
Abstract
The homeostatic function of endothelial cells (EC) is critical for a number of physiological processes including vascular integrity, immunity, and wound healing. Indeed, vascular abnormalities resulting from EC dysfunction contribute to the development and spread of malignancies. The alternative SDF-1/CXCL12 receptor CXCR7 is frequently and specifically highly expressed in tumor-associated vessels. In this study, we investigate whether CXCR7 contributes to vascular dysfunction by specifically examining the effect of CXCR7 expression on EC barrier function and motility. We demonstrate that CXCR7 expression in EC results in redistribution of CD31/PECAM-1 and loss of contact inhibition. Moreover, CXCR7+ EC are deficient in barrier formation. We show that CXCR7-mediated motility has no influence on angiogenesis but contributes to another motile process, the invasion of CXCR7+ EC into ligand-rich niches. These results identify CXCR7 as a novel manipulator of EC barrier function via alteration of PECAM-1 homophilic junctions. As such, aberrant expression of CXCR7 in the vasculature has the potential to disrupt vascular homeostasis and could contribute to vascular dysfunction in cancer systems.
Introduction
Endothelial cells (EC) line the vasculature of both the blood and lymphatic circulatory systems. As the “gate-keepers” of the vasculature, EC play critical roles in a variety of physiological processes including development, tissue and vascular fluid homeostasis, immune surveillance, and wound healing. In normal adult vasculature there is very little growth or turnover of EC because stable cell–cell and cell–substrate contacts regulate long-term survival and homeostatic functions. Moreover, these contacts, as well as their regulation and signaling outcomes, dictate EC barrier function and the maintenance of vascular homeostasis. The formation and maintenance of the EC barrier as well as its disruption in inflammation is a subject of continuing investigation.Citation1 In contrast to normal vessels, the vasculature found in solid tumors is structurally and functionally abnormal. This phenomenon, termed tumor vascular dysfunction, is linked to abnormalities in EC barrier function and ultimately results in tissues with poor oxygenation, low pH, and high interstitial pressure, thus producing a microenvironment that reduces immune surveillance and drives tumor growth and metastasis.Citation2,Citation3
The chemokine SDF-1/CXCL12 and its canonical receptor CXCR4 are among the most highly studied chemokine/receptor pairs in cancer biology.Citation4,Citation5 CXCR4/SDF-1 interactions are known to drive cellular motility and metastasis in a variety of malignancies. Interestingly, a second receptor for CXCL12 was recently discovered and designated CXCR7.Citation6 The existence of a second receptor for SDF-1/CXCL12 necessitates a re-evaluation of the effects of SDF-1 in cancer models. Moreover, CXCR7 binding to its alternate ligand ITAC/CXCL11 also has the potential to influence tumor progression.Citation7 Studies of CXCR7 expression in tumor tissues reveal only sporadic expression on malignant cells in a variety of cancers.Citation8-Citation13 However, several studies have reported that CXCR7 is specifically expressed in EC of tumor vasculatureCitation9,Citation14,Citation15 and CXCR7 may be a specific marker for tumor vessels.Citation10 Our laboratory has observed that CXCR7 is uniformly overexpressed in Kaposi sarcoma (KS) tumor cells, which are of EC originCitation16 as well as in normal EC following infection with Kaposi sarcoma herpesvirus (KSHV) in vitro.Citation17 Despite the compelling evidence that CXCR7 is specifically expressed on tumor-associated EC, the functional consequences of CXCR7 expression are ill-defined. We have previously reported that CXCR7 expression in EC results in aberrant post-confluent proliferation of EC cultures via proteasomal degradation of the Retinoblastoma protein (pRb).Citation18 Moreover, in that previous study, we obtained data indicating that CXCR7+ EC are not contact inhibited and form abnormal monolayers.
Much of the current literature addressing the CXCL12 receptors characterizes CXCR7 as a decoy receptor. Decoy receptors typically lack intrinsic signaling activity and exert their physiological effects by sequestering chemokine ligands, thereby altering chemotactic gradients at alternate receptors. As such, several studies have shown that binding of CXCR7 to its ligands produces no G-protein-mediated signalingCitation19 but that CXCR7-expressing cells modulate the free concentration of SDF-1/CXCL12 and can thereby alter responses from CXCR4 and CXCR3.Citation9,Citation20,Citation21 Several recent studies have also reported a non-scavenging function of CXCR7 in specific cell types.Citation22 Similarly, our previous experiments in EC showed that (1) endogenously produced CXCR7 ligands were required for CXCR7-mediated degradation of pRb and (2) CXCR3 and CXCR4 protein levels were low or absent (respectively) in this context and were not affected by CXCR7 expression.Citation18 Therefore, we concluded that our gain-of-function phenotypes were a result of CXCR7 signaling and that CXCR7 can function as a bona fide chemokine receptor in EC, making its EC-specific expression in solid tumors of particular interest and significance.
In the current study, we extend our previous findings by specifically examining the effect of CXCR7 on EC barrier function and motility. We demonstrate that CXCR7 expression alone is sufficient to remove CD31/PECAM-1 from cell–cell junctions in confluent EC cultures. We present evidence that CD31 relocalization is associated with alterations in EC morphology and present further evidence that CXCR7+ EC have lost contact inhibition. Using Electrical Cell-Substrate Impedance Sensing (ECIS) to allow real-time electrical monitoring of barrier function, we demonstrate that CXCR7+ EC are defective in barrier formation and display characteristics of increased micromotion. CXCR7-mediated motility does not contribute to tubule formation in vitro but affects another motile process, invasion of CXCR7+ cells into ligand-rich niches. Taken together, these data demonstrate that CXCR7 expression has profound effects on EC homeostatic functions and suggest that CXCR7 may contribute to vascular dysfunction in cancer.
Results
CXCR7 expression results in removal of CD31/PECAM-1 from cell–cell junctions
For the studies described herein, we chose to use primary or HPV E6/E7-immortalized human lymphatic EC (pLEC and iLEC, respectively) due to the fact that they (1) display robust growth and junctional stability in the absence of shear stress and (2) have uniformly low endogenous levels of CXCR7 under normal culture conditions (data not shown). However, key findings of this study were verified in blood vascular-lineage EC (BEC) to verify that phenotypes were not lineage-specific. CXCR7 expression was achieved via transduction of LEC with our previously described tet-inducible adenovirus vector expressing a full-length version of human CXCR7 containing an N-terminal HA-tag.Citation18 CXCR7 expression from this vector is controlled by co-infection with a second adenovirus vector expressing the tet-transactivator (herein referred to as Trans). In all experiments, infection with Trans only is used to control for non-specific effects of adenovirus transduction.
