Abstract
Aberrant and/or cumulative amyloid-beta (Aβ) production, resulting from proteolytic processing of the amyloid precursor protein (APP) by β and γ-secretases, have been postulated to be a main etiological basis of Alzheimer disease (AD). A number of proteins influence the subcellular trafficking itinerary of APP and the b-site APP-cleaving enzyme (BACE1) between the cell surface, endosomes and the trans-Golgi network (TGN). Available evidence suggests that co-residence of APP and BACE1 in the endosomal compartments promotes amyloidogenesis. Retrograde transport of APP out of the endosome to the TGN reduces Aβ production, while APP routed to and kept at the cell surface enhances its non-amyloidogenic, α-secretase-mediated processing. Changes in post-Golgi membrane trafficking in aging neurons that may influence APP processing is particularly relevant to late-onset, idiopathic AD. Dystrophic axons are key features of AD pathology, and impaired axonal transport could play crucial roles in the pathogenesis of idiopathic AD. Recent evidence has also indicated that Aβ-induced synaptic defects and memory impairment could be explained by a loss of both AMPA and NMDA receptors through endocytosis. Detail understanding of factors that influence these neuronal trafficking processes will open up novel therapeutic avenues for preventing or delaying the onset of symptomatic AD.
The Amyloid Cascade Hypothesis of Alzheimer Disease
An estimate 24.3 million people in the world have dementia in 2005.Citation1 With an extrapolated 4.6 million new cases of dementia every year, the number of dementia patient will double every 20 years, reaching 81.1 million by 2040. The neurodegenerative disease first reported by Alois AlzheimerCitation2 a hundred years agoCitation3,Citation4 is the most common course of dementia in the elderly above 65 years. Alzheimer disease (AD), as it has become known, is therefore the neurodegenerative disease that has attracted the most research focus in the past decades.
Over 95% of AD cases are late-onset, sporadic and have a complex, idiopathic etiology. A small number of early-onset AD cases has hereditary monogenic defects, and investigations on the underlying mutations had provided important clues for understanding AD pathology. AD presents two distinct pathological features—intracellular neurofibrillary tangles (NFTs) and extracellular amyloid plaques. Both features have clear connections to neuronal demise and consequential onset of dementia symptoms. NFTs comprise of bundles of filaments twisted about each other in pairs (known as paired helical filaments, PHF), and contain abnormally hyperphosphorylated forms of the microtubule-binding protein tau.Citation5 Amyloid plaques are made up of insoluble aggregates of amyloid β (Aβ) peptides.Citation6,Citation7 NFTs and amyloid plaques are functionally related, and the latter could potentially promote tau hyperphosphorylation and NFT pathogenesis.Citation8,Citation9
The amyloid cascade hypothesisCitation10 posits that accumulation of Aβ peptide, generated from proteolytic cleavages of the amyloid precursor protein (APP) in the brain, is the source of extracellular plaques, and the primary etiological agent of AD. However, extracellular amyloid plaque burden does not necessarily correlate with clinical AD symptoms, and it is known that Aβ exists in multiple assembly states, with each exhibiting different physiological or pathophysiological effects.Citation11–Citation13 Accumulating evidence also suggests that both intracellular and extracellular soluble oligomeric forms of Aβ are capable of inducing synaptic defects and the onset of AD symptoms.Citation14–Citation17
As summarized in , Aβ is generated from APP (but not its paralogues APLP1 and APLP2) by a sequential two-step proteolytic process involving β-and γ-secretases.Citation18,Citation19 APP is first cleaved by the β-site APP cleaving enzyme 1 (BACE1),Citation20,Citation21 a member of the pepsin family of aspartyl proteases, to generate a membrane bound C-terminal fragment (C99, CTFβ). A subsequent γ-secretase activity further generates peptides mainly of 40 or 42 amino acids in length, termed Aβ40 and Aβ42, as well as the remaining APP intracellular domain (AICD). Minor forms of Aβ peptides, including Aβ35, Aβ37, Aβ38 and others with less clear neuropathological roles, can also be generated. Aβ40 is more abundant than Aβ42 and although both could be found in amyloid plaques, Aβ42 is apparently more directly neurotoxic,Citation22 and has a greater propensity to oligomerize and to form fibrils.Citation23 Interestingly, Aβ40 appears to be able to reverse the above effect and properties of Aβ42.Citation24–Citation26 Alternative APP proteolytic processing pathways exist, most notably by α-secretases of the A Disintegrin and Metalloprotease (ADAM) family of surface metalloproteases.Citation27 α-secretase cleavage occurs at a site that effectively abolishes BACE1 recognition. The α-secretase cleaved C-terminal fragment (CTFα) is also subsequently cleaved by γ-secretase. However, the small fragment generated as such, named p3, is non-amyloidogenic and has no recognized role in AD. α-secretase cleavage also generates a soluble sAPPα fragment that has some neuroprotective properties.Citation28–Citation31
The γ-secretaseCitation32 responsible for Aβ generation upon β-secretase-mediated cleavage of APP is not a single polypeptide. Rather, the proteolytic activity resides in a complex which mediates a unique process of regulated intramembrane proteolysis (RIP)Citation33-Citation24 of several proteins, such as APP, NotchCitation35 and cadherin.Citation36 The active complex consists of at least four proteins—presenilins (PS)-1 or -2 at the catalytic core, nicastrin, anterior pharynx-defective phenotype 1 (APH1, two isoforms—APH1a and APH1b) and PS-enhancer 2 (PEN-2).Citation37–Citation39 A fifth component, p23/TMP21, a member of the p24 cargo protein family, has recently been identified as part of the γ-secretase complex.Citation40 As alluded to above, γ-secretase cleavage also generates the AICD fragment, with possible transcriptional functions.Citation41,Citation42 Other than cleavages by the conventional α, β- and γ-secretase, the intracellular domain of APP is also processed by caspases to a small 31-amino acid fragment that was shown to induce apoptosis.Citation43,Citation44 APP is also cleave by the mitochondrial serine protease HtrA2, generating a C161 fragment (encompassing amino acids 535–695 of APP695), which function is yet unclear.Citation45
Known genetic predispositionsCitation46,Citation47 to early-onset AD are primarily associated with mutations in APPCitation48 and the presenilins 1/2 component of the γ-secretase complex.Citation49 All these mutations increase somewhat the generation of Aβ, or the Aβ42/Aβ40 ratio.Citation50,Citation51 Endowment with the apolipoprotein E (ApoE) η4 allele is a known risk factor for late-onset AD. ApoE η4 appears to influence Aβ physiology in the aging brain in terms of its oligomerization and clearance, perhaps through its roles in lipid and cholesterol homeostasis.Citation52,Citation53 On that note, there is evidence for high cholesterol levels in mid-life being a risk for ADCitation54,Citation55 and preliminary evidence have suggested that cholesterol-lowering statins could reduce this risk.Citation56 The low density lipoprotein receptor (LDLR) family of proteins, which binds ApoE, modulates APP trafficking and proteolytic processing (see section 3 below). The neuronal sortilin related receptor 1 (SorL1, also named LR11 or SorLA) has in fact been recently found to be genetically associated with late-onset AD.Citation57 Several other potential genetic links to AD are known, including components of all three classes of the APP secretases, but not robustly confirmed. Excellent web information resources for these potential AD genes and loci could be found at www.alzforum.org.
