer M, Marquardt H, et al. beta ig-h3: a transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol 1994; 13:571 - 84; http://dx.doi.org/10.1089/dna.1994.13.571; PMID: 8024701
- Shelton L, Rada JA. Inhibition of human scleral fibroblast cell attachment to collagen type I by TGFBIp. Invest Ophthalmol Vis Sci 2009; 50:3542 - 52; http://dx.doi.org/10.1167/iovs.09-3460; PMID: 19387070
- Nam JO, Jeong HW, Lee BH, Park RW, Kim IS. Regulation of tumor angiogenesis by fastatin, the fourth FAS1 domain of betaig-h3, via alphavbeta3 integrin. Cancer Res 2005; 65:4153 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-04-2705; PMID: 15899806
- Becker J, Erdlenbruch B, Noskova I, Schramm A, Aumailley M, Schorderet DF, Schweigerer L. Keratoepithelin suppresses the progression of experimental human neuroblastomas. Cancer Res 2006; 66:5314 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-05-3049; PMID: 16707457
- Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, et al. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev 2008; 22:308 - 21; http://dx.doi.org/10.1101/gad.1632008; PMID: 18245446
- Reinboth B, Thomas J, Hanssen E, Gibson MA. Beta ig-h3 interacts directly with biglycan and decorin, promotes collagen VI aggregation, and participates in ternary complexing with these macromolecules. J Biol Chem 2006; 281:7816 - 24; http://dx.doi.org/10.1074/jbc.M511316200; PMID: 16434404
- Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol 2009; 10:843 - 53; http://dx.doi.org/10.1038/nrm2799; PMID: 19904298
- Zhang Y, Wen G, Shao G, Wang C, Lin C, Fang H, Balajee AS, Bhagat G, Hei TK, Zhao Y. TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res 2009; 69:37 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-08-1648; PMID: 19117985
- Nabokikh A, Ilhan A, Bilban M, Gartner W, Vila G, Niederle B, Nielsen JH, Wagner O, Base W, Luger A, et al. Reduced TGF-beta1 expression and its target genes in human insulinomas. Exp Clin Endocrinol Diabetes 2007; 115:674 - 82; http://dx.doi.org/10.1055/s-2007-984477; PMID: 18058603
- Ma J, Cui W, He SM, Duan YH, Heng LJ, Wang L, Gao GD. Human U87 astrocytoma cell invasion induced by interaction of βig-h3 with integrin α5β1 involves calpain-2. PLoS One 2012; 7:e37297; http://dx.doi.org/10.1371/journal.pone.0037297; PMID: 22629380
- Hei TK, Zhao Y, Zhou H, Ivanov V. Mechanism of radiation carcinogenesis: role of the TGFBI gene and the inflammatory signaling cascade. Adv Exp Med Biol 2011; 720:163 - 70; http://dx.doi.org/10.1007/978-1-4614-0254-1_13; PMID: 21901626
- Zhao YL, Piao CQ, Hei TK. Overexpression of Betaig-h3 gene downregulates integrin alpha5beta1 and suppresses tumorigenicity in radiation-induced tumorigenic human bronchial epithelial cells. Br J Cancer 2002; 86:1923 - 8; http://dx.doi.org/10.1038/sj.bjc.6600304; PMID: 12085188
- Shao G, Berenguer J, Borczuk AC, Powell CA, Hei TK, Zhao Y. Epigenetic inactivation of Betaig-h3 gene in human cancer cells. Cancer Res 2006; 66:4566 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-05-2130; PMID: 16651406
- Zhao Y, El-Gabry M, Hei TK. Loss of Betaig-h3 protein is frequent in primary lung carcinoma and related to tumorigenic phenotype in lung cancer cells. Mol Carcinog 2006; 45:84 - 92; http://dx.doi.org/10.1002/mc.20167; PMID: 16329146
- Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr Opin Cell Biol 2011; 23:569 - 78; http://dx.doi.org/10.1016/j.ceb.2011.07.003; PMID: 21807492
- Maijenburg MW, Noort WA, Kleijer M, Kompier CJ, Weijer K, van Buul JD, van der Schoot CE, Voermans C. Cell cycle and tissue of origin contribute to the migratory behaviour of human fetal and adult mesenchymal stromal cells. Br J Haematol 2010; 148:428 - 40; http://dx.doi.org/10.1111/j.1365-2141.2009.07960.x; PMID: 19863541
- Bergert M, Chandradoss SD, Desai RA, Paluch E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci U S A 2012; 109:14434 - 9; http://dx.doi.org/10.1073/pnas.1207968109; PMID: 22786929
- Gu Y, Williams DA. RAC2 GTPase deficiency and myeloid cell dysfunction in human and mouse. J Pediatr Hematol Oncol 2002; 24:791 - 4; http://dx.doi.org/10.1097/00043426-200212000-00027; PMID: 12468931