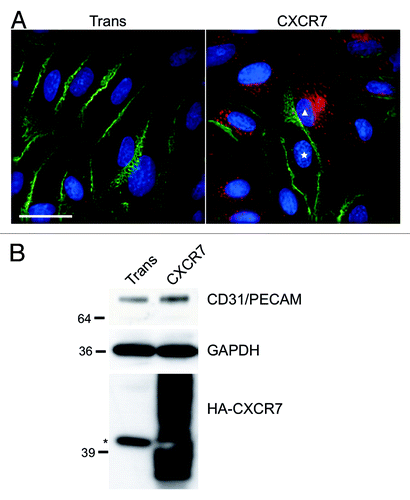
Platelet endothelial cell adhesion molecule-1 PECAM-1/CD31 is highly enriched at EC cell–cell junctions and, although it is not required for junction formation,Citation23 PECAM-1 has been shown to play important roles in EC barrier function,Citation24 the response of EC to inflammatory stimuliCitation25,Citation26 and the transendothelial migration of lymphocytes.Citation27 Therefore, we wanted to determine whether CXCR7 expression affected PECAM-1 homophilic junctions in confluent EC monolayers. For these experiments, we allowed pLEC cultures to grow 2 d past confluence to facilitate formation of a monolayer with mature junctions. We then infected monolayers with Trans only or Trans+CXCR7 adenovirus. At 20 h post-infection, cultures were fixed and stained for CXCR7 and PECAM-1 using indirect immunofluorescence (IFA). While transduction with the Trans adenovirus alone did not disrupt PECAM-1 junctions compared with uninfected pLEC (data not shown), we observed decreased junctional PECAM-1 staining in CXCR7-expressing EC to a degree that was inversely correlated with the intensity of CXCR7 staining on a per cell basis (, white triangle). Moreover, adjacent CXCR7-negative cells in the same cultures (, white star) retained peripheral PECAM-1 staining suggesting that this effect is cell-autonomous and not due to the release of paracrine factors. Given that intercellular homophilic PECAM-1 interaction is important for stable junctional retention of PECAM-1,Citation28,Citation29 we believe that the peripherally expressed PECAM-1 in CXCR7-negative cells lying adjacent to CXCR7-positive cells that lack peripheral PECAM-1 constitutes PECAM-1 molecules that are continuously recycling between the junctional space and the lateral border recycling compartment (LBRC).Citation30 This PECAM-1 expression pattern was reproducible in multiple fields in five independent experiments and similar results were obtained in primary blood vascular EC (pBEC) cultures, indicating that the phenotype is not restricted to the lymphatic EC lineage (data not shown). Next, we examined total PECAM-1 levels via western blot in identical pLEC cultures transduced with either Trans or Trans+CXCR7. These experiments revealed no reduction in total PECAM-1 expression in CXCR7-expressing EC relative to the Trans control (), consistent with PECAM-1 relocalization as opposed to degradation.
Figure 1. CXCR7 expression results in removal of CD31/PECAM-1 from cell-cell junctions. pLEC were infected with Trans or Trans+CXCR7. (A) At 20 h post-infection cultures were fixed and stained by IFA for PECAM-1 (green), CXCR7 (red), and DAPI. The white star marks the nucleus of an internal control CXCR7 negative cell in contrast to CXCR7+ cell denoted by white triangle. Scale bar is 30 µm. (B) Identical cultures were lysed and analyzed by western blot for GAPDH and CD31/PECAM. Data are representative of three replicate experiments. Membranes were then reprobed for HA to verify adenovirus transduction efficiency. The star (*) denotes a non-specific band commonly detected by HA antibody in pLEC lysates. As with many proteins containing multiple transmembrane domains, HA-CXCR7 in boiled lysates appears as a smear due to aggregation of the hydrophobic domains.
CXCR7+ EC display altered morphology and lack contact inhibition
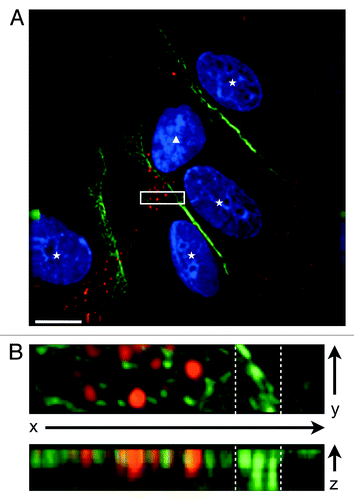
Careful examination of CXCR7-expressing pLEC cultures by IFA revealed a large number of CXCR7+ cells with elongated morphology that were apparently growing on top of CXCR7-negative cells. Multi-layered growth is remarkable in EC cultures, which are highly contact inhibited. In order to verify these observations, we performed 3-dimensional microscopic analysis of high magnification z-stacks on the CXCR7+ cells that were growing on top of the monolayer. shows a representative analysis of one such field acquired via a 40-image z-stack at 0.1 µM step size and 100X magnification and subjected to deconvolution analysis. In this image, a single CXCR7+ cell (, white triangle) with an elongated projection staining for CXCR7 but lacking any peripheral PECAM-1 staining apparently lies on top of four CXCR7-negative EC (, white stars), which display normal junctional PECAM-1. In order to verify that the CXCR7+ cell was indeed in a different z-plane than the cells with junctional PECAM-1 staining, we chose a region of interest within the field where both CXCR7 and PECAM staining were evident (, white box) and performed 3-dimensional rotational analysis (). Indeed, rotation of the image around the x-axis reveals that the intense junctional PECAM-1 staining (indicated by white dashed lines) resides predominantly in the bottom half of the z-stack whereas the CXCR7 staining resides in the top half of the z-stack together with diffuse, non-junctional PECAM-1 staining (, bottom). These data indicate that the loss of junctional PECAM-1 in CXCR7+ EC is associated with the ability to grow on top of an ordered monolayer, a characteristic of EC that have lost contact inhibition. We did not observe a redistribution of PECAM-1 staining in CXCR7+ EC at 60X magnification (), probably due to the relative intensity of the tightly clustered junctional PECAM-1 staining at this magnification. However, at 100x magnification, disordered PECAM-1 staining is visible in both the x-y plane (, top) and is specifically localized to the top half of the z-stack, coincident with CXCR7 staining (, bottom). Taken together with our western blot data demonstrating that CXCR7 expression does not reduce total PECAM-1 expression (), the data indicate that CXCR7 expression is mediating redistribution of PECAM-1 to extra-junctional sites.
Figure 2. CXCR7+ EC display altered morphology and lack contact inhibition. pLEC were infected with Trans+CXCR7. At 20 h post-infection cultures were fixed and stained by IFA for PECAM-1 (green), CXCR7 (red), and DAPI. (A) A field of interest was selected and a z-stack series of 40 images with 0.1 µM step size was photographed at 100X magnification. The image was subjected to deconvolution analysis. Nuclei of CXCR7 negative cells are marked with white stars, the nucleus of the CXCR7+ cell is marked with a white triangle. Scale bar is 20 µm. A region of interest (white box) containing both CXCR7 staining and junctional CD31 staining was extracted and (B) subjected to 3-dimensional analysis. White dashed lines denote the region in the x-y plane containing junctional CD31 staining.
