
Abstract
Leukocyte transendothelial migration (TEM) is one of the crucial steps during inflammation. A better understanding of the key molecules that regulate leukocyte extravasation aids to the development of novel therapeutics for treatment of inflammation-based diseases, such as atherosclerosis and rheumatoid arthritis. The adhesion molecules ICAM-1 and VCAM-1 are known as central mediators of TEM. Clustering of these molecules by their leukocytic integrins initiates the activation of several signaling pathways within the endothelium, including a rise in intracellular Ca2+, activation of several kinase cascades, and the activation of Rho-GTPases. Activation of Rho-GTPases has been shown to control adhesion molecule clustering and the formation of apical membrane protrusions that embrace adherent leukocytes during TEM. Here, we discuss the potential regulatory mechanisms of leukocyte extravasation from an endothelial point of view, with specific focus on the role of the Rho-GTPases.
Introduction
Efficient and tightly controlled leukocyte transendothelial migration (TEM) is of key importance in physiological processes such as immune surveillance and acute inflammation. Uncontrolled and excessive TEM is characteristic for various disorders such as chronic inflammatory diseases (e.g., rheumatoid arthritis, atherosclerosis, asthma) and tumor cell metastasis.Citation1,Citation2 In order to specifically interfere with excessive leukocyte or tumor cell TEM, a detailed understanding of endothelial signaling that regulates TEM is required.
It is believed that the TEM process occurs through different steps. Butcher and Springer proposed in timeless reviews the multi-step model for the process of TEM.Citation3,Citation4 Currently, the basis of this model is still accurate and some additional steps have been included (). Importantly, the active contribution of endothelial signaling in TEM has been recognized. The group of Alon described the need for the presence of immobilized chemokines on the surface of the endothelium.Citation5 Recently, they showed that the endothelium itself generates chemokines and presents those at the apical surface to promote TEM.Citation6 The same group also put forward the importance of shear flow during TEM.Citation7 Barreiro and colleagues, together with Carman and co-workers, showed the contribution of cup-like membrane structures created by the endothelium that surround adherent leukocytes in order to facilitate directional transmigration.Citation8-Citation10 Nevertheless, the main steps of TEM, namely rolling, adhesion, and transmigration, as proposed by Butcher and Springer more than 20 years ago, still constitute the central processes that drive leukocyte extravasation ().Citation3,Citation4
Figure 1. The multistep process of leukocyte transendothelial migration, divided in five consecutive steps. Step 1 represents the rolling and tethering phase; step 2 shows the initial adhesion of the leukocytes to the endothelium. Step 3 is the firm adhesion and crawling part. In step 4, the cup-like structures are formed, resulting in step 5; actual transmigration, either para- or transcellular.
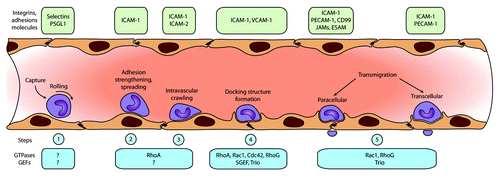
In this review, we discuss the regulatory mechanisms that control the different steps of leukocyte extravasation () from an endothelial point of view, with specific focus on the role of the Rho-GTPases and their activators guanine-nucleotide exchange factors (GEFs).
Step 1: Capture and Rolling
Initially, transient leukocyte–endothelial cell interactions are mediated by the endothelial adhesion molecules E- and P-selectin with their leukocyte counterparts L-selectin and PSGL-1. These interactions comprise the so-called “rolling step;” slowing down the leukocytes on the endothelial cell surface (, step 1). P-selectin, also known as CD62P, is present in Weibel-Palade bodies and can be quickly released and presented at the luminal side of the endothelium upon activation with histamine/thrombin or with pharmacological compounds such as Ca2+ ionophores or phorbol esters.Citation11-Citation14 Maximal expression of P-selectin on the endothelial surface is seen after 10 min of activation, after which (30–60 min) the protein is being downregulated, either by internalization or shedding. CD63 was found to be an essential co-factor for P-selectin, since endothelial cells deficient for CD63 showed a loss of P-selectin-mediated adhesion function.Citation15
E-selectin (CD62E) is not found in Weibel-Palade bodies,Citation16 but is rapidly upregulated by inflammatory stimuli, such as TNF-α and IL1β. As for P-selectin, the upregulation involves the small Rho-GTPases RhoA, RhoB, and Rac1.Citation17 Maximum expression for E-selectin is reached after approximately 3–4 h of stimulation. Interestingly, clustering of E- and also P-selectin, using crosslinking of antibodies, induced intracellular signals into the endothelium, including a remodeling of actin stress fibers.Citation18 The authors also reported that initial leukocyte adhesion to the endothelium induced an immediate increase in calcium concentrations in the endothelium, in line with the role of selectins, mediating initial interaction of the leukocytes with the endothelium. Additionally, clustering of E-selectin has been shown to redistribute E-selectin to caveolin-rich membrane domains and promote its interaction with and activation of phospholipase Cγ.Citation19 Moreover, E-selectin clustering also triggers the activation of Erk1/2 and expression of c-fos,Citation20,Citation21 as well as changes in the endothelial cell morphology and F-actin distribution, in line with the work by Lorenzon and colleagues.Citation18 In fact, clustering of E-selectin induced by monocytes was dependent on upstream RhoA activation.Citation22 Additionally, E-selectin clustering induced a linkage to the actin cytoskeleton through its intracellular tail.Citation22-Citation24 Yoshida and co-workers showed the presence of actin-associated proteins α-actinin, vinculin, filamin, FAK, and paxillin, but not talin, in E-selectin-clustering precipitation assays.Citation23 In addition, blocking actin polymerization reduced the adhesive capacity of E-selectin. This was measured by applying mechanical stress to the endothelial cells using anti-E-selectin antibody-coated ferromagnetic beads with a magnetical twisting cytometer. These data suggested that actin remodeling is instrumental for proper E-selectin function and strongly suggests a prominent role for small Rho-GTPases. And although Rho-GTPases have been implicated upstream from P- and E-selectin, so far no role for Rho-GTPase signaling downstream from either P- or E-selectin during step 1 of TEM has been reported.
In the next section, we will discuss the endothelial signaling during step 2 and 3 in more detail.