APP interacts with multiple proteins, and these myriad of interacting partners could influence APP processing and AD pathology. Several diverse groups of molecules which superficially appear unrelated to AD pathology have also recently been shown to influence Aβ levels in vitro and in vivo through interactions with APP or the secretases.Citation58 These include the reticulon family members,Citation59,Citation60 the Nogo-66 receptor,Citation61,Citation62 the Rho family GTPases and their effectors,Citation63–Citation65 as well as the peptidyl-prolyl isomerase Pin1.Citation66 Another important and interesting aspect of AD pathology concerns metals ions. Metal ions such as zinc and copper can interact with both APP and Aβ to potentiate AD by enhancing their aggregation as well as the generation of reactive oxygen species.Citation67–Citation69 Oxidative stress has indeed been proposed to be a major mechanism for chronic neuronal death in AD.Citation70,Citation71
Neuronal membrane protein traffic, regulated or deregulated, plays vital roles in AD pathology. Although the etiology of AD appears complex, the pathological roots could be traced to Aβ generation from APP. APP and BACE1 have rather similar subcellular trafficking patterns and itineraries. Effective meeting and reaction between enzyme and substrate as they traverse the cell surface, endosome/lysosome, the trans-Golgi network (TGN) and the early secretory pathway would therefore determine the extent of Aβ production. Emerging evidence suggests that colocalization and interaction between APP and BACE1 in the endosomal compartments promotes amyloidogenesis. Retrograde transport of APP out of the endosome to the TGN reduces Aβ production, while APP routed to and kept at the cell surface enhances its non-amyloidogenic, α-secretase-mediated processing. Changes in post-Golgi membrane trafficking in aging neurons that may influence APP processing are therefore particularly relevant to late-onset, sporadic AD. Recent evidence also indicates that Aβ induced synaptic defects and memory impairment could be explained by loss of both the ionotropic glutamate receptors (AMPA and NMDA receptors) through localized dendritic spine endocytosis, involving fundamental mechanisms that are responsible for synaptic plasticity. This chapter focuses on how membrane traffic processes (and their defects thereof) may influence APP processing and Aβ production, as well as how Aβ could in turn cause synaptic defects by influencing the homeostatic trafficking of glutamate receptors.
Membrane Traffic Itinerary and Localization of APP, BACE1 and γ-secretase—Sites of APP Cleavage and Aβ Production
Both APP and BACE1 are type I membrane proteins with rather similar membrane traffic itineraries. Made at the cell body, they traverse both the exocytic and endocytic pathways of intracellular membrane traffic, and assume at least transient residence in various membrane compartments (summarized in ). Productive interaction between APP and BACE1 resulting in β-secretase cleavage could potentially occur at multiple points in the ER, TGN, cell surface, endosome and lysosomes.Citation72,Citation73 The γ-secretase complex, on the other hand, has a different mode of biogenesis (which includes functional subunit assembly) and membrane traffic dynamics from APP/BACE1. Identifying the predominant site (or sites) of β- and γ-secretase cleavages and consequential Aβ production has been controversial. The issues have been complicated by cell line and methodological differences, and the fact that localization or cleavage site may be altered as a result of mutations in the components concerned. Experimental overexpression of one component may also affect the trafficking of another. For example, high BACE1 expression could shift the subcellular location of APP cleavage to more proximal parts of the secretory pathway.Citation74
β-secretase cleavage sites.