CXCR7+ EC are defective in barrier formation
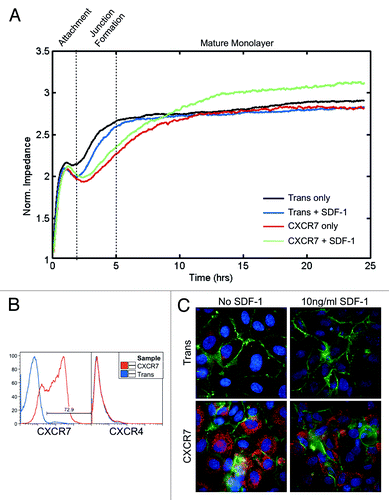
We next wanted to determine whether loss of junctional PECAM-1 in CXCR7+ EC resulted in defects in the ability of these EC to form a tight barrier. For these experiments, we used a technique called Electrical Cell-Substrate Impedance Sensing (ECIS) in which cells are plated into multi-well arrays that contain electrodes on the growth surface. The growth media and the electrodes comprise an electrical circuit. As the cells spread and grow over the electrodes they form an insulating layer, thereby increasing the impedance of the system, which is measured in real-time without damaging the cells.Citation31,Citation32 In the case of a confluent layer of EC, the impedance value is a real-time, quantitative measure of the barrier integrity, with the magnitude and stability of impedance values being indicative of junctional integrity.Citation33 We infected pLEC with Trans or Trans+CXCR7. At 20 h post-infection, cells were trypsinized and 3 × 105 cells per well were replated into duplicate wells on ECIS arrays and allowed to adhere and spread. Because cell adherence and barrier formation requires coordinated cell movement, we also included SDF-1/CXCL12 at 10 ng/ml in replicate wells to determine whether CXCR7 ligands affect this process. At the cell concentration used, we expect the formation of a confluent monolayer without the need for cells to proliferate on the ECIS array. Impedance measurements were recorded every 8 s for a further 25 h. In order to get an overall view of barrier formation in these cultures, we used ECIS arrays that collect and average the impedance recordings of 40 electrodes distributed across the culture growth surface. Data from at least two independent wells representing 80 individual impedance readings at each timepoint were averaged to generate the plotted impedance curves. As expected, control Trans cells attached to the substrate within 2 h post-seeding, as indicated by an initial plateau of impedance values during this time period. Impedance values increased again between 2 and 5 h post-seeding as cells spread and cell-cell junctions were formed, after which impedance values reached a second plateau and remained stable throughout the rest of the timecourse, coincident with junction maturation (, black curve). Treatment of Trans cells with SDF-1/CXCL12 did not significantly affect either the timecourse or final impedance values of barrier formation and maturation (, blue curve). The timecourse of initial attachment for CXCR7+ EC was comparable to Trans (, red curve). However, in contrast to Trans controls, the kinetics of junction formation and maturation was significantly slowed in CXCR7+ EC over the timecourse with CXCR7+ EC taking 5–7 h longer to reach the same impedance levels achieved in Trans controls. This delay in barrier formation occurred regardless of ligand stimulation (, red and green curves). Despite the delay in junction formation and maturation untreated CXCR7+ cultures stabilized at the same impedance level as Trans controls (, red curve). In contrast, the SDF-1/CXCL12-treated CXCR7+ cultures displayed increased impedance compared with Trans or untreated CXCR7+ conditions (, green curve). Similar trends were observed in replicate experiments. A subset of cells was reserved prior to seeding in ECIS arrays and stained by flow cytometry for surface expression of CXCR7 and the alternate SDF-1/CXCL12 receptor CXCR4 (). As expected, CXCR7 transduction was very efficient and no CXCR4 was present at the cell surface in either condition. Therefore, the observed phenotypes are a product of CXCR7 gain-of-function and are not influenced by CXCR4. IFA analysis of duplicate cultures were fixed and stained for PECAM-1 and CXCR7 at 25 h post-seeding revealing abnormal monolayers in the CXCR7+ conditions (), consistent with our previously published data.Citation18 Moreover, our previous report demonstrated that CXCR7+ EC are hyperproliferative in response to CXCR7 ligands.Citation18 Taken together with our data from this study indicating that CXCR7+ EC are able to grow on top of a confluent monolayer (), we believe that the increased impedance observed in the SDF-1/CXCL12-treated cultures (, green curve) is representative of aberrant proliferation and the formation of multi-layered structures rather than the formation of a functional quiescent barrier. Higher doses of SDF-1/CXCL12 did not alter the observed phenotype (data not shown). Similar experiments in which cells were stimulated with ITAC/CXCL11 revealed non-specific effects of this chemokine on barrier formation in Trans controls, possibly due to low levels of CXCR3 expressionCitation18 (data not shown). These results demonstrate that CXCR7 expression in EC compromises their ability to form cell-cell junctions and establish barrier function, a phenotype that could have significant implications for vascular function.
Figure 3. CXCR7+ EC are defective in barrier formation. pLEC were infected with Trans or Trans+CXCR7. (A) At 20 h post-infection cells were trypsinized and replated onto ECIS arrays with or without SDF-1/CXCL12 at 10 ng/ml and allowed to attach for 25 h. Impedance readings at 4000 Hz were taken every 8 s throughout the timecourse. Individual wells were normalized to impedance at t = 0 and duplicate wells were averaged to create the impedance curves. Data are representative of duplicate wells from three independent experiments. (B) At the time of ECIS seeding, a subset of cells was reserved for analysis of CXCR7 and CXCR4 expression by flow cytometry. Necrotic cells were excluded from the analysis via propidium iodide staining and scatter characteristics. (C) Duplicate multi-well plates seeded identically to ECIS arrays were fixed at 25 h post-seeding and stained for CXCR7 (red), PECAM-1 (green), and DAPI and analyzed by deconvolution microscopy.
CXCR7+ EC display increased micromotion by ECIS
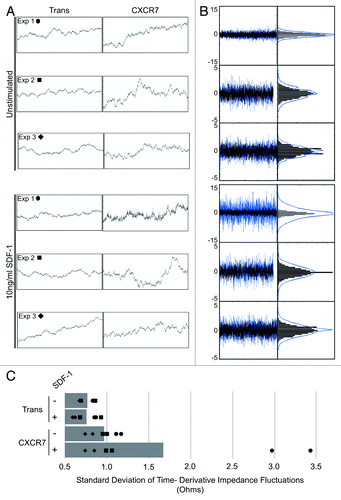
In our ECIS experiments, the impedance measurement curves were highly unstable in CXCR7+ conditions compared with Trans controls when short time intervals were inspected at narrow impedance ranges. In order to better illustrate these small-scale fluctuations, we extracted 100 ohm impedance windows for 2-h time periods following barrier formation for a replicate well from each of three independent experiments (designated Exp 1, 2, and 3) wherein each curve represents the average of 40 individual electrode measurements every 8 s (). At this scale, we can clearly see increased small-scale variability of impedance measurements exclusively in the CXCR7+ conditions. These impedance fluctuations were consistent throughout the timecourse following the period of attachment and spreading (data not shown). In order to quantitatively analyze this phenotype and determine its ligand-dependence, we developed an algorithm in which the time derivative of the impedance measurements was calculated for each condition. The timepoints following attachment and spreading were used for the analysis in order to quantify fluctuations irrespective of the total impedance values (, left). To allow for relative comparison, the time derivative data for Trans (black) was overlayed onto the CXCR7 data (blue). From this data, a binned distribution of the magnitudes of short-timescale impedance variability for each sample was created (, right) and the standard deviation of this distribution was used as a measure of the overall impedance fluctuation for each condition (). We can clearly see in the overlayed time derivative data that in all conditions CXCR7 expression increases the magnitude of impedance variability. In one experiment, there was a highly significant increase in impedance fluctuations in CXCR7+ EC treated with SDF-1/CXCL12. This effect was reproducible in additional experimental replicates but not to the same magnitude; this variability is likely due to varying levels of endogenous ligand in the individual wells. This analysis reveals that (1) the visual quality of the curves corresponds to a mathematically demonstrable increase in short-timescale variability and (2) that there is evidence for some degree of ligand-dependence with this phenotype. Such small-scale impedance fluctuations are characteristic of cellular micromotionCitation31 and have been used to distinguish malignant cells from non-malignant cellsCitation34 and can also be correlated with the invasive and metastatic potential of cancer cell lines in vitro.Citation35 We thus conclude that this aspect of our ECIS data are similarly indicative of increased motility in CXCR7+ EC.