Step 2 and 3: Adhesion, Strengthening, Spreading, and Intravascular Crawling
Firm adhesion of leukocytes is initiated upon binding of activated integrins to their endothelial ligands Intercellular Adhesion Molecule 1 (ICAM-1) and Vascular Cell Adhesion Molecule (VCAM-1). To demonstrate the importance of ICAM-1 in firm adhesion and TEM, Chinese Hamster Ovary (CHO), or HeLa cells that expressed no or very little endogenous ICAM-1 were artificially transfected with ICAM-1. This was sufficient to recapitulate the entire process of neutrophil adhesion and migration across these cellsCitation25 (van Buul JD, data not shown). The group of Silverstein showed that leukocyte adhesion to endothelial cells induced the release of intracellular calcium in endothelial cells although the upstream signals, i.e., selectin- or CAM-mediated, were unknown at that time.Citation26 Nevertheless, they were the first to show the importance of intracellular signaling in endothelial cells during the adhesion step of leukocyte TEM.
Clustering of the adhesion molecules ICAM-1 and VCAM-1 is crucial to induce endothelial signaling. This is both a passive and an active event. ICAM-1 and VCAM-1 reside in preformed membrane nanodomains (also termed endothelial adhesive platforms) that are controlled by several members of the tetraspanin family of integral membrane proteins.Citation27 Leukocyte binding to the endothelium through the engagement of integrins induces these nanodomains to coalesce into higher order clusters, leading to the activation of several signaling pathways in endothelial cells. It is now well recognized that this signaling, in turn, activates positive feedback loops, promoting additional clustering of Ig-CAMs like ICAM-1 and VCAM-1 into ring-like structures around adherent leukocytes, which subsequently amplifies signaling through a positive feedback loop.Citation9,Citation28-Citation31
The carboxyl (C)-terminal intracellular domain of ICAM-1 is relatively small (28 amino acids) compared with its extracellular region (481 aa). Nevertheless, signaling by ICAM-1 was shown to be dependent on this small intracellular domain.Citation30,Citation32-Citation34 Also, VCAM-1 shows a relatively small C-terminal intracellular domain compared with its extracellular domain (19 vs. 699 aa, respectively). Since the C-terminal domains of these proteins do not contain any apparent signaling motifs, signaling is likely relayed via adaptor proteins. Several adaptor proteins have been reported to interact with the intracellular domains of ICAM-1 and VCAM-1, including α-actinin, cortactin, filamin, and members of the ERM protein family.Citation8,Citation25,Citation35-Citation38 Besides acting as scaffolding proteins, these adapters are also able to bind actin, and can therefore anchor ICAM-1 and VCAM-1 physically to the actin cytoskeleton.Citation39-Citation42
Early studies showed that leukocyte adhesion and clustering of ICAM-1 promote an increase in intracellular Ca2+ levels.Citation26 Others followed up on this crucial finding and showed that the change in calcium concentration leads to activation of the tyrosine kinase Src by protein kinase C (PKC).Citation43 In turn, Src induces tyrosine phosphorylation of focal adhesion proteins, such as paxillin, cortactin, and FAK. The group of Luscinskas underscored the importance of endothelial Src-mediated phosphorylation of cortactin in leukocyte TEM. They showed that a non-phosphorylatable mutant of cortactin, expressed in endothelial cells, blocked leukocyte TEM.Citation44 In addition, ICAM-1 clustering leads to the activation of the small GTPase RhoA, which stimulates the formation of F-actin stress fibers.Citation45,Citation46 Moreover, RhoA activity was also demonstrated to be required for efficient ICAM-1 recruitment around adherent monocytes, suggesting an upstream role for RhoA within the ICAM-1-induced signaling cascade.Citation22 Interestingly, others have shown that thrombin-induced RhoA activation, resulting in increased stress fibers, showed loss of cell–cell contacts and increased gap formation in endothelial cells.Citation47 However, high RhoA activity and increased stress fibers downstream from ICAM-1 clustering did not necessarily result in endothelial gap formation.Citation46,Citation48 This indicates that the consequences of the intracellular signals that lead from RhoA activation to cell–cell junctions in endothelial cells specifically depend on the extracellular stimulus.
Although guanine–nucleotide exchange factors (GEFs) are likely candidates to activate RhoA downstream from ICAM-1 engagement, no direct RhoA–GEF interaction has been identified so far. Etienne and colleagues showed that the Rap1–GEF C3G binds to Cas upon antibody-induced clustering of ICAM-1.Citation45 They additionally indicated no role for the Ras- and Rac1-GEF SOS-1 downstream from ICAM-1 in their model system. Interestingly, RhoA can also be directly activated in a GEF-independent manner by reactive oxygen species (ROS).Citation49 Aghajanian and co-workers demonstrated that ROS can directly target two critical cysteine residues that are located in a unique redox-sensitive motif within the phosphoryl binding loop of RhoA, resulting in RhoA-GTP loading.Citation49
Clustering of VCAM-1 was shown to promote activation of the GTPase Rac1, leading to the production of ROS.Citation50,Citation51 VCAM-1-dependent ROS production was demonstrated to regulate the activation of matrix metalloproteases, which may contribute to the local breakdown of the endothelial adherens junctions.Citation52 In addition, VCAM-1 clustering was shown to regulate lymphocyte TEM by activation of the kinase PKCα and the tyrosine phosphatase PTP1B in a ROS-dependent manner.Citation53 More recently, it was shown by the group of Vestweber that VCAM-1-induced Rac1 activation and subsequent ROS production was also involved in the dissociation of the endothelial receptor phosphatase VE-PTP from the junctional adhesion molecule VE-cadherin, which is an obligatory event during leukocyte TEM.Citation54
More recent advances in confocal microscopy have allowed detailed analysis of leukocyte–endothelium interactions in three dimensions. This revealed that upon clustering, both ICAM-1 and VCAM-1 were recruited to cup-like, F-actin-rich membrane protrusions that surround adherent leukocytes (step 4).Citation8-Citation10 In the next section, we will discuss the signaling and function that underlies the formation of these structures.