β-secretase cleavage is the critical first step in amyloidogenic APP processing, and brain Aβ load correlated well with increased β-secretase activity in sporadic AD patients.Citation75,Citation76 As BACE1 knockout mice appeared healthy,Citation86 BACE1 has emerged as a primed therapeutic target for AD.Citation77–Citation79 Understanding BACE1 trafficking in relation to APP and APP cleavage is therefore important. Early studies have indicated that Aβ production involves the endocytic pathway,Citation80 and recent studies have strengthened the notion that endosomes are the principle sites of β-cleavage.Citation81 APP has at least two putative endocytic signaling motifs at its cytoplasmic tail, and detail analysis by scanning mutagenesis indicated that the tetrapeptide sequence of YENP is the dominant signal for APP internalization.Citation82 Upregulation of the endocytic pathway stimulated by the endosomal Rab5 increased Aβ production,Citation83 while inhibition of dynamin-dependent endocytosis reduced it.Citation84
The β-secretase activity has been known for some time, and the particularly sticky problem of investigating an enzyme activity without an identity was eventually resolved with the cloning of BACE1.Citation20 Mature BACE1 is found on the cell surface and endosomes, but not ER or lysosomes.Citation85 Sensitive fluorescence microscopy analysis by fluorescence resonance energy transfer (FRET) indicated that APP and BACE1 interact at the cell surface and endosomes, but not lysosomes.Citation86 Since biochemical studies indicate that BACE1 has an acidic pH optimum,Citation87 it follows that the acidic endosomes are the main compartments in which β-cleavage of APP occurs. This notion is further supported by the fact that cellular factors which influence APP and BACE1's trafficking in and out of endosomes also modulate Aβ production (see section 3 below).
γ-secretase cleavage sites
Attempts to identify the main γ-secretase cleavage site(s) have generated a good deal of controversy. Inhibition of γ-cleavage resulting from presenilin (PS)-1 knockoutCitation88 and subsequent demonstration that it contains the catalytic site,Citation89 established it as an essential component of the γ-secretase activity. Unlike APP and BACE1, however, both PS1 and PS2 were localized to compartments of the early secretory pathway (ER and Golgi),Citation90 and there is evidence for “active” γ-secretase activity in a pre-Golgi compartment.Citation91 The exact localization of γ-secretase activity is also undoubtedly complicated by the fact that we are looking at a multi-protein complex that needs to be assembledCitation92,Citation93 and also by the fact that active presenilin is itself an endoproteolyzed, membrane bound polypeptide. γ-secretase activity have been demonstrated in multiple cellular locations, even in the mitochondria.Citation94 More recent analysis is consistent with functional γ-secretase activity concentrating on the cell surface and in early endosomes.Citation95
A potential function of presenilin that has not received as much attention as its catalytic activity is its role in protein trafficking,Citation96 in particular that of APP.Citation97 Surface APP transport is defective in cells overexpressing PS1 mutants which cause familial AD.Citation97 PS1 interacts with phospholipase D1 (PLD1).Citation98 Overexpression of PLD1 in wild type cells promotes generation of APP-containing vesicles from the TGN, corrects impaired neurite outgrowth in PS1 mutant neurons, and reduced Aβ generation.Citation99 Presenilin/γ-secretase activity is also apparently required for normal endosomal recycling of soluble and membrane-associated proteins, and treatment with γ-secretase inhibitors resulted in the accumulation of APP and APP-CTFs in the recycling endosome.Citation100
Where is Aβ produced in the neurons? β cleavage versus α cleavage.
Many cell types in the body express APP. In the brain, BACE1 expression is observed in most neurons. Glia cells expressed little or undetectable BACE1 under normal conditions, although activated astrocytes may have elevated BACE1 expression.Citation20,Citation101 Neurons are therefore believed to be the major source of Aβ produced in the brain. Mature neurons are highly polarized cells with distinct axonal and somatodendritic plasma membrane domains. Endosomal compartments can be found in both axons and dendrite in isolation from those at the cell body. The dendrite is also endowed with rather elaborate internal membranes of the ER and Golgi.Citation102 In view of the highly polarized and complex traffic processes that occur in a neuron, it would be of considerable interest to know if there is a main site of Aβ generation, especially in connection with excess generation during AD pathology.
Another point of interest stems from the fact that proteolytic processing of APP by α-secretases would essentially preclude β-cleavage and Aβ generationCitation103 and Aβ generation is therefore influenced by APP's close encounters with either secretases in its trafficking itinerary. The plasma membrane was shown to be the major site for α-secretase cleavage of APP.Citation104,Citation105 In polarized Madin-Darby canine kidney (MDCK) cells, APP is predominantly targeted to the basolateral membrane where α-secretases are found, but BACE1 is instead targeted to the apical membrane.Citation106,Citation107 APP processing in MDCK cells therefore occurs mainly by α-cleavage.Citation107 In hippocampal neurons, BACE1 is mainly targeted to the axonsCitation107 (analogous to the epithelial apical domain). On the other hand, APP has been shown in early studies to undergo axonal transport,Citation108 but could be transcytose to dendrites.Citation109 Recent work also suggests that APP can be transported in a non-signal dependent manner to both axons and dendrites.Citation110 It appears that APP could be processed by both BACE1 and α-secretases on axonal plasma membranes. However, keeping in mind that BACE1 has an acidic pH optimum and is most active in endosomes, it would logically follow that axonal presynaptic endosomes may be a major site of Aβ production. This notion is supported by findings that endoproteolytic fragments of PS1 can be found in synaptic organelles and growth cone membranes,Citation111 but requires further experimental confirmation. In any case, it should be intuitively clear from the discussions above that the time spent by APP in the endosome versus that of other compartments (such as TGN and the plasma membrane) could have a major influence on Aβ production. Alterations in APP processing in favor of β-secretase cleavage resulting from changes in its trafficking dynamics and resident time in the endosomes, perhaps due to neuronal aging and cumulative oxidative damage, could therefore be relevant for late-onset, sporadic AD. This notion is explore further below as we look at how APP traffic and Aβ production is influenced by multiple cellular factors.
Factors Participating in or Influencing APP/BACE1/γ-Secretase Traffic and Aβ Production
A myriad of cellular factors and processes are known to influence APP/BACE1/γ-secretase traffic and consequential Aβ production. These include general molecular components of exocytic and endocytic traffic, as well as cargo adaptors that may specifically associate with APP. Their mode of function and interactions are briefly summarized below.
General protein trafficking components in APP/BACE1/γ-secretase traffic.