Figure 4. CXCR7+ EC display increased micromotion by ECIS. (A) Extracted impedance readings from ECIS traces scaled to 2 h by 100 ohms. Curves shown are impedance traces of pLEC infected with Trans or Trans+CXCR7 and treated with media only (Unstimulated, top 3 panels) or 10 ng/ml SDF-1/CXCL12 (bottom three panels). Symbols indicated for each experiment correspond to the data points shown in (C). (B) Overlayed time differential (left) and binned histograms of time differential data (right) for Trans (black, overlayed) and Trans+CXCR7 (blue, underlayed) corresponding to the same panels in (A). (C) Quantitation of the standard deviation of impedance fluctuations in Trans or Trans+CXCR7 cultures with or without 10 ng/ml SDF-1/CXCL12. Analysis includes duplicate wells from three independent experiments (black data points) and the average for each condition (gray bar).
CXCR7 expression does not affect angiogenesis
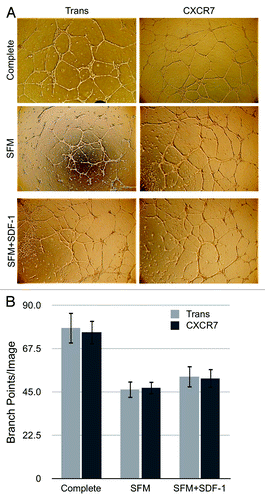
In order to confirm whether the increased variability of ECIS measurements correlates with increased motility in CXCR7+ EC, we next performed specific functional assays for EC motility. First, we tested whether CXCR7 expression and/or SDF-1/CXCL12 treatment affected the ability of EC to form tubules in an in vitro angiogenesis assay. Accordingly, Trans or Trans+CXCR7-infected pLEC were replated on to a matrigel matrix in medium lacking serum or growth factors but supplemented with or without 50 ng/ml SDF-1/CXCL12. We used higher concentrations of SDF-1/CXCL12 in these assays in order to allow for diffusion of effective concentrations of SDF-1/CXCL12 into the matrigel matrix. Tubule formation was photographed at 24 h post-seeding () and branch points per field were quantified for three to five biological replicates in four independent experiments (). Appreciable tubule formation was seen in all conditions, and although there was a slight trend toward higher numbers of branch points in the SDF-1/CXCL12-treated conditions, there was no statistically significant effect of either CXCR7 expression or SDF-1/CXCL12 treatment on in vitro tubule formation. This data suggested that CXCR7 signaling does not affect the coordinated EC motility necessary for tubule formation. We obtained similar results in duplicate conditions performed with different doses of SDF-1/CXCL12 (data not shown), demonstrating that the lack of an SDF-1/CXCL12 response is not a result of the ligand dosage.
Figure 5. CXCR7 expression does not affect angiogenesis. pLEC were infected with Trans or Trans+CXCR7. At 20 h post-infection cells were trypsinized and replated onto matrigel in complete medium or serum-free medium with or without SDF-1 at 50 ng/ml. Tubule formation was analyzed at 24 h post-plating by (A) light microscopy and (B) quantitation of branch points in 3–5 wells per condition from four independent experiments. P > 0.8 for Trans vs. CXCR7 in all conditions. P > 0.2 for SFM vs. SDF-1 for both Trans and CXCR7.
CXCR7 expression enhances EC invasion toward CXCR7 ligands
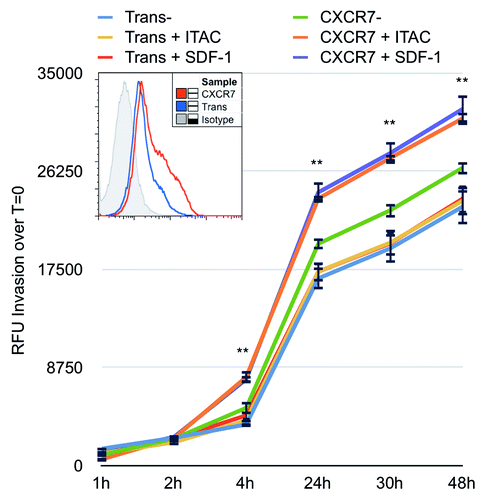
We next utilized an in vitro invasion assay to determine whether CXCR7 enhances single-cell chemotactic motility in EC. For this assay, Fluoroblok™ invasion chambers were coated with Matrigel (BD) and allowed to solidify overnight at 37 °C. Fluoroblok™ plates contain membranes that are impermeable to fluorescent light, allowing sequential reads of fluorescence in the bottom chamber without disassembling the invasion plate. iLEC were infected with Trans or Trans+CXCR7. At 20 h post-infection, cells were labeled with the membrane-permeable dye Calcein-AM, trypsinized and replated at 2 × 104 cells/well into the top chambers of the invasion plate. Media only, 50 ng/ml SDF-1/CXCL12 or 50 ng/ml ITAC/CXCL11 was added to the bottom chambers to provide the chemotactic stimulus. As in the Matrigel angiogenesis experiments, these higher concentrations of chemokine (50 ng/ml) were used to allow for diffusion of the chemotactic gradient through the Matrigel barrier to the target cells. We assayed invasion by reading bottom chamber fluorescence at 1, 2, 4, 24, 30, and 48 h post-seeding. The background fluorescence was subtracted for each well and invasion was calculated based on the average RFU increase over baseline fluorescence values. We observed that control Trans EC invade at the same rate regardless of the presence or absence of a chemotactic stimulus. CXCR7+ EC in the absence of ligand invaded slightly faster than Trans controls between 4 and 24 h post-seeding. This difference was reproducible in multiple experiments, but was not statistically significant compared with unstimulated Trans controls. However, addition of either SDF-1/CXCL12 or ITAC/CXCL11 as a chemotactic stimulus significantly increased the invasion of CXCR7+ EC compared with untreated Trans controls (). Increased invasion in the ligand-treated conditions was statistically significant compared with unstimulated Trans controls as early as 4 h post-seeding. These data demonstrate that CXCR7+ EC are indeed hypermotile, as suggested by our ECIS data (), and that this motility is sufficient to mediate ligand-dependent but not ligand-specific invasion of CXCR7+ EC through a matrigel barrier.
Figure 6. CXCR7 expression enhances EC invasion toward CXCR7 ligands. iLEC were infected with Trans or Trans+CXCR7. At 20 h post-infection cells were labeled with Calcein-AM dye, trypsinized, and transferred to matrigel-coated Fluoroblok™ invasion plates with ITAC/CXCL11 at 50 ng/ml, SDF-1/CXCL12 at 50 ng/ml, or no ligand in the bottom chamber. Invasion was measured via fluorescence accumulation in the bottom chamber at the indicated timepoints. Individual wells were normalized to fluorescence intensity at t = 0 and RFU increases were averaged for each condition. n = 16 wells per condition and data are representative of three independent experiments. **P < 0.001 for both CXCR7+SDF-1 and CXCR7+ITAC compared with unstimulated Trans control. A subset of cells was stained prior to seeding for HA-CXCR7 by flow cytometry to control for adenovirus transduction efficiency (histogram, inset).