Step 4: Cup-Like Structures
Barreiro and co-workers were the first to report on the induction of these F-actin rich “cups.”Citation8 Using live-cell imaging, they showed that adhesion and spreading of lymphoblasts on the endothelial cell surface induced the recruitment of VCAM-1 and the ERM-family member moesin, whereas ICAM-1 and moesin recruitment were primarily observed during TEM. In addition, adhesion of K562 cells that were stably transfected with α4 integrin (4M7 cells) resulted in an actin-rich endothelial cup structure embracing the 4M7 cells, and contained ICAM-1, VCAM-1, moesin, ezrin, α-actinin, vinculin, and VASP. Phospho-inositides and the Rho-ROCK-pathway were involved in the generation and maintenance of these so-called docking structures.Citation8 Initially, Barreiro and colleagues proposed that the structures are essential during the docking or adhesion phase and may protect leukocytes from detachment by shear flow, hence the term “docking structures.”Citation8,Citation55 This was underscored by Samson et al., who demonstrated using in vivo studies that removal of one of the crucial players involved in docking structure formation reduced leukocyte adhesion.Citation56
However, although docking structures are formed around adhering leukocytes, it is still under debate whether or not these structures function in strengthening the adhesion of the leukocyte to the endothelium. Several reports have shown that inhibition of ICAM-1 signaling, and thus, preventing docking structure formation in vitro, affects leukocyte TEM, but not adhesion.Citation30,Citation32,Citation33 In fact, Carman and colleagues revealed that the endothelium pro-actively generates microfilament, microtubule, and calcium-dependent ICAM-1-enriched cup-like structures within minutes of binding to LFA-1-bearing leukocytes.Citation9 Interestingly, disruption of endothelial projections by blocking actin polymerization (cytochalasin D) or microtubule polymerization (colchicine), or by chelating calcium (BAPTA) did not affect firm adhesion of leukocytes. Thus, from this work, these structures appear not to function in adhesion strengthening, but may in fact play a more direct role in the final diapedesis step.
To assess the role of Rho-GTPases in the formation of these endothelial actin-rich projections, Carman and colleagues treated the endothelial cells with Clostridium diffucile toxin-B to inhibit Rho, Rac1, and Cdc42.Citation9 Toxin-B treatment was associated with a 2-fold reduction in total projections and TEM. In contrast to the docking structures proposed by Barreiro and co-workers, blocking Rho by C3 transferase had no effect on either projections or TEM. These data suggest an active role for the Rho-GTPases Rac1 and Cdc42 in projection formation and provide correlative support for a functional role of projections in leukocyte diapedesis.
A year later, the Carman lab demonstrated a cup-like structure that is formed around transmigrating leukocytes in both the paracellular and transcellular migration pathway.Citation10 Disruption of these projections was highly correlated with inhibition of transmigration. Again, blocking Rho-kinase (by Y27632) or Rho (by C3) did not prevent cup formation downstream from ICAM-1 engagement. The structure contained high ICAM-1 and VCAM-1 and was enriched for vertical microvilli-like structures. Leukocyte integrins were redistributed into linear tracks oriented in parallel to the direction of diapedesis. Carman and co-workers proposed that docking structures may promote diapedesis by providing additional membrane surface to provide directional guidance to leukocytes for transmigration, and hence, proposed the term “transmigratory cups.”Citation10,Citation57
Alternatively, the group of Kubes demonstrated in vivo that during neutrophil TEM, docking structures develop into dome-like structures, which completely encapsulate the neutrophil.Citation58,Citation59 They additionally showed that when dome formation was inhibited by silencing the expression of the F-actin-binding protein Lsp1, vascular leakage during neutrophil diapedesis was increased.Citation58 They therefore proposed that endothelial domes may function to seal off the transmigrating leukocyte in order to minimize vascular leakage during extravasation. It is interesting to note that several molecular players in the formation of endothelial cup structures, such as Rac1, cortactin, and filamin, were also reported to be important for maintaining endothelial monolayer integrity.Citation38,Citation60,Citation61 It is therefore possible that activation or recruitment of these molecules during leukocyte TEM may have a dual purpose by promoting diapedesis, and at the same time maintaining endothelial barrier integrity, although proof for this hypothesis is currently lacking.
The formation of the endothelial cup-like structure shows several similarities with the formation of the so-called phagocytic cup. Also, the role of the Rho-GTPase RhoG in the phagocytosis of apoptotic cellsCitation62 and its specific exchange factor SH3-containing GEF (SGEF) in macropinocytosisCitation63 show similar functions in both processes. This was the rationale to examine if RhoG and SGEF may contribute to the formation of endothelial cups and participate in TEM. Our group showed that ICAM-1 binds SGEF through its intracellular tail.Citation34 Subsequently, this results in Src-dependent activation of RhoG, leading to the formation of apical cup assembly. Specifically, SGEF binds to ICAM-1 via its SH3 domain and silencing of endothelial SGEF or RhoG decreased cup formation and inhibited leukocyte TEM, but did not affect leukocyte adhesion. Recently, Schnoor and co-workers showed that ICAM-1-induced activation of RhoG required cortactin.Citation38 Interestingly, the ICAM-1/SGEF interaction was independent of ICAM-1 clustering. However, using nucleotide-free GST mutants of RhoG to measure GEF activity,Citation64 we found that ICAM-1 clustering did activate SGEF (JDvB, data not shown). Using a SGEF-deficient mouse line, Samson and colleagues showed that SGEF deficiency resulted in reduced on-set of atherosclerosis, most likely by the inability of the vasculature to form proper cup-like structures and prevent leukocyte TEM.Citation56 These data suggest that the RhoG/SGEF signaling axis is one of the central mediators of cup-structure formation.
The work by Carman and colleagues suggested a role for the small GTPase Rac1 downstream from leukocyte adhesion. When measuring Rac1 activation downstream from ICAM-1 clustering, in parallel with RhoG activation, it became clear that Rac1 activity preceded RhoG activation.Citation30 This indicated that, next to SGEF, which activates RhoG, another GEF is activated downstream from ICAM-1 clustering. Our initial data showed that ICAM-1, when clustered, interacted with the actin-adaptor protein filamin.Citation35 Interestingly, the exchange factor Trio was shown to recruit filamin.Citation65 Also, deBakker and colleagues showed that Trio activates RhoG to allow phagocytosis of apoptotic cells.Citation62 Moreover, next to RhoG, this GEF was also able to activate Rac1 and RhoA,Citation66 two GTPases known to be involved in downstream ICAM-1 signaling.Citation30,Citation45,Citation46,Citation67 Additional data from our laboratory showed that depletion of Rac1 or RhoG reduced TEM of primary neutrophils.Citation30 Rac1 depletion showed defects in actual ICAM-1 clustering, and RhoG depletion impaired the induction of cup structures.Citation30 These data implicate that Rac1 and RhoG have separate functions to induce endothelial cup structures upon leukocyte binding ().