Vesicular transport of APP and BACE1 is governed by the basic molecular machinery of protein traffic in eukaryotic cells. Both molecules are transported in their post-Golgi routes by clathrin-coated vesicles through its associated adaptor proteins. Polarized transport of APP in epithelial cells is mediated by the adaptor protein complex AP1-1B,Citation112 and a similar complex likely interacts with APP in neurons. APP, particularly the cytoplasmic NPXY motif, is known to interact with a host of molecules with implicated functions as cargo adaptors. Some of these molecules also interact with and regulate the trafficking of the ApoE receptor 2 (ApoER2). These are considered collectively in the following section. We first look at two known coat adaptor complexes that mediate BACE1 and APP traffic.
The cytoplasmic domain of BACE1 contains an acid cluster dileucine motif that binds the Vps-27, Hrs and STAM (VHS) domain of Golgi-localized gamma-ear-containing ARF-binding (GGA) proteins.Citation113 Overexpression of GGA1 in cells was reported to increase the level of APP C-terminal fragment (CTFβ) resulting from β cleavage but paradoxically reduced Aβ levels, which suggest that GGA1 somehow prevents CTFβ from being subjected to subsequent γ-secretase cleavage.Citation114 Another report, which showed that GGA1 is enriched in neurons of human brain, demonstrated that overexpression of GGA1, or a dominant-negative variant, reduced CTFβ generation and Aβ secretion.Citation115 On the other hand, silencing of GGA1 increased Aβ secretion. The general picture provided by these reports showed that GGAs have a pivotal influence on the cellular trafficking of BACE1 (and APP) between the TGN and the endosomal compartments, which in turn affects Aβ biogenesis. How GGA-mediated processes may change during AD pathology is still unclear. Interestingly, BACE1 levels appear to be regulated by that of GGA3. GGA3 protein levels were significantly decreased in AD brains, and this inversely correlated with increased levels of BACE1.Citation116
Another coat complex implicated in the transport of BACE1 is the retromer complex, which has an evolutionarily conserved function in endosome-to-Golgi transport.Citation117 Retromer components are highly expressed in the brain, and silencing of the retromer component VPS26 incurred a change in BACE1 trafficking that was similar to GGA silencing.Citation118 Both VPS26 and VPS35 levels are reduced in brain regions selectively vulnerable to AD.Citation119 Like the GGAs, the retromer complex likely modulates BACE1 and APP partitioning between the endosome and the TGN. Although a role for the retromer complex in AD pathology is not supported by the lack of an association between its genetic polymorphism and AD risk,Citation120 changes in retromer subunit levels with age could contribute to late onset AD.
Other than coat proteins, the other two major classes of components of the mammalian protein traffic machinery, namely the Rabs and SNAREs, are less implicated in the modulation of APP or BACE1 traffic. Rab1B and Rab6, which regulates transport at the ER-Golgi boundary and the Golgi-TGN, respectively, appeared to influence APP processing.Citation121,Citation122 One would also expect SNAP (soluble NSF attachment protein) and NSF (N-ethylmaleimide-sensitive factor) attachment receptors (SNAREs) operating at the TGN and endosomes of neuronsCitation123 to participate in APP and BACE1 trafficking. Changes in the function and activities of these SNAREs should influence Aβ biogenesis, but have not yet been reported as such.
APP-interacting adaptor proteins influence APP trafficking and Aβ generation.
Tyrosine phosphorylation of the YENPTY sorting motif at APP's C-terminus resulted in its interaction with several cytosolic phosphotyrosine binding (PTB) domain or Src homology 2 (SH2) domain-containing proteins.Citation124 Some of these, such as the munc-18 interacting proteins (Mints)/X11s, mammalian Disabled (DAB), the Fe65 family of proteins and the JNK-interacting proteins (JIPs), have been extensively implicated in the modulation of APP trafficking and Aβ production.Citation125 Almost invariably, these adaptors also influence trafficking of the ApoE receptor, ApoER2. It was recently shown that binding of ApoE to ApoER2 triggers the endocytosis of APP, BACE1, and ApoER2 in neuroblastoma cells, thereby increasing the production of Aβ. This is apparently mediated by X11α or X11β,Citation126 which bind both APP and the cytosolic domain of ApoER2.Citation127 Another report showed that X11γ/Mint3 and APP are found in the same purified transport vesicles. Silencing of X11γ enhanced endosomal/lysosomal sorting of APP and increased Aβ secretion.Citation128
Mammalian disabled (mDAB)-1 likewise interacts with both APP and ApoER2, and decreased both CTFβ and secreted Aβ in a reelin-dependent manner.Citation129 Interestingly, X11α and mDab-1 exert opposing effects on APP processing, and when both proteins are co-expressed, the effect of X11α overrides that of mDab-1.Citation130 Fe65 is involved in APP signaling through the AICDCitation131 as well as several aspects of APP-processing, and it also binds ApoER2.Citation132,Citation133 The JIPs are scaffold proteins that could link APP to JNK associated apoptotic functionsCitation134,Citation135 and kinesin that mediate APP (as well as ApoER2) axonal transport.Citation136 The interactions between APP and these binding proteins, and the resulting influences on APP trafficking and processing are exceedingly complex, and the links between APP and ApoER2 pertaining to AD pathology needs further clarification.