Discussion
In the current study, we sought to determine whether CXCR7 expression in the vasculature could contribute to tumor vascular dysfunction by affecting EC barrier function. We demonstrate that CXCR7 expression in EC has the capacity to remove PECAM-1 from cell–cell junctions resulting in a loss of contact inhibition and defects in endothelial barrier formation. The lack of contact inhibition and aberrant barrier function seen in this study is consistent with our previous results demonstrating that CXCR7+ EC are hyperproliferative and form abnormal monolayers in culture.Citation18 In the current study, we further demonstrate that CXCR7+ EC are hypermotile and that this motility is sufficient to direct invasion of CXCR7+ EC into niches containing CXCR7 ligands. Taken together, these results provide strong evidence that aberrant CXCR7 expression results in significant disruption of EC homeostatic functions. As such, we believe this study identifies CXCR7 as a potential mediator of vascular dysfunction and a target for therapeutic intervention.
A variety of studies in PECAM-1-knockout mice have revealed the critical contribution of PECAM-1 expression to vascular integrity, particularly under conditions of physiological stress.Citation25,Citation26 PECAM-1 signaling via tyrosine phosphorylation of its Immunoreceptor tyrosine inhibitory motifs (ITIMs) has been linked to a variety of EC functions, including interactions with immune cells and cellular motility.Citation36,Citation37 Interestingly, a recent study found that the localization of PECAM-1 to cell–cell junctions and homophilic PECAM-1 interactions mediated EC barrier formation and plasticity even in the absence of PECAM-1 signaling.Citation38 Our current study establishes CXCR7 as a novel manipulator of EC barrier function via manipulation of PECAM-1 localization. Recently, PECAM-1 signaling in EC was shown to affect the proliferation of advanced metastatic tumor cells via the release of paracrine factors.Citation39 Intriguingly, this study showed that administration of a PECAM-1-targeting antibody could slow the formation and growth of advanced metastases in mice, identifying PECAM-1 as a potential target for the treatment of advanced cases of cancer. However, the specific effect of this therapeutic PECAM-1 antibody on PECAM-1 localization, expression, and signaling remains unclear. It would be interesting to determine whether, in advanced metastatic lesions, CXCR7 expression in vascular EC results in a redistribution of PECAM-1 from EC junctions to other cell compartments such as the LBRC, where it then participates in the release of paracrine factors or other activities that affect tumor cell growth. Moreover, future studies of the effect of CXCR7 expression on other EC junction and adhesion molecules are warranted. The signaling mechanisms underlying CXCR7-mediated redistribution of PECAM-1 will also be a critical subject for future research. Given that CXCR7 does not exhibit classic G-protein-mediated signaling in most cellular contexts, the downstream signaling molecules activated by ligand-dependent CXCR7 signaling remain obscure. Our results presented herein justify studies into effects of CXCR7 on signaling mechanisms in EC known to modulate PECAM-1 localization and enhance cell motility such as mitogen-activated protein (MAP) kinase signaling.Citation40
Tumor vascular dysfunction exacerbates the development and spread of cancer by selecting for tumor cells that can survive and proliferate under adverse conditions, thereby enhancing malignancy and driving the development of metastases. As such, treatments that normalize the tumor vasculature have the potential to simultaneously slow the emergence of metastatic disease and improve delivery of chemotherapeutic agents into the primary tumor.Citation2 Despite promising results from animal models, the response of human cancers to vascular normalization therapy via VEGF-targeted agents has been modest and has achieved only moderate short-term efficacy in combination therapy. Moreover, the current VEGF-targeting agents have significant drawbacks in several cancers including neoplasms of the central nervous system and pediatric solid tumors.Citation3,Citation41,Citation42 These studies have validated vascular normalization as a new paradigm for chemotherapy, yet underscore the need for new therapies that more specifically target the tumor vasculature. In this study, we demonstrate that CXCR7 expression in EC results in abnormalities in barrier function and enhanced invasion. Taken together with the compelling observation that tumor-associated endothelium is almost uniformly CXCR7+ regardless of the cancer type,Citation10 we believe that CXCR7 represents a potential therapeutic target for tumor vascular normalization therapy.
When it was still the orphan GPCR RDC-1, we found that CXCR7 is a critical mediator of EC transformation by the oncogenic γ-herpesvirus KSHV.Citation17 The tumor cells, known as spindle cells, of Kaposi sarcoma (KS) lesions are uniformly infected with KSHV and are of EC-origin.Citation43-Citation45 As such, our data demonstrating that CXCR7 expression alters EC homeostatic functions has significant implications for the pathogenesis of KS. Indeed, the enlongated morphology of CXCR7+ EC we observed growing on top of CXCR7 negative EC () is reminiscent of the spindle cell phenotype acquired by KSHV-infected EC in vitro as well as KS tumor cell morphology in vivo. The molecular factors that drive the preferential incidence and recurrence of KS lesions in the skin and at sites of injury and inflammation have never been adequately cataloged. Interestingly, KSHV-infected cells have been shown to undergo transendothelial migration in response to chemotactic gradients of SDF-1/CXCL12.Citation46 Taken together with the results of our current study showing that CXCR7+ EC invade in response to an SDF-1/CXCL12 gradient, these data support the hypothesis that the induction of CXCR7 by KSHV is a critical factor for the dissemination of KS. As such, we believe that the therapeutic value of CXCR7-targeted therapy for KS deserves further study.
Materials and Methods
Cell lines and reagents
Primary adult lymphatic endothelial cells (pLEC) and primary adult blood vascular endothelial cells (pBEC) were commercially obtained (Lonza CC-2810 and CC-2811, respectively) and iLEC were immortalized by transduction with the recombinant retrovirus LXSN16 E6E7 as described previously.Citation47 EC lines were maintained in EGM-2 medium (Lonza #CC-3024) containing bullet kit growth supplements (Lonza #CC-3124), 10% fetal bovine serum (FBS, Hyclone SH30396.03), and Penicillin/Streptomycin/L-Glutamine (PSG, Hyclone SV30082.01). Primary EC cultures were used between passage 4 and 10; immortalized EC were used between passage 7 and 14. MouseIgG1-anti-CXCR7 antibody (11G8) was kindly provided by Chemocentryx. MouseIgG2a-anti-CD31/PECAM-1 antibody for indirect immunofluorescence (IFA) and western blot was from Thermo/Fisher (MA3100). AlexaFlour647-conjugated-rabbit-anti-HA antibody (sc-805) and HRP-conjugated anti-mouse (sc-2306) were from Santa Cruz Biotechnology. GAPDH antibody was from Abcam (ab8245). Anti-mouseIgG2a-AlexaFluor488 (A21131), anti-mouseIgG1-AlexaFluor594 (A21125), anti-rabbit-AlexaFluor488 (A110088), and Calcein-AM (C3100MP) were from Invitrogen. HA-HRP direct conjugate was from Roche (12013819011). Recombinant human SDF-1/CXCL12 (350-NS-050) and recombinant human ITAC/CXCL11 (672-IT-025) were from R&D Systems. Growth Factor Reduced High Concentration Matrigel was from BD Biosystems (354263). Propidium Iodide was from Sigma (P4170).