Figure 2. The cup-like structure. Signaling routes that are initiated by clustering of ICAM-1 and result in the remodeling of the actin cytoskeleton and the formation of cup-like structures. Signaling includes the activation of the small GTPases Rac1and RhoG downstream from ICAM-1 clustering and involvement of at least two GEFs, Trio and SGEF. Additionally, Rac1 activation may result in a positive feedback loop, increasing the clustering of ICAM-1.
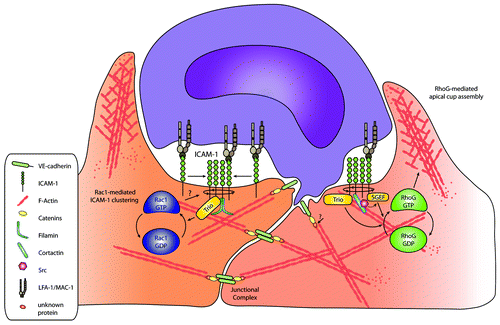
Additionally, Trio binds to the intracellular tail of ICAM-1 via regions located in the N-terminal GEF domain.Citation30 Surprisingly, the binding of Trio to ICAM-1 is independent of filamin. However, reduced filamin expression in endothelial cells did prevent ICAM-1-induced Rac1 and RhoG activation.Citation30 Moreover, also ICAM-1-induced Trio activation, measured through its binding to the nucleotide-free GTPase mutants (Rac1-G15A/RhoG-G15A), was impaired in filamin-deficient cells. So, although filamin did not regulate the binding of Trio to ICAM-1, it did control downstream GTPase activity, probably through other signaling molecules such as Src-family kinases.Citation40
One of the drawbacks of the above-described study is the use of artificial anti-ICAM-1 antibody-coated beads to cluster ICAM-1. It is therefore good to note that the kinetics of ICAM-1 clustering in these experiments may differ from ICAM-1 signaling during leukocyte diapedesis. In fact, from the time-point of firm adhesion, neutrophils take approximately 100 s to cross the endothelium.Citation68 Thus, the real-time temporal and also spatial activation of Rac1 and RhoG during neutrophil TEM remains an open question.
As mentioned above, the different steps in the formation of endothelial cup-like structures are reminiscent of the process of phagocytic cup formation by phagocytic cells.Citation69 Formation of a phagocytic cup is initiated by a local coalescence of F-actin in a ring-like structure, followed by membrane protrusion around the opsonized cell or particle.Citation70 Finally, actomyosin contractility forces ingestion and membrane fusion completes the process.Citation64,Citation65 Although we never observed complete closure and fusion of membrane protrusions during docking structure formation in vitro, others have reported in vivo docking structures advancing into dome-like structures, completely covering the transmigrating leukocyte.Citation59 Interestingly, similar molecular players, including the GTPases RhoG, Rac1/2, Ccd42, RhoA and GEFs Trio, and SGEF were reported to regulate phagocytosis or related processes such as macropinocytosis, suggesting that cells use analogous signaling pathways.Citation62,Citation63,Citation71 Using FRET-based biosensors for Rac1, Rac2, and Cdc42, these GTPases were shown to have different spatial activation patterns within the phagocytic cup.Citation72 It is therefore tempting to speculate that also within a docking structure RhoG, Rac1, and RhoA are distinctly spatially activated, with RhoA activation at the base of the cup, followed by Rac1 at the base and leading edge of the cup, and RhoG within the membrane protrusions. Previous work to delineate the role of GTPase signaling in leukocyte TEM was primarily based on inhibitor studies, knockdown studies, or biochemical assays such as classical GTPase pull-down or pull-down assays using nucleotide-free GTPase mutants. To date, genetically encoded FRET-based biosensors of Rho-GTPases would be the best tool to delineate the fast and local kinetics of Rho-GTPase activation in endothelial cells during leukocyte TEM.Citation73 Overall, it is evident that endothelial signaling is essential to initiate the final step of the TEM process: transmigration.
Step 5: Transmigration
To breech the endothelial barrier, leukocytes can take two different routes: (1) paracellular, i.e., through the endothelial cell–cell junctions or (2) transcellular, i.e., through the endothelial cell body (, step 5).Citation57,Citation74,Citation75 However, what signal determines to finally transmigrate across the endothelium and by which pathway?
First, shear stress has been suggested to be an important initiator of diapedesis.Citation7 Although shear clearly plays an important role in TEM and triggers diapedesis, robust diapedesis is also observed in the absence of shear. For more information on the role of shear stress on TEM, the reader is referred to reviews, listed here.Citation28,Citation76,Citation77
Second, central-to-peripheral expression gradients of adhesion molecules, such as PECAM-1, CD99, and JAMs, have been proposed to direct leukocytes to those sides to provide the required traction to drive diapedesis at these locations.Citation78-Citation80 In addition, chemokines presented at the luminal surface of the endothelium may also drive diapedesis through cell–cell junctions by using similar gradients as the adhesion molecules. For instance, IL-8 and RANTES have been shown to be distributed apically on endothelial microvilli in vivo.Citation79,Citation81,Citation82 Recently, it has been shown that perivascular macrophages, lining pericytes that cover blood vessels, locally secrete chemokines that cause local “hotspots” for neutrophil diapedesis in vivo.Citation83
Third, the spatial and temporal recruitment of adhesion molecules upon leukocyte binding, followed by their interactions with cytoskeletal components, induce signals in the endothelium, resulting in actin-rich structures that embrace adherent leukocyte, i.e., cup-like structures (step 4), and may provide directional guidance to leukocytes for diapedesis.Citation57
The dynamic distribution of endothelial and leukocytic adhesion molecules with the actin cytoskeleton during TEM was first studied by Martin Sandig and co-workers.Citation84 They identified a circular structure in endothelial cell–cell contact regions that facilitates leukocyte diapedesis. Additionally, they noticed that leukocytes induced LFA-1-containing pseudopodia that penetrated in between endothelial cells. At sites of diapedesis, high levels of LFA-1 and F-actin were found, suggesting a major role for these molecules in TEM. This work was supported by work from the Luscinskas lab.Citation85 They showed that upon transmigration, LFA-1 was rapidly redistributed in a ring-like structure together with ICAM-1. The redistribution and co-clustering of ICAM-1 and LFA-1 was also demonstrated by the group of Carman.Citation9,Citation86 In fact, they showed that invasive podosomes from leukocytes initiate transcellular migration, i.e., through the endothelial cell body. Sandig and co-workers furthermore showed that endothelial adherence junction proteins were locally redistributed at sites of leukocyte diapedesis, but the VE-cadherin complex, judged by VE-cadherin and α-catenin co-localization, remained intact during diapedesis.Citation84 Our previous work underscored that junctional proteins like VE-cadherin are locally dispersed upon passage of leukocytes.Citation48 Additionally, Su and co-workers demonstrated differential movement of VE-cadherin and PECAM-1 upon leukocyte passage. Whereas these authors underscored the local redistribution of VE-cadherin, they additionally showed that PECAM-1 forms a ring-like structure upon diapedesis.Citation87 These data show that, next to endothelial cell adhesion molecules, junctional proteins can be differently regulated during the final TEM step, and therefore, may be involved in the final decision when and where leukocytes cross.