ApoER2 belongs to the family of LDL receptor-related (LRP) proteins, several of which have been implicated in APP processing and AD.Citation137–Citation139 LRP1, a neuronal receptor for ApoE, binds to APP (either directly or through Fe65) affects Aβ generation and clearance in the brain. APP processing is mediated by LRP1 on multiple levels. Enhanced APP internalization through LRP expression decreased cell surface APP levels, thereby reducing APP-shedding and enhanced Aβ secretion.Citation140 In a recent report, LRP1 expression was shown to regulate ApoE and cholesterol levels within the CNS. Interestingly, deletion of APP, APLP2, or components of the γ-secretase complex, enhanced the expression and function of LRP1 in a way that is reversible by forced overexpression of AICD. The latter, together with Fe65 and Tip60, interacts with the LRP1 promoter and suppresses its transcription. The cellular interaction between APP and LRP1 thus goes beyond the physical interactions during protein trafficking, but occurs at the level of gene transcription as well. Several genetic polymorphisms of LRP1 have indeed been weakly associated with AD. On the other hand, LRP1B, a homologue of LRP1, appears to retain APP at the cell surface and reduced Aβ production due to its slower rate of endocytosis.Citation141 Clearly, a detail understanding of LRP1 and LRP1B expression in AD-affected brain regions would be important, and may need to novel therapeutic approaches.
The neuronal sorting protein receptor-related containing LDLR class A repeats (SorLA) (also named SorL1 or LR11), is another neuronal sorting protein that plays pivotal roles in APP processing. SorLA has received much recent attention as a clear demonstration of genetic linkage of its variants to late-onset, sporadic AD has now been made,Citation66 consistent with earlier findings that its expression is reduced in brains of sporadic AD but not in familial AD cases.Citation142 SorLA interacts with both APP and BACE1, and its interaction with APP apparently reduced the latter's interaction with BACE1.Citation143 Overexpression of sorLA in neurons redistribution surface APP to the Golgi and decreased its processing to Aβ, whereas ablation of sorLA expression in knockout mice results in increased levels of Aβ in the brain.Citation143–Citation145 SorLA may act to retain APP in TGN, and its sorting activity is dependent on functional interaction with adaptor proteins involved in protein transport to and from the trans-Golgi network, such as GGA and phosphoprotein acidic cluster sorting protein-1 (PACS-1).Citation146 SorLA is indeed an interesting and promising target for AD prevention or delay of late-onset AD symptoms. Recent findings with brain samples from antemortem diagnosis showed that SorLA levels are in fact already reduced in individuals with the pre-AD condition of mild cognitive impairment, and cognitive impairment is more severe for those with lower SorLA.Citation147 Another interesting report showed that docosahexaenoic acid feeding increases SorLA levels in multiple systems, including brain.Citation148 Dietary modulation of SorLA levels therefore provides a plausible explanation for the association of ω-3 fatty acids with improved cognition and reduced AD risk.
Other factors influencing APP trafficking and Aβ generation.
New factors that modulate APP traffic continue to emerge. Genetic variants in the ubiquilin 1 (UBQLN1) gene, which encodes a ubiquitin-like protein on chromosome 9q22 increased the risk for AD, possibly through altered expression and alternative splicing in the brain.Citation149 Ubiquilin 1 knockdown seemed to increase the cellular maturation and trafficking of APP, enhancing the appearance of full-length APP on the cell surface. Secretion of sAPPα/β and Aβ40/42 were also increased.Citation150 Fluorescence resonance energy transfer (FRET) analysis demonstrated that APP and ubiquilin 1 do come into close contact in vivo. Another APP sorting signal interacting protein is PAT1, a microtubule binding protein.Citation151 PAT1 appears to modulate the trafficking of not just APP, but also its homologues APLP1 and APLP2.Citation152 Modulation of PAT1 levels by overexpression and siRNA-mediated silencing also changes APP surface expression and Aβ production.
Another factor that could modulate APP traffic is p23/TMP21, a member of the p24 family type I transmembrane proteins that was recently identified as a presenilin complex component capable of modulating γ-secretase cleavage.Citation40 TMP21 silencing increased secretion of sAPP and Aβ production. The effect of TMP21 depletion and expression of a familial AD-linked PS1 mutant on Aβ production appeared to be additive, and immunoisolation analyses showed a decrease in co-residence of APP and X11γ/Mint3.Citation153 Other than modulating γ-secretase activity, TMP21 may therefore have a separate modulation effect on APP traffic.
All the findings discussed above provide a general picture of how modulation of trafficking of APP and its secretases by various cellular factors could modulate Aβ generation. Broadly speaking, enhanced retrograde transport of APP out of the endosome to the TGN reduces Aβ production. Enhanced APP routing to, or reduction of its internalization from the cell surface, facilitates its non-amyloidogenic, α-secretase-mediated processing. Changes in post-Golgi membrane trafficking in aging neurons that may influence APP processing may therefore precipitate late-onset, idiopathic AD.
Axonal Transport and Alzheimer Disease
Efficient and tightly regulated axonal transport from and to the neuronal cell body is essential for neuronal survival and function. Bidirectional transport of vesicles, organelles, cargo carriers and protein complexes occurs along specifically polarized microtubule tracks by engaging microtubule-dependent motor proteins of the kinesin and dynein superfamilies.Citation154 Impair axonal transport as a result of gene mutations is known to predispose individuals to pathogenesis of several neurodegenerative diseases.Citation155,Citation156 Perhaps the most prominent of these are CNS motor neuron diseases such as amyotrophic lateral sclerosis (ALS).Citation157 Mutation in the dynactin subunit, p150Glued, for example, is a genetic risk factor for ALS.Citation158 In Huntington's disease, the mutant huntingtin protein has an expanded polyglutamine tract and impairs axonal trafficking in mammalian neurons in vivo and in vitro.Citation159 Certain forms of the peripheral neuropathy Charcot-Marie-Tooth type II could also result from mutations in traffic components such as the microtubule motor KIF1β,Citation160 regulator of vesicular traffic such as Rab7,Citation161 and neurofilamentCitation162 that cause defects in axonal transport. There are many histopathological as well as cellular and molecular links between AD and defects in axonal transport, as elaborated in more detail below.
Impairment of axonal transport in AD.