CXCR7 adenovirus vectors
CXCR7-expressing vectors with an N-terminal HA tag were described previously.Citation18 This vector contains the tet-responsive enhancer within a minimal CMV promoter followed by the SV40 late poly(A) cassette, adenovirus E1A, and a single loxP site to increase recombination frequency. Recombinant adenoviruses were produced by co-transfection of 293 cells expressing the Cre recombinase with adenovirus DNA (Ad5-ψ5) that contains an E1A/E3-deleted adenovirus genome and pAdtet7-HA-CXCR7. Recombinant adenoviruses were expanded on 293-Cre cells, purified by sucrose gradient ultracentrifugation, and titered on 293 cells by limiting dilution. Expression was driven by co-infection with Ad-Trans expressing the Tet-off transactivator.
Infection of EC with adenovirus vectors
Adenovirus infections were performed on confluent EC cultures in endothelial SFM medium (endoSFM, Gibco 11111) supplemented with 1% FBS, PSG, and 37.5 µg/ml endothelial cell growth supplement (ECGS, BD Biosystems 356006).
Indirect immunofluorescence (IFA) for EC junctions
EC were plated on collagen-coated coverslips (BD Biosystems 354089) and infected with either the adenovirus expressing Trans alone at MOI 100, or Trans at MOI 100 and CXCR7 at MOI 100. At 20 h post-infection, cells were washed once with PBS+ and fixed in PBS+ containing 2% paraformaldehyde (PFA) and 1% TritonX-100 for 15 min at room temperature (RT). Coverslips were then post-fixed for a further 5 min at RT in PBS+ containing 2% PFA only. Coverslips were blocked in PBS+ with 1% TritonX-100 and 2% normal goat serum (NGS) for 15 min at RT. All further incubations were performed in PBS+ with 1% TritonX-100 and 0.2% NGS. Coverslips were incubated with MouseIgG2a-anti-CD31/PECAM and MouseIgG1-anti-CXCR7 primary antibodies at 1:100 dilution for 30 min at RT in a humidity chamber. Coverslips were washed twice for 10 min and incubated with anti-mouseIgG2a-AlexaFluor488 and anti-mouseIgG1-AlexaFluor594 secondary antibodies and DAPI at 1:1000 dilution for 30 min at RT in a humidity chamber. Following secondary antibody incubation, coverslips were washed three times for 10 min and mounted on glass slides using Fluoromount G (Southern Biotech).
Indirect immunofluorescence image acquisition and analysis
Image acquisition and analysis was performed on a Deltavision Real-Time Deconvolution Microscope using Softworx analysis software (Applied Precision). Unless otherwise indicated, z-stacks with a 0.2 µm z-step size were taken at 60X magnification. Stacks were subjected to deconvolution analysis and 2–3 section projections were made superimposing representative z-planes to generate the final image. For 3-dimensional analysis, 40-section z-stacks with a 0.1 µm z-step were taken at 100X magnification, a region of interest was selected and the entire 40-image z-stack was processed by rotational transformation around the x-axis to generate the x-z image.
Western blot analysis
Cells were lysed in 200 µl modified radioimmunoprecipitation (RIPA) buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1mM EDTA, 1%Triton-X-100, 1% Sodium Deoxycholate, 0.1% SDS) containing protease inhibitors (Roche 11836145001) for 10 min on ice. Lysates were freeze-thawed once at -20 °C and protein was quantified by BCA protein assay (Pierce, 23225). 0.5 µg of total lysate was boiled for 5 min in 1X LDS protein sample buffer containing reducing agent (Invitrogen, NP0009), loaded on NuPAGE 4–12% Bis-Tris gradient protein gels (Invitrogen, NP0335), and run in 1X NuPAGE MOPS buffer (Invitrogen, NP0001). Protein was transferred to Immobilon-P blotting membrane (Millipore, IPVH00010) and membranes were blocked by drying overnight at RT. Membranes were rehydrated in 2% ECL advance blocking buffer (Amersham, CPK1075) with 0.2% Tween-20 (Block) for 15 min. All subsequent incubations were performed in Block. PECAM-1/CD31 primary antibody was at 1:1000 dilution and GAPDH primary antibody was at 1:50 000 dilution for 1 h at RT. Anti-mouse-HRP-conjugated secondary antibody was diluted 1:40 000 and incubated for 20 min at RT. Blots were developed using ECL Advance (Amersham, RPN2135) chemiluminescent reagent and autoradiography. Membranes were then air-dried overnight to inactivate bound HRP secondary antibodies. Membranes were re-blocked as above and HRP-inactivation was verified by incubation of membranes with ECL reagents and autoradiography. Membranes were then blotted as above with a direct conjugate anti-HA-HRP to verify the efficacy of adenovirus transduction.
Flow cytometry
Cells were dissociated with Cellstripper (Cellgro, 25-056-CI) and resuspended in cold PBS+ containing 2% NGS and 0.1% sodium azide (NaN3) (Surface Block) for 15 min on ice. Cells were then incubated for 15 min on ice with mouse anti-CXCR7 (11G8) and CXCR4 (12G5) antibodies diluted 1:100 in cold PBS+ containing 0.2% NGS and 0.1% NaN3 (Surface Wash) followed by 15 min on ice with anti-mouseIgG1-Alexa647 and anti-mouseIgG2a-Alexa488 secondary antibodies at 1:1000 dilution and 1 µM propidium iodide (PI) in 100 µl Surface Wash. Analysis was on a BD FACS Calibur flow cytometer. Live, non-necrotic cells were gated based on scatter characteristics and negative PI staining.
ECIS barrier formation assay
pLEC were infected with either the adenovirus expressing Trans alone at MOI 100, or Trans at MOI 100 and CXCR7 at MOI 200. 8W10E+ ECIS arrays (Applied Biophysics) were cleaned with 200 µl per well of 10 mM l-cysteine for 15 min at RT, rinsed twice with water, coated with 1% gelatin for 15 min at RT, and equilibrated with complete EGM-2 medium for 30 min at 37 °C. At 20 h post-infection, cells were trypsinized, counted, and replated in complete EGM-2 at 3 × 105 cells/well in duplicate on ECIS arrays with or without SDF-1/CXCL12 at 10 ng/ml. Real-time impedance measurements at 4000 Hz were recorded for each well every 8 s for 12 h on an ECIS Z unit (Applied Biophysics). Impedance measurements for duplicate wells were averaged and plotted using the ECIS software.
ECIS micromotion analysis
Following the statistical analysis presented in,Citation48 cellular micromotion was quantified using a custom written MATLAB (The MathWorks Inc.) program to numerically determine the time derivative of impedance values yielding a temporal fluctuation profile. The fluctuation profile was then binned into a histogram and fit with a normal distribution. The full-width half-max of the normal fit quantified the range of fluctuations irrespective of the total impedance values. This procedure was performed for each well individually over the portion of the timecourse that followed attachment and spreading.