Forces during TEM
Remarkable little vascular leakage occurs when leukocytes cross the endothelium through the cell–cell junctions.Citation88,Citation89 However, under certain patho-physiological conditions such as chronic inflammation, leakage is increased.Citation90 To be able to interfere with this effect, it is important to understand how junctions are restored after leukocyte passage. Recently, Martinelli and co-workers provide a mechanism by which endothelial cells maintain their barrier function during leukocyte transendothelial migration.Citation91 In response to µm-scale disruptions induced by transmigrating leukocytes, endothelial cells generate unique ventral lamellipodia that close these small gaps in the endothelium. These ventral lamellipodia were enriched in Rac1 effectors cortactin, IQGAP, and p47phox. They propose that barrier disruptions are detected as local release of isometric tension and unloading of force, which is directly coupled to ROS-dependent self-restorative membrane protrusions.Citation91
Closing the gap after leukocyte passage is one thing, opening the junctions to allow crossing is another. Although several papers have described the importance of ICAM-1-induced phosphorylation of VE-cadherin, possibly through Src-like kinases, leading to the opening of VE-cadherin-mediated cell–cell junctions, and thereby, facilitating TEM (reviewed in refs. Citation92–Citation95), we focused in this section on the potential role of endothelial tension in the final step of TEM.
Hixenbaugh and colleagues showed that fMLP-stimulated neutrophils induced phosphorylation of endothelial myosin light chain (MLC) on T18/S19 that was mediated by the calcium/calmodulin-regulated enzyme MLC-kinase.Citation96 In turn, MLC-kinase facilitated the interaction of myosin with the actin cytoskeleton, leading to the actomyosin–contractile responses and increased tension in endothelial cells. They proposed that increased endothelial contraction and generation of isometric tension may function as a mechanism by which endothelial cells open their intercellular junctions, thereby facilitating leukocyte TEM (). Adherent neutrophils measured under static conditions induce cytoskeletal rearrangements in the form of stress fibers and increase pMLC after 30 min.Citation97 Inhibition of the calcium–calmodulin-regulated enzyme MLC-kinase (by KT-5926 or ML-9) or treatment with the calmodulin antagonist Trifluoperazine reduced PMN transendothelial migration toward a leukotriene B4 chemotactic gradient, indicating that the cytoskeletal rearrangements were depending on calcium signaling. When endothelial cells were treated with calyculin, a myosin-phosphatase inhibitor, increasing MLC phosphorylation, this resulted in increased endothelial cell contraction, paracellular gap formation, and enhanced neutrophil TEM.Citation98 In addition, inhibition of the endothelial Rho/ROCK pathway, using C3 or Y-27632, significantly reduced actin polymerization, myosin II filament formation, MLC phosphorylation, and neutrophil TEM, suggesting that Rho/ROCK regulate TEM through the control of MLC activity.Citation99 However, one should take in account that these assays were all done under static conditions. Also, the initial trigger to induce MLC phosphorylation downstream from leukocyte adhesion is not known.
Figure 3. Leukocyte adhesion-induced endothelial tension. Leukocyte adhesion results in the induction of actin-myosin contractility, as depicted in the figure, through the Rho-ROCK pathway or calcium signaling, resulting in MLC phosphorylation. This may finally result in the opening of cell–cell junctions, and thus, be involved in facilitating leukocyte TEM.
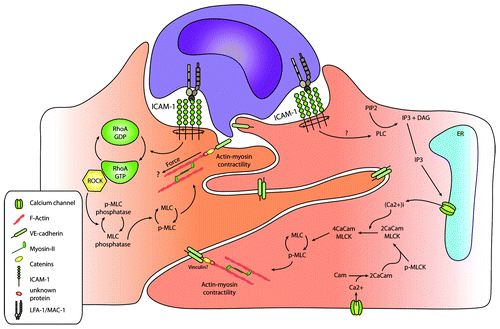
Recently, the group of Chen showed that monocytes induced traction force onto the endothelial cells.Citation100 Interestingly, the forces were measured in the endothelial cell to which a single monocyte was adhering to. Moreover, the direction of these forces aligned centripetally to the migrating monocytes. The increased endothelial traction force was measured in the sub-cellular zone of monocytes adhesion. Thus, the above-discussed results suggest that leukocytes can directly induce signals into a single endothelial cell that in turn induce actin-mediated forces. In fact, Liu and colleagues used beads that were coated with anti-ICAM-1 and anti-VCAM-1 antibodies. Using these beads they noticed similar traction forces, indicating that these signals are indeed submitted through ICAM-1 and VCAM-1 and induced by clustering. Potentially, this process can be involved in opening endothelial cell–cell junctions, and thus, in facilitating leukocyte TEM.
Conclusion
From the above-discussed data, it is clear that endothelial Rho-GTPases play an essential role in the process of transendothelial migration by remodeling the actin cytoskeleton. In particular in step 2 to 5, Rho-GTPase signaling is critical to remodel the actin cytoskeleton to induce membrane protrusions, i.e., cup-like structures and support leukocyte-induced tension force in order to open or close the cell–cell junctions or gaps through which leukocytes have crossed. Future challenges remain to investigate the spatial and temporal activation of the Rho-GTPases during the different steps of TEM, and which GEFs are locally activating these GTPases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof Dr P.L. Hordijk for critically reading the manuscript. Heemskerk N is supported by a Landsteiner Foundation for Blood Transfusion Research (LSBR) fellowship (grant #1028). van Buul JD is a NHS Dekker fellow (grant #2005T039).