The two major pathological hallmark features of AD are amyloid plaques consisting of aggregated Aβ and neurofibrillary tangles (NFT) containing hyperphosphorylated tau. Another prominent histopathological manifestation of AD is the presence of abnormal and dystrophic axons and dendrites. These may be associated with amyloid plaques and NFTs,Citation163,Citation164 but are in some cases found unassociated with either plaques or tangles.Citation165,Citation166 Mouse models overexpressing wild type and mutant forms of AD-related proteins such as APP,Citation167 BACE1,Citation168 PS1,Citation169,Citation170 tauCitation171,Citation172 and ApoE4,Citation173 have all been shown to exhibit axonal defects as in human AD. Interestingly, co-expression of the Drosophila homologue of APP, APPL, together with tau, also disrupts axonal transport in flies.Citation174 Selective white matter changes indicative of axonopathy are readily detected by magnetic resonance imaging (MRI) of AD patients.Citation175,Citation176 All the above indicate that AD pathogenesis causes axonal defects. That dystrophic axons could be found before clear plaque deposition or tangle formation in transgenic animals, and that white matter changes could be detected in pre-AD patients, would also suggest that axonal defects occurs rather early in AD pathogenesis.
How exactly does onset of AD pathology result in axonal defects? It is worth noting that many proteins associated with the pathogenesis of AD are transported to the axons and the presynaptic compartment,Citation177,Citation178 and that synaptic release of Aβ contributes significantly to extracellular amyloid deposits.Citation179 APP has been shown to undergo fast axonal transportCitation108 through direct binding with kinesin-1.Citation180 It was in fact suggested that BACE1 and PS-1 are also co-transported with APP in a membranous vesicular compartment, with the latter functioning like a membrane receptor for kinesin-1.Citation181 These earlier findings have recently been disputed,Citation182 but there is little doubt that axonal/presynaptic trafficking of APP and its processing enzymes occurs in normal and dystrophic neurons. Another point to note is that APP (and BACE1 and PS-1) are often found to accumulate in damaged axons following traumatic brain injury,Citation183,Citation184 probably resulting from axolemmal disruption and impaired axonal transport.Citation185 Traumatic brain injury is a known epigenetic risk factor for AD,Citation186 and one plausible link could be aberrant production of neurotoxic Aβ with a detrimental positive feedback mechanism that further damages the surviving axons.
In understanding the basis of axonal transport defects in AD, a particularly relevant point to consider is the involvement of abnormally phosphorylated tau in AD pathology. Tau is an axonally transported microtubule-associated protein.Citation187 Hyperphosphorylation of tau not only results in a loss of function in terms of impaired microtubule binding, but probably also a toxic gain of functionCitation188 that may disrupt axonal transport.Citation189,Citation190 Tau is known to be involved in regulating axonal transport, and this notion is augmented by recent evidence showing that tau interacts with the dynactin complex.Citation191 Interestingly, in spite of the frequent presence of tau-associated NTFs in AD, no tau mutations have been identified for familial cases of AD.
The ontogenic relationships between amyloid plaques and NFTs have been enigmatic. Aβ may interact with tau, either directly or indirectly, resulting in the latter's hyperphosphorylationCitation192,Citation193 and drives NFT formation.Citation8,Citation194 Aβ may also induce proteosomal impairments that affect tau degradation, leading to its accumulation.Citation195 Aberrant activation of the tau-phosphorylating cyclin-dependent kinase 5 (Cdk5) has also been heavily implicated in AD pathogenesis.Citation196 Aβ and other neuronal stress factors could activate calpain, thereby enhancing Cdk5 activation. Conversely, the neuroprotective sAPPα generated by the non-amyloidogenic processing of APP, suppresses Cdk5 activation. Recent advances also suggest that activated p25/Cdk5 could induce the production and intraneuronal accumulation of Aβ in vivo, prior to any apparent tau or amyloid-associated neuropathology.Citation197 Aberrant Cdk5 activation could therefore lie upstream of amyloidogenic Aβ production, with the latter feeding forward to further activate Cdk5, as well as glycogen synthase kinase 3-β (GSK3-β), resulting in tau hyperphosphorylation at later stages.
Could aging and brain injury-related axonal transport defects play a causal role in AD pathogenesis?
While it is clear that axonal defect is a key disease manifestation of AD and is responsible for its symptoms, could it also play a causal role in precipitating idiopathic AD? Available data indicate that axonal defects can be found in both pre-AD and AD brains, either in association or not in association with amyloid pathology. The latter, in particular, points to the possibility that axonal defects could coincide with the earliest stages of AD pathogenesis. Particularly strong evidence for axonal defects underlying AD pathogenesis has been presented by Goldstein and colleagues.Citation167 The authors showed that reducing kinesin-1 gene dosage by half in APP transgenic mice enhanced axonal defects, Aβ generation and amyloid deposition. The latter likely resulted from increased APP amyloidogenic processing in transport retarded axons.Citation198
Idiopathic, late-onset AD is an age-dependent disease, and is therefore expected to be influenced and precipitated by chronic cellular defects that accumulate as the individual ages. An example of such contributive factors to AD that has received popular attention is oxidative stress and related mitochondrial dysfunction.Citation199 The potential of cumulative focal blockages in axonal transport resulting from a myriad of factors such as physical trauma and neurochemical toxicity in initiating AD pathogenesis are difficult to assess, but are definitely plausible. Injury to axons causes the formation of axonal spheroids—focal swellings that likely result from block transport.Citation200 Speculatively, what could possibly happen is that the accumulation of stalled APP and its processing enzymes in these focal injuries, resulting in increased APP processing and Aβ generations, perhaps even in a way that favors amyloidogenic ratios of Aβ40/Aβ42. These, together with other factors associated with brain injury (such as neuroinflammation), triggers a cascade of events that might increase tau phosphorylation and NFT formation. The notion of chronic and cumulative axonal transport defects leading to AD pathogenesis is attractive, and is an avenue for potential therapeutic interventions. However, further understanding of the underlying processes involved in the transition from axonal defect to AD is needed.