Tubule formation assay
Matrigel was thawed on ice and diluted with cold serum-free Medium 199 to a final protein concentration of 7 mg/ml. Three hundred µl of liquid matrigel was added to a chilled 24-well tissue culture plate and allowed to solidify for 30 min at 37 °C. pLEC were infected with either the adenovirus expressing Trans alone at MOI 100, or Trans at MOI 100 and CXCR7 at MOI 200. At 20 h post-infection, matrigel was overlayed with 200 µl serum-free Medium 199 only, or medium supplemented with 50 ng/ml SDF-1/CXCL12. Cells were trypsinized, counted, and overlayed onto matrigel and media at 5 × 104 cells per well in 200 µl of serum-free Medium 199. Each condition was performed in three to five replicate wells per experiment. Tubule formation was allowed to proceed for 20 h at 37 °C after which analysis was performed by light microscopy at 5x magnification using a Zeiss Axiovert 25 microscope and a Sony Cyber-shot digital camera. Manual quantitation of branch points was performed on images using ImageJ software.
Invasion assay
Matrigel was thawed on ice and diluted with cold serum-free Medium 199 to a final protein concentration of 5 mg/ml. 25µl of liquid matrigel was added to each well of a 96-well Fluoroblok™ invasion plate (BD Falcon, 351164) and allowed to solidify overnight at 37 °C. iLEC were infected with either the adenovirus expressing Trans alone at MOI 100, or Trans at MOI 100 and CXCR7 at MOI 200. At 20 h post-infection, cells were labeled with 2 µg/ml Calcein-AM in 1% endoSFM+ECGS for 30 min at 37 °C. During labeling, SDF-1/CXCL12 at 50 ng/ml or ITAC/CXCL11 at 50 ng/ml was diluted in 1% endoSFM+ECGS and added to bottom chambers at 200 µl per well. Calcein-AM-labeled, cells were trypsinized, counted and replated into the top chamber of the invasion plate at 2 × 104 cells per well in 50µl of 1% endoSFM+ECGS. Baseline fluorescence was read immediately on a Molecular Devices Flexstation fluorescence plate reader set for 485 nm excitation and 525 nm emission monochromators and a 515 nm cutoff filter. Invasion plates were kept at 37 °C between timepoints and read at 1, 2, 4, 24, 30, and 48 h post-seeding. The baseline fluorescence reading for each well was subtracted from each timepoint and RFU increases for biological replicates were averaged. P values for were obtained via Student's T-test with two tails and unequal variance using Apple Numbers software. Values less than 0.01 were considered significant.
| Abbreviations: | ||
| EC | = | endothelial cell(s) |
| SDF-1 | = | stromal cell-derived factor-1 |
| PECAM-1 | = | platelet/endothelial cell adhesion molecule-1 |
| ITAC | = | interferon-inducible T Cell Alpha Chemoattractant |
| ECIS | = | Electrical Cell Substrate Impedance Sensing |
| LEC | = | lymphatic endothelial cells |
| BEC | = | blood vascular endothelial cells |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Mark Penfold of Chemocentryx, Inc. for providing the 11G8 CXCR7 monoclonal antibody and valuable advice. This work was supported by a grant to Moses AV from the National Cancer Institute (grant 5R01CA099906-07) and the National Institutes of Health Core Grant to ONPRC (8P510D011092-53). Totonchy J was supported by fellowship funding from the National Institutes of Health (grant 5T32AI007472-17). Phillips KG and McCarty OJT were supported by a grant from the National Institutes of Health (U54CA143906) and Phillips KG was supported by a Medical Research Foundation Early Clinical Investigator Award.
References
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. [Internet] Physiol Rev 2006; 86:279 - 367; http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16371600&retmode=ref&cmd=prlinks http://dx.doi.org/10.1152/physrev.00012.2005; PMID: 16371600
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10:417 - 27; http://dx.doi.org/10.1038/nrd3455; PMID: 21629292
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307:58 - 62; http://dx.doi.org/10.1126/science.1104819; PMID: 15637262
- Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol 2004; 14:171 - 9; http://dx.doi.org/10.1016/j.semcancer.2003.10.003; PMID: 15246052
- Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 2010; 16:2927 - 31; http://dx.doi.org/10.1158/1078-0432.CCR-09-2329; PMID: 20484021
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold MET, Sunshine MJ, Littman DR, Kuo CJ, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 2006; 203:2201 - 13; http://dx.doi.org/10.1084/jem.20052144; PMID: 16940167
- Singh AK, Arya RK, Trivedi AK, Sanyal S, Baral R, Dormond O, Briscoe DM, Datta D. Chemokine receptor trio: CXCR3, CXCR4 and CXCR7 crosstalk via CXCL11 and CXCL12. Cytokine Growth Factor Rev 2013; 24:41 - 9; http://dx.doi.org/10.1016/j.cytogfr.2012.08.007; PMID: 22989616
- Gahan JC, Gosalbez M, Yates T, Young EE, Escudero DO, Chi A, Garcia-Roig M, Satyanarayana R, Soloway MS, Bird VG, et al. Chemokine and chemokine receptor expression in kidney tumors: molecular profiling of histological subtypes and association with metastasis. J Urol 2012; 187:827 - 33; http://dx.doi.org/10.1016/j.juro.2011.10.150; PMID: 22245330
- Luker KE, Lewin SA, Mihalko LA, Schmidt BT, Winkler JS, Coggins NL, Thomas DG, Luker GD. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene 2012; 31:4750 - 8; http://dx.doi.org/10.1038/onc.2011.633; PMID: 22266857
- Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A 2007; 104:15735 - 40; http://dx.doi.org/10.1073/pnas.0610444104; PMID: 17898181
- Zheng K, Li H-Y, Su X-L, Wang X-Y, Tian T, Li F, Ren G-S. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J Exp Clin Cancer Res 2010; 29:31; http://dx.doi.org/10.1186/1756-9966-29-31; PMID: 20380740
- Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ, Wang J. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev 2010; 29:709 - 22; http://dx.doi.org/10.1007/s10555-010-9256-x; PMID: 20839032
- Hattermann K, Held-Feindt J, Lucius R, Müerköster SS, Penfold MET, Schall TJ, Mentlein R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res 2010; 70:3299 - 308; http://dx.doi.org/10.1158/0008-5472.CAN-09-3642; PMID: 20388803
- Monnier J, Boissan M, L’Helgoualc’h A, Lacombe M-L, Turlin B, Zucman-Rossi J, Théret N, Piquet-Pellorce C, Samson M. CXCR7 is up-regulated in human and murine hepatocellular carcinoma and is specifically expressed by endothelial cells. Eur J Cancer 2012; 48:138 - 48; http://dx.doi.org/10.1016/j.ejca.2011.06.044; PMID: 21778049
- Würth R, Barbieri F, Bajetto A, Pattarozzi A, Gatti M, Porcile C, Zona G, Ravetti J-L, Spaziante R, Florio T. Expression of CXCR7 chemokine receptor in human meningioma cells and in intratumoral microvasculature. J Neuroimmunol 2011; 234:115 - 23; http://dx.doi.org/10.1016/j.jneuroim.2011.01.006; PMID: 21316111
- Vomaske J, Clepper L, Douglas J, Pantanowitz L, Früh K, Moses AV. KSHV infection of endothelial cells manipulates CXCR7-mediated signaling: implications forKaposi’s Sarcoma progression and intervention. Infect Agent Cancer 2012; 7:Suppl 1 O6; http://dx.doi.org/10.1186/1750-9378-7-S1-O6