References
- Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res 2009; 105:223 - 30; http://dx.doi.org/10.1161/CIRCRESAHA.109.200717; PMID: 19644057
- Vestweber D. Novel insights into leukocyte extravasation. Curr Opin Hematol 2012; 19:212 - 7; http://dx.doi.org/10.1097/MOH.0b013e3283523e78; PMID: 22395664
- Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 1991; 67:1033 - 6; http://dx.doi.org/10.1016/0092-8674(91)90279-8; PMID: 1760836
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 1994; 76:301 - 14; http://dx.doi.org/10.1016/0092-8674(94)90337-9; PMID: 7507411
- Cinamon G, Shinder V, Shamri R, Alon R. Chemoattractant signals and beta 2 integrin occupancy at apical endothelial contacts combine with shear stress signals to promote transendothelial neutrophil migration. J Immunol 2004; 173:7282 - 91; PMID: 15585851
- Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, Klein E, Shinder V, Stoler-Barak L, Feigelson SW, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol 2012; 13:67 - 76; http://dx.doi.org/10.1038/ni.2173; PMID: 22138716
- Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol 2001; 2:515 - 22; http://dx.doi.org/10.1038/88710; PMID: 11376338
- Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 2002; 157:1233 - 45; http://dx.doi.org/10.1083/jcb.200112126; PMID: 12082081
- Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol 2003; 171:6135 - 44; PMID: 14634129
- Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 2004; 167:377 - 88; http://dx.doi.org/10.1083/jcb.200404129; PMID: 15504916
- Bonfanti R, Furie BC, Furie B, Wagner DD. PADGEM (GMP140) is a component of Weibel-Palade bodies of human endothelial cells. Blood 1989; 73:1109 - 12; PMID: 2467701
- McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest 1989; 84:92 - 9; http://dx.doi.org/10.1172/JCI114175; PMID: 2472431
- McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol 2002; 14:581 - 6; http://dx.doi.org/10.1016/S0955-0674(02)00367-8; PMID: 12231353
- Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev 1999; 79:181 - 213; PMID: 9922371
- Doyle EL, Ridger V, Ferraro F, Turmaine M, Saftig P, Cutler DF. CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 2011; 118:4265 - 73; http://dx.doi.org/10.1182/blood-2010-11-321489; PMID: 21803846
- Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone MA Jr.. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc Natl Acad Sci U S A 1987; 84:9238 - 42; http://dx.doi.org/10.1073/pnas.84.24.9238; PMID: 2827173
- Cernuda-Morollón E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res 2006; 98:757 - 67; http://dx.doi.org/10.1161/01.RES.0000210579.35304.d3; PMID: 16574916
- Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol 1998; 142:1381 - 91; http://dx.doi.org/10.1083/jcb.142.5.1381; PMID: 9732297
- Kiely JM, Hu Y, García-Cardeña G, Gimbrone MA Jr. Lipid Raft Localization of Cell Surface E-Selectin Is Required for Ligation-Induced Activation of Phospholipase C gamma. J Immunol 2003; 171:3216 - 24; PMID: 12960351
- Hu Y, Kiely JM, Szente BE, Rosenzweig A, Gimbrone MA Jr.. E-selectin-dependent signaling via the mitogen-activated protein kinase pathway in vascular endothelial cells. J Immunol 2000; 165:2142 - 8; PMID: 10925300
- Hu Y, Szente B, Kiely JM, Gimbrone MA Jr.. Molecular events in transmembrane signaling via E-selectin. SHP2 association, adaptor protein complex formation and ERK1/2 activation. J Biol Chem 2001; 276:48549 - 53; PMID: 11602579
- Wójciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol 1999; 145:1293 - 307; http://dx.doi.org/10.1083/jcb.145.6.1293; PMID: 10366600
- Yoshida M, Westlin WF, Wang N, Ingber DE, Rosenzweig A, Resnick N, Gimbrone MA Jr.. Leukocyte adhesion to vascular endothelium induces E-selectin linkage to the actin cytoskeleton. J Cell Biol 1996; 133:445 - 55; http://dx.doi.org/10.1083/jcb.133.2.445; PMID: 8609175
- Kaplanski G, Farnarier C, Benoliel AM, Foa C, Kaplanski S, Bongrand P. A novel role for E- and P-selectins: shape control of endothelial cell monolayers. J Cell Sci 1994; 107:2449 - 57; PMID: 7531200
- Celli L, Ryckewaert JJ, Delachanal E, Duperray A. Evidence of a functional role for interaction between ICAM-1 and nonmuscle alpha-actinins in leukocyte diapedesis. J Immunol 2006; 177:4113 - 21; PMID: 16951376
- Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol 1993; 120:1371 - 80; http://dx.doi.org/10.1083/jcb.120.6.1371; PMID: 8449983
- Barreiro O, Zamai M, Yáñez-Mó M, Tejera E, López-Romero P, Monk PN, Gratton E, Caiolfa VR, Sánchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol 2008; 183:527 - 42; http://dx.doi.org/10.1083/jcb.200805076; PMID: 18955551
- Alcaide P, Auerbach S, Luscinskas FW. Neutrophil recruitment under shear flow: it’s all about endothelial cell rings and gaps. Microcirculation 2009; 16:43 - 57; http://dx.doi.org/10.1080/10739680802273892; PMID: 18720226
- van Buul JD, van Rijssel J, van Alphen FP, Hoogenboezem M, Tol S, Hoeben KA, van Marle J, Mul EP, Hordijk PL. Inside-out regulation of ICAM-1 dynamics in TNF-alpha-activated endothelium. PLoS One 2010; 5:e11336; http://dx.doi.org/10.1371/journal.pone.0011336; PMID: 20596527
- van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FP, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell 2012; 23:2831 - 44; http://dx.doi.org/10.1091/mbc.E11-11-0907; PMID: 22696684
- van Buul JD, van Rijssel J, van Alphen FP, van Stalborch AM, Mul EP, Hordijk PL. ICAM-1 clustering on endothelial cells recruits VCAM-1. J Biomed Biotechnol 2010; 2010:120328; http://dx.doi.org/10.1155/2010/120328; PMID: 20300427
- Lyck R, Reiss Y, Gerwin N, Greenwood J, Adamson P, Engelhardt B. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood 2003; 102:3675 - 83; http://dx.doi.org/10.1182/blood-2003-02-0358; PMID: 12893765
- Greenwood J, Amos CL, Walters CE, Couraud PO, Lyck R, Engelhardt B, Adamson P. Intracellular domain of brain endothelial intercellular adhesion molecule-1 is essential for T lymphocyte-mediated signaling and migration. J Immunol 2003; 171:2099 - 108; PMID: 12902516
- van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, García-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol 2007; 178:1279 - 93; http://dx.doi.org/10.1083/jcb.200612053; PMID: 17875742