Aβ-induced Synaptic Defects and Glutamate Receptor Trafficking
Dementia, which is generally preceded by memory deficits and a decline in cognitive functions, is the primary neurological defect associated with AD. The amyloid cascade hypothesis as it was originally proposed suggests that accumulation of fibrillar β-amyloid and their deposition as insoluble extracellular plaques underlies these neurological deficits. A major caveat for the above notion is that amyloid burden does not necessarily correlate well with clinical severity of AD. In some cases AD symptoms and neuronal loss could be traced to brain regions that are amyloid free. Furthermore, memory and cognitive function deficits could become obvious much earlier than plaques. An opposite notion in fact suggests that the insoluble amyloid deposits are sinks, rather than sources of Aβ toxicity. Recent findings have indicated that non-fibrillar, soluble oligomers of Aβ could cause cognitive deficits by impairing synaptic function.Citation12,Citation13 Aβ oligomers could induce aberrations in synapse composition, shape and density, particularly in AD relevant excitatory pyramidal neurons.Citation201,Citation202 Aβ oligomers may act presynaptically, suppressing spontaneous synaptic activity by inhibition of P/Q-type calcium currents,Citation203 or by disrupting synaptic vesicle endocytosis.Citation204 Soluble Aβ interacts with several receptor molecules, and have been postulated to disrupt synaptic memory mechanisms via stress-activated kinases and mediators of oxidative stress.Citation205 Excitatory transmissions could be depressed by both presynaptic and postsynaptic mechanisms.Citation206 A particularly relevant aspect Aβ's effect on synapses in AD is the impairment of hippocampal LTP.Citation207 It has in fact been shown that neuronal activity could modulate the formation and secretion of Aβ in hippocampal slices, with the peptide in turn depressing excitatory synaptic transmission.Citation208
An important underlying mechanism by which Aβ suppress LTP (and presumably induce consequential memory impairment), appears to be its influence on synaptic AMPA and NMDA receptors. AMPA receptors are the major excitatory neurotransmitter receptors in the brain, and its expression levels at the synapses are highly dynamic. Regulation of AMPA receptor to and from the synaptic plasma membrane, and localized recycling of AMPA receptors at dendritic spines are major mechanism underlying synaptic plasticity.Citation209,Citation210 Aβ could interact directly with AMPA receptors and influence receptor function and channel activity,Citation211 and this may occur to varying degrees depending on Aβ levels. A more eminent mechanism by which Aβ induces synaptic depression involves enhanced loss of AMPA receptor from dendritic spine surfaces via endocytosis. Interestingly, this is mediated by signaling pathways similar to that occurring during long term depression (LTD), involving calcineurin and p38 MAP kinase activity.Citation212
Aβ-induced depression of excitatory transmission and synaptic spine loss is dependent on NMDA receptor activity.Citation213,Citation214 Although not perceived to be as dynamic as AMPA receptor, activity modulated alterations in NMDA receptor trafficking contributes to homeostatic plasticity.Citation215 Intriguingly, Aβ also promotes the endocytosis of NMDA receptors,Citation216,Citation217 and does this via binding to α7 nicotinic acetylcholine receptor, initiating subsequent signaling processes that require calcineurin and the tyrosine phosphatase STEP (striatal-enriched phosphatase). Although Aβ's direct binding to α7 nicotinic acetylcholine receptor is a controversial issue,218 Aβ treatment reduced surface NR2B and NR1, but not GABA receptor subunits.
Concluding Remarks
Advances in our basic understanding of biology are sometimes driven by intense efforts galvanized by the need to resolve a clinical problem. AD exemplifies such cases, as it is an exceptionally fascinating disease in terms of molecular and cellular etiology. Much of our understanding of the importance and mechanism of intramembrane proteolysis, for example, could be attributed to investigations on APP processing. In a similar way, studies on the subcellular trafficking and dynamics of the various membrane proteins associated with AD pathology are also pushing the frontiers of our understanding of protein trafficking in neurons. Much is now known, but there is much more to be learned. A detail understanding of factors that influence neuronal trafficking processes will open up novel therapeutic avenues for AD, as well as other neurodegenerative disorders.
Figures and Tables
Figure 1 Proteolytic processing of APP. A schematic depiction of APP proteolytic processing. APP is a type I transmembrane protein. The Aβ peptide domain is located partially within the membrane spanning hydrophobic segment. In amyloidogenesis, the β-secretase recognition site is cleaved by BACE1 to a soluble fragment N-terminal fragment sAPPα, a C-terminal fragment CTFβ. The latter is then cleaved intramembranously by γ-secretase to yield Aβ and the APP intracellular domain (AICD). In the non-amyloidogenic pathway, α-secretases cleave at a site within the Aβ peptide sequence. This cleavage essentially disrupts the β-secretase recognition site. α-secretases cleavage yields the soluble N-terminal sAPPα, and the C-terminal CTFα. The latter can also be cleave by γ-secretase to yield a non-toxic 3 kDa fragment known as p3, and the APP intracellular domain (AICD).