- Raggo C, Ruhl R, McAllister S, Koon H, Dezube BJ, Früh K, Moses AV. Novel cellular genes essential for transformation of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Cancer Res 2005; 65:5084 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-04-2822; PMID: 15958552
- Totonchy JE, Osborn JM, Botto S, Clepper L, Moses AV. Aberrant proliferation in CXCR7+ endothelial cells via degradation of the retinoblastoma protein. PLoS One 2013; 8:e69828; http://dx.doi.org/10.1371/journal.pone.0069828; PMID: 23894550
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A 2010; 107:628 - 32; http://dx.doi.org/10.1073/pnas.0912852107; PMID: 20018651
- Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes H-G, Rot A, Thelen M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One 2010; 5:e9175; http://dx.doi.org/10.1371/journal.pone.0009175; PMID: 20161793
- Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 2010; 29:4599 - 610; http://dx.doi.org/10.1038/onc.2010.212; PMID: 20531309
- Puchert M, Engele J. The peculiarities of the SDF-1/CXCL12 system: in some cells, CXCR4 and CXCR7 sing solos, in others, they sing duets. Cell Tissue Res 2014; 355:239 - 53; http://dx.doi.org/10.1007/s00441-013-1747-y; PMID: 24292718
- Duncan GS, Andrew DP, Takimoto H, Kaufman SA, Yoshida H, Spellberg J, de la Pompa JL, Elia A, Wakeham A, Karan-Tamir B, et al. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol 1999; 162:3022 - 30; PMID: 10072554
- Ferrero E, Ferrero ME, Pardi R, Zocchi MR. The platelet endothelial cell adhesion molecule-1 (PECAM1) contributes to endothelial barrier function. FEBS Lett 1995; 374:323 - 6; http://dx.doi.org/10.1016/0014-5793(95)01110-Z; PMID: 7589563
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest 2002; 109:383 - 92; http://dx.doi.org/10.1172/JCI0213595; PMID: 11827998
- Carrithers M, Tandon S, Canosa S, Michaud M, Graesser D, Madri JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am J Pathol 2005; 166:185 - 96; http://dx.doi.org/10.1016/S0002-9440(10)62243-2; PMID: 15632011
- Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med 1993; 178:449 - 60; http://dx.doi.org/10.1084/jem.178.2.449; PMID: 8340753
- Sun QH, DeLisser HM, Zukowski MM, Paddock C, Albelda SM, Newman PJ. Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J Biol Chem 1996; 271:11090 - 8; http://dx.doi.org/10.1074/jbc.271.19.11090; PMID: 8626652
- Sun J, Williams J, Yan HC, Amin KM, Albelda SM, DeLisser HM. Platelet endothelial cell adhesion molecule-1 (PECAM-1) homophilic adhesion is mediated by immunoglobulin-like domains 1 and 2 and depends on the cytoplasmic domain and the level of surface expression. J Biol Chem 1996; 271:18561 - 70; http://dx.doi.org/10.1074/jbc.271.31.18561; PMID: 8702505
- Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature 2003; 421:748 - 53; http://dx.doi.org/10.1038/nature01300; PMID: 12610627
- Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A 1991; 88:7896 - 900; http://dx.doi.org/10.1073/pnas.88.17.7896; PMID: 1881923
- Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res 2000; 259:158 - 66; http://dx.doi.org/10.1006/excr.2000.4919; PMID: 10942588
- Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci U S A 1992; 89:7919 - 23; http://dx.doi.org/10.1073/pnas.89.17.7919; PMID: 1518814
- Lovelady DC, Richmond TC, Maggi AN, Lo C-M, Rabson DA. Distinguishing cancerous from noncancerous cells through analysis of electrical noise. Phys Rev E Stat Nonlin Soft Matter Phys 2007; 76:041908; http://dx.doi.org/10.1103/PhysRevE.76.041908; PMID: 17995027
- Han A, Yang L, Frazier AB. Quantification of the heterogeneity in breast cancer cell lines using whole-cell impedance spectroscopy. Clin Cancer Res 2007; 13:139 - 43; http://dx.doi.org/10.1158/1078-0432.CCR-06-1346; PMID: 17200348
- Jackson DE. The unfolding tale of PECAM-1. FEBS Lett 2003; 540:7 - 14; http://dx.doi.org/10.1016/S0014-5793(03)00224-2; PMID: 12681475
- O’Brien CD, Cao G, Makrigiannakis A, DeLisser HM. Role of immunoreceptor tyrosine-based inhibitory motifs of PECAM-1 in PECAM-1-dependent cell migration. Am J Physiol Cell Physiol 2004; 287:C1103 - 13; http://dx.doi.org/10.1152/ajpcell.00573.2003; PMID: 15201144
- Privratsky JR, Paddock CM, Florey O, Newman DK, Muller WA, Newman PJ. Relative contribution of PECAM-1 adhesion and signaling to the maintenance of vascular integrity. J Cell Sci 2011; 124:1477 - 85; http://dx.doi.org/10.1242/jcs.082271; PMID: 21486942
- DeLisser H, Liu Y, Desprez P-Y, Thor A, Briasouli P, Handumrongkul C, Wilfong J, Yount G, Nosrati M, Fong S, et al. Vascular endothelial platelet endothelial cell adhesion molecule 1 (PECAM-1) regulates advanced metastatic progression. Proc Natl Acad Sci U S A 2010; 107:18616 - 21; http://dx.doi.org/10.1073/pnas.1004654107; PMID: 20926749
- Wu J, Sheibani N. Modulation of VE-cadherin and PECAM-1 mediated cell-cell adhesions by mitogen-activated protein kinases. J Cell Biochem 2003; 90:121 - 37; http://dx.doi.org/10.1002/jcb.10600; PMID: 12938162
- Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT, Sorensen AG. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol 2009; 6:327 - 38; http://dx.doi.org/10.1038/nrclinonc.2009.63; PMID: 19483739
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011; 91:1071 - 121; http://dx.doi.org/10.1152/physrev.00038.2010; PMID: 21742796
- Li JJ, Huang YQ, Cockerell CJ, Friedman-Kien AE. Localization of human herpes-like virus type 8 in vascular endothelial cells and perivascular spindle-shaped cells of Kaposi’s sarcoma lesions by in situ hybridization. Am J Pathol 1996; 148:1741 - 8; PMID: 8669460
- Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev 2003; 67:175 - 212; http://dx.doi.org/10.1128/MMBR.67.2.175-212.2003; PMID: 12794189
- Hengge UR, Ruzicka T, Tyring SK, Stuschke M, Roggendorf M, Schwartz RA, Seeber S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect Dis 2002; 2:281 - 92; http://dx.doi.org/10.1016/S1473-3099(02)00263-3; PMID: 12062994
- Yao L, Salvucci O, Cardones AR, Hwang ST, Aoki Y, De La Luz Sierra M, Sajewicz A, Pittaluga S, Yarchoan R, Tosato G. Selective expression of stromal-derived factor-1 in the capillary vascular endothelium plays a role in Kaposi sarcoma pathogenesis. Blood 2003; 102:3900 - 5; http://dx.doi.org/10.1182/blood-2003-02-0641; PMID: 12907452
- Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, Chandran B, Nelson JA. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol 1999; 73:6892 - 902; PMID: 10400787
- Phillips KG, Kolatkar A, Rees KJ, Rigg R, Marrinucci D, Luttgen M, Bethel K, Kuhn P, McCarty OJT. Quantification of cellular volume and sub-cellular density fluctuations: comparison of normal peripheral blood cells and circulating tumor cells identified in a breast cancer patient. Front Oncol 2012; 2:96; PMID: 22934287