- Williams DA, Zheng Y, Cancelas JA. Rho GTPases and regulation of hematopoietic stem cell localization. Methods Enzymol 2008; 439:365 - 93; http://dx.doi.org/10.1016/S0076-6879(07)00427-2; PMID: 18374178
- Chauhan BK, Lou M, Zheng Y, Lang RA. Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc Natl Acad Sci U S A 2011; 108:18289 - 94; http://dx.doi.org/10.1073/pnas.1108993108; PMID: 22021442
- Chae HD, Lee KE, Williams DA, Gu Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood 2008; 111:2597 - 605; http://dx.doi.org/10.1182/blood-2007-06-093237; PMID: 18089848
- Ishibe S, Joly D, Zhu X, Cantley LG. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell 2003; 12:1275 - 85; http://dx.doi.org/10.1016/S1097-2765(03)00406-4; PMID: 14636584
- Roskoski R Jr.. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 2012; 66:105 - 43; http://dx.doi.org/10.1016/j.phrs.2012.04.005; PMID: 22569528
- Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem 2006; 99:35 - 52; http://dx.doi.org/10.1002/jcb.20956; PMID: 16823799
- Sawhney RS, Cookson MM, Omar Y, Hauser J, Brattain MG. Integrin alpha2-mediated ERK and calpain activation play a critical role in cell adhesion and motility via focal adhesion kinase signaling: identification of a novel signaling pathway. J Biol Chem 2006; 281:8497 - 510; http://dx.doi.org/10.1074/jbc.M600787200; PMID: 16461767
- Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci 2004; 117:4619 - 28; http://dx.doi.org/10.1242/jcs.01481; PMID: 15371522
- Nguyen DH, Catling AD, Webb DJ, Sankovic M, Walker LA, Somlyo AV, Weber MJ, Gonias SL. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J Cell Biol 1999; 146:149 - 64; PMID: 10402467
- Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer 2002; 2:91 - 100; http://dx.doi.org/10.1038/nrc727; PMID: 12635172
- Lämmermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol 2009; 21:636 - 44; http://dx.doi.org/10.1016/j.ceb.2009.05.003; PMID: 19523798
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol 2010; 188:11 - 9; http://dx.doi.org/10.1083/jcb.200909003; PMID: 19951899
- Sahin AO, Buitenhuis M. Molecular mechanisms underlying adhesion and migration of hematopoietic stem cells. Cell Adh Migr 2012; 6:39 - 48; http://dx.doi.org/10.4161/cam.18975; PMID: 22647939
- Cancelas JA. Adhesion, migration, and homing of murine hematopoietic stem cells and progenitors. Methods Mol Biol 2011; 750:187 - 96; http://dx.doi.org/10.1007/978-1-61779-145-1_13; PMID: 21618092
- Vagima Y, Lapid K, Kollet O, Goichberg P, Alon R, Lapidot T. Pathways implicated in stem cell migration: the SDF-1/CXCR4 axis. Methods Mol Biol 2011; 750:277 - 89; http://dx.doi.org/10.1007/978-1-61779-145-1_19; PMID: 21618098
- Zhao Y, Malinin NL, Meller J, Ma Y, West XZ, Bledzka K, Qin J, Podrez EA, Byzova TV. Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J Biol Chem 2012; 287:40012 - 20; http://dx.doi.org/10.1074/jbc.M112.380469; PMID: 23012377
- Peled A, Hardan I, Trakhtenbrot L, Gur E, Magid M, Darash-Yahana M, Cohen N, Grabovsky V, Franitza S, Kollet O, et al. Immature leukemic CD34+CXCR4+ cells from CML patients have lower integrin-dependent migration and adhesion in response to the chemokine SDF-1. Stem Cells 2002; 20:259 - 66; http://dx.doi.org/10.1634/stemcells.20-3-259; PMID: 12004084
- Jing D, Oelschlaegel U, Ordemann R, Hölig K, Ehninger G, Reichmann H, Ziemssen T, Bornhäuser M. CD49d blockade by natalizumab in patients with multiple sclerosis affects steady-state hematopoiesis and mobilizes progenitors with a distinct phenotype and function. Bone Marrow Transplant 2010; 45:1489 - 96; http://dx.doi.org/10.1038/bmt.2009.381; PMID: 20098455
- Forsberg EC, Prohaska SS, Katzman S, Heffner GC, Stuart JM, Weissman IL. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet 2005; 1:e28; http://dx.doi.org/10.1371/journal.pgen.0010028; PMID: 16151515
- Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG. Calcium signaling regulates translocation and activation of Rac. J Biol Chem 2003; 278:39413 - 21; http://dx.doi.org/10.1074/jbc.M302083200; PMID: 12888567