- Kanters E, van Rijssel J, Hensbergen PJ, Hondius D, Mul FP, Deelder AM, Sonnenberg A, van Buul JD, Hordijk PL. Filamin B mediates ICAM-1-driven leukocyte transendothelial migration. J Biol Chem 2008; 283:31830 - 9; http://dx.doi.org/10.1074/jbc.M804888200; PMID: 18809679
- Oh HM, Lee S, Na BR, Wee H, Kim SH, Choi SC, Lee KM, Jun CD. RKIKK motif in the intracellular domain is critical for spatial and dynamic organization of ICAM-1: functional implication for the leukocyte adhesion and transmigration. Mol Biol Cell 2007; 18:2322 - 35; http://dx.doi.org/10.1091/mbc.E06-08-0744; PMID: 17429072
- Romero IA, Amos CL, Greenwood J, Adamson P. Ezrin and moesin co-localise with ICAM-1 in brain endothelial cells but are not directly associated. Brain Res Mol Brain Res 2002; 105:47 - 59; http://dx.doi.org/10.1016/S0169-328X(02)00392-3; PMID: 12399107
- Schnoor M, Lai FP, Zarbock A, Kläver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med 2011; 208:1721 - 35; http://dx.doi.org/10.1084/jem.20101920; PMID: 21788407
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 2002; 3:586 - 99; http://dx.doi.org/10.1038/nrm882; PMID: 12154370
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2001; 2:138 - 45; http://dx.doi.org/10.1038/35052082; PMID: 11252955
- van Buul JD, Hordijk PL. Endothelial adapter proteins in leukocyte transmigration. Thromb Haemost 2009; 101:649 - 55; PMID: 19350107
- Kirkbride KC, Sung BH, Sinha S, Weaver AM. Cortactin: a multifunctional regulator of cellular invasiveness. Cell Adh Migr 2011; 5:187 - 98; http://dx.doi.org/10.4161/cam.5.2.14773; PMID: 21258212
- Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol 2000; 165:3375 - 83; PMID: 10975856
- Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res 2006; 98:394 - 402; http://dx.doi.org/10.1161/01.RES.0000201958.59020.1a; PMID: 16385081
- Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol 1998; 161:5755 - 61; PMID: 9820557
- Thompson PW, Randi AM, Ridley AJ. Intercellular adhesion molecule (ICAM)-1, but not ICAM-2, activates RhoA and stimulates c-fos and rhoA transcription in endothelial cells. J Immunol 2002; 169:1007 - 13; PMID: 12097408
- van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res 2000; 87:335 - 40; http://dx.doi.org/10.1161/01.RES.87.4.335; PMID: 10948069
- van Buul JD, Voermans C, van den Berg V, Anthony EC, Mul FP, van Wetering S, van der Schoot CE, Hordijk PL. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J Immunol 2002; 168:588 - 96; PMID: 11777950
- Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 2009; 4:e8045; http://dx.doi.org/10.1371/journal.pone.0008045; PMID: 19956681
- Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J 2004; 378:539 - 47; http://dx.doi.org/10.1042/BJ20030794; PMID: 14594451
- van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol 2003; 285:C343 - 52; http://dx.doi.org/10.1152/ajpcell.00048.2003; PMID: 12700137
- Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood 2004; 104:2385 - 93; http://dx.doi.org/10.1182/blood-2004-02-0665; PMID: 15265790
- Deem TL, Abdala-Valencia H, Cook-Mills JM. VCAM-1 activation of endothelial cell protein tyrosine phosphatase 1B. J Immunol 2007; 178:3865 - 73; PMID: 17339486
- Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood 2013; 122:2512 - 22; http://dx.doi.org/10.1182/blood-2013-04-499228; PMID: 23908467
- Barreiro O, Vicente-Manzanares M, Urzainqui A, Yáñez-Mó M, Sánchez-Madrid F. Interactive protrusive structures during leukocyte adhesion and transendothelial migration. Front Biosci 2004; 9:1849 - 63; http://dx.doi.org/10.2741/1285; PMID: 14977592
- Samson T, van Buul JD, Kroon J, Welch C, Bakker EN, Matlung HL, van den Berg TK, Sharek L, Doerschuk C, Hahn K, et al. The guanine-nucleotide exchange factor SGEF plays a crucial role in the formation of atherosclerosis. PLoS One 2013; 8:e55202; http://dx.doi.org/10.1371/journal.pone.0055202; PMID: 23372835
- Carman CV, Springer TA. Trans-cellular migration: cell-cell contacts get intimate. Curr Opin Cell Biol 2008; 20:533 - 40; http://dx.doi.org/10.1016/j.ceb.2008.05.007; PMID: 18595683
- Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood 2011; 117:942 - 52; http://dx.doi.org/10.1182/blood-2010-02-270561; PMID: 21030556
- Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One 2008; 3:e1649; http://dx.doi.org/10.1371/journal.pone.0001649; PMID: 18297135
- Singleton PA, Dudek SM, Chiang ET, Garcia JGN. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J 2005; 19:1646 - 56; http://dx.doi.org/10.1096/fj.05-3928com; PMID: 16195373
- Van Rijssel J, Timmerman I, Van Alphen FP, Hoogenboezem M, Korchynskyi O, Geerts D, Geissler J, Reedquist KA, Niessen HW, Van Buul JD. The Rho-GEF Trio regulates a novel pro-inflammatory pathway through the transcription factor Ets2. Biol Open 2013; 2:569 - 79; http://dx.doi.org/10.1242/bio.20134382; PMID: 23789107
- deBakker CD, Haney LB, Kinchen JM, Grimsley C, Lu M, Klingele D, Hsu PK, Chou BK, Cheng LC, Blangy A, et al. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol 2004; 14:2208 - 16; http://dx.doi.org/10.1016/j.cub.2004.12.029; PMID: 15620647
- Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell 2004; 15:3309 - 19; http://dx.doi.org/10.1091/mbc.E04-02-0146; PMID: 15133129
- García-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 2006; 406:425 - 37; http://dx.doi.org/10.1016/S0076-6879(06)06031-9; PMID: 16472675
- Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol 2000; 2:888 - 92; http://dx.doi.org/10.1038/35046533; PMID: 11146652
- van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol 2004; 24:824 - 33; http://dx.doi.org/10.1161/01.ATV.0000122854.76267.5c; PMID: 14976004
- Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol 1999; 162:2964 - 73; PMID: 10072547
- Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 2011; 12:761 - 9; http://dx.doi.org/10.1038/ni.2062; PMID: 21706006
- Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 2008; 9:639 - 49; http://dx.doi.org/10.1038/nrm2447; PMID: 18612320
- Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol Rev 2013; 256:222 - 39; PMID: 24117824
- Sayedyahossein S, Dagnino L. Chapter Six - Integrins and Small GTPases as Modulators of Phagocytosis in International Review of Cell and Molecular Biology (ed. Kwang,W.J.) 321-354 (Academic Press, 2013).
- Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell 2004; 15:3509 - 19; http://dx.doi.org/10.1091/mbc.E03-11-0847; PMID: 15169870
- Welch CM, Elliott H, Danuser G, Hahn KM. Imaging the coordination of multiple signalling activities in living cells. Nat Rev Mol Cell Biol 2011; 12:749 - 56; http://dx.doi.org/10.1038/nrm3212; PMID: 22016058
- Sage PT, Carman CV. Settings and mechanisms for trans-cellular diapedesis. Front Biosci. (Landmark. Ed) 14, 5066-5083 (2009).
- Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci 2009; 122:3025 - 35; http://dx.doi.org/10.1242/jcs.047522; PMID: 19692589
- Cinamon G, Grabovsky V, Winter E, Franitza S, Feigelson S, Shamri R, Dwir O, Alon R. Novel chemokine functions in lymphocyte migration through vascular endothelium under shear flow. J Leukoc Biol 2001; 69:860 - 6; PMID: 11404368
- Stroka KM, Aranda-Espinoza H. A biophysical view of the interplay between mechanical forces and signaling pathways during transendothelial cell migration. FEBS J 2010; 277:1145 - 58; http://dx.doi.org/10.1111/j.1742-4658.2009.07545.x; PMID: 20121945
- Weber KSC, von Hundelshausen P, Clark-Lewis I, Weber PC, Weber C. Differential immobilization and hierarchical involvement of chemokines in monocyte arrest and transmigration on inflamed endothelium in shear flow. Eur J Immunol 1999; 29:700 - 12; http://dx.doi.org/10.1002/(SICI)1521-4141(199902)29:02<700::AID-IMMU700>3.0.CO;2-1; PMID: 10064088
- Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood 2002; 100:3853 - 60; http://dx.doi.org/10.1182/blood.V100.12.3853; PMID: 12433694
- Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol 2007; 7:467 - 77; http://dx.doi.org/10.1038/nri2096; PMID: 17525755
- Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 1997; 91:385 - 95; http://dx.doi.org/10.1016/S0092-8674(00)80422-5; PMID: 9363947
- Whittall C, Kehoe O, King S, Rot A, Patterson A, Middleton J. A chemokine self-presentation mechanism involving formation of endothelial surface microstructures. J Immunol 2013; 190:1725 - 36; http://dx.doi.org/10.4049/jimmunol.1200867; PMID: 23325889
- Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol 2014; 15:45 - 53; http://dx.doi.org/10.1038/ni.2769; PMID: 24270515
- Sandig M, Negrou E, Rogers KA. Changes in the distribution of LFA-1, catenins, and F-actin during transendothelial migration of monocytes in culture. J Cell Sci 1997; 110:2807 - 18; PMID: 9427289
- Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med 2004; 200:1571 - 80; http://dx.doi.org/10.1084/jem.20040965; PMID: 15611287
- Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity 2007; 26:784 - 97; http://dx.doi.org/10.1016/j.immuni.2007.04.015; PMID: 17570692
- Su WH, Chen HI, Jen CJ. Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood 2002; 100:3597 - 603; http://dx.doi.org/10.1182/blood-2002-01-0303; PMID: 12393634
- He P. Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled?. Cardiovasc Res 2010; 87:281 - 90; http://dx.doi.org/10.1093/cvr/cvq140; PMID: 20472564
- Vestweber D. Relevance of endothelial junctions in leukocyte extravasation and vascular permeability. Ann N Y Acad Sci 2012; 1257:184 - 92; http://dx.doi.org/10.1111/j.1749-6632.2012.06558.x; PMID: 22671605
- Chavez A, Smith M, Mehta D. Chapter six - New Insights into the Regulation of Vascular Permeability in International Review of Cell and Molecular Biology (ed. Kwang,W.J.) 205-248 (Academic Press, 2011).
- Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer TA, et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol 2013; 201:449 - 65; http://dx.doi.org/10.1083/jcb.201209077; PMID: 23629967
- Aghajanian A, Wittchen ES, Allingham MJ, Garrett TA, Burridge K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J Thromb Haemost 2008; 6:1453 - 60; http://dx.doi.org/10.1111/j.1538-7836.2008.03087.x; PMID: 18647230
- Vestweber D. Molecular mechanisms that control leukocyte extravasation through endothelial cell contacts. Ernst. Schering. Found. Symp. Proc.151-167 (2007).
- Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 2009; 19:8 - 15; http://dx.doi.org/10.1016/j.tcb.2008.10.001; PMID: 19010680
- Wittchen ES. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci (Landmark Ed) 2009; 14:2522 - 45; http://dx.doi.org/10.2741/3395; PMID: 19273217
- Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ. Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am J Physiol 1997; 273:H981 - 8; PMID: 9277518
- Saito H, Minamiya Y, Kitamura M, Saito S, Enomoto K, Terada K, Ogawa J. Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J Immunol 1998; 161:1533 - 40; PMID: 9686621
- Garcia JGN, Verin AD, Herenyiova M, English D. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol (1985) 1998; 84:1817 - 21; PMID: 9572834
- Saito H, Minamiya Y, Saito S, Ogawa J. Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leukoc Biol 2002; 72:829 - 36; PMID: 12377953
- Liu Z, Sniadecki NJ, Chen CS. Mechanical Forces in Endothelial Cells during Firm Adhesion and Early Transmigration of Human Monocytes. Cell Mol Bioeng 2010; 3:50 - 9; http://dx.doi.org/10.1007/s12195-010-0105-3; PMID: 20862208