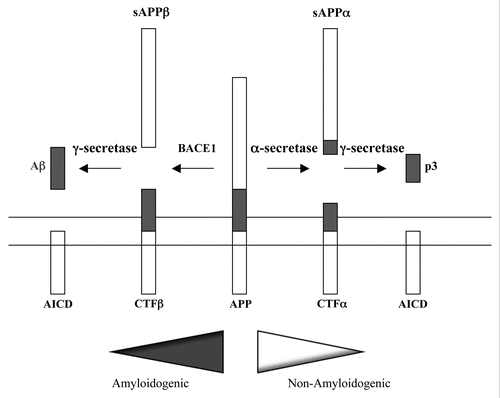
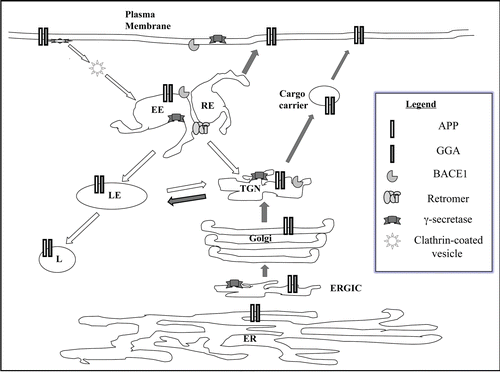
Figure 2 Trafficking itinerary of APP and BACE1. A schematic diagram of the trafficking itinerary of APP and BACE1. Both proteins have very similar itinerary and can be found in compartments of the exocytic pathway and endocytic pathway. Locations of general components of membrane traffic known to affect APP/BACE1 traffic, such as gamma-ear-containing ARF-binding (GGA) proteins and the retromer complex are also shown. Although presenilins are largely localized to the ER-Golgi region at steady state, these and other components of the γ-secretase complex also have widespread presence. Note that this is a general scheme which does not include detail depiction of neuronal domains such as axons and dendrites. Dark arrows indicate exocytic or anterograde transport and white arrow endocytic or retrograde trafficking. ER, endoplasmic reticulum; ERGIC, ER-Golgi intermediate compartment; TGN, trans-Golgi network; EE, early endosome; RE, recycling endosome; LE, late endosome; L, lysosome.
Acknowledgements
The author declares no financial conflict of interest. The exceptionally vast relevant literature had prevented direct citation of many excellent works, and I apologize for this glaring inadequacy in this short overview.
References
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Alzheimer's Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet 2005; 366:2112 - 2117
- Goedert M, Ghetti B. Alois Alzheimer: his life and times. Brain Pathol 2007; 17:57 - 62
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006; 314:777 - 781
- Hardy J. A hundred years of Alzheimer's disease research. Neuron 2006; 52:3 - 13
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 2005; 1739:198 - 210
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885 - 890
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald B, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985; 82:4245 - 4249
- Blurton-Jones M, Laferla FM. Pathways by which Aβ facilitates tau pathology. Curr Alzheimer Res 2006; 3:437 - 448
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol 2006; 175:541 - 546
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002; 297:353 - 356
- Lee HG, Casadesus G, Zhu X, Joseph JA, Perry G, Smithm MA. Perspectives on the amyloid-β cascade hypothesis. J Alzheimers Dis 2004; 6:137 - 145
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006; 440:352 - 357
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8:79 - 84
- Hoozemans JJ, Chafekar SM, Baas F, Eikelenboom P, Scheper W. Always around, never the same: pathways of amyloid beta induced neurodegeneration throughout the pathogenic cascade of Alzheimer's disease. Curr Med Chem 2006; 13:2599 - 2605
- Wirths O, Multhaup G, Bayer TA. A modified β-amyloid hypothesis: intraneuronal accumulation of the β-amyloid peptide—the first step of a fatal cascade. J Neurochem 2004; 91:513 - 520
- Cuello AC. Intracellular and extracellular Aβ, a tale of two neuropathologies. Brain Pathol 2005; 15:66 - 71
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci 2007; 8:499 - 509
- Haass C. Take five—BACE1 and the γ-secretase quartet conduct Alzheimer's amyloid β-peptide generation. EMBO J 2004; 23:483 - 488
- Wilquet V, De Strooper B. Amyloid-beta precursor protein processing in neurodegeneration. Curr Opin Neurobiol 2004; 14:582 - 588
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE1. Science 1999; 286:735 - 741
- Vassar R. BACE1: the β-secretase enzyme in Alzheimer's disease. J Mol Neurosci 2004; 23:105 - 114
- Zhang Y, McLaughlin R, Goodyear C, LeBlanc A. Selective cytotoxicity of intracellular amyloid-β peptide 1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol 2002; 156:519 - 529
- Kim W, Hecht MH. Sequence determinants of enhanced amyloidogenicity of Alzheimer Aβ42 peptide relative to Aβ40. J Biol Chem 2005; 280:35069 - 35076
- Awasthi A, Matsunaga Y, Yamada T. Amyloid-β causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid-β-derived short peptides. Exp Neurol 2005; 196:282 - 289
- Zou K, Kim D, Kakio A, Byun K, Gong JS, Kim J, et al. Amyloid β-protein (Aβ)1–40 protects neurons from damage induced by Aβ1-42 in culture and in the brain. J Neurochem 2003; 87:609 - 619
- Yan Y, Wang C. Aβ40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol 2007; 369:909 - 916
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein α-secretases. J Neurosci Res 2003; 74:342 - 352
- Mattson MP. Secreted forms of β-amyloid precursor protein modulate dendritic outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons. J Neurobiol 1994; 25:439 - 450
- Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, et al. Memory-enhancing effects of secreted forms of the β-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci USA 1998; 95:12683 - 12688
- Tang BL. Alzheimer's disease: channeling APP to non-amyloidogenic processing. Biochem Biophys Res Commun 2005; 331:375 - 378
- Fahrenholz F, Postina R. Alpha-secretase activation—an approach to Alzheimer's disease therapy. Neurodegener Dis 2006; 3:255 - 261
- Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane?. Nat Rev Mol Cell Biol 2004; 5:499 - 504
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 2000; 100:391 - 398
- Weihofen A, Martoglio B. Intramembrane-cleaving proteases: Controlled liberation of functional proteins and peptides from membranes. Trends Cell Biol 2003; 13:71 - 78
- Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 2003; 26:565 - 597
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, et al. A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 2002; 2:1948 - 1956
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999; 398:513 - 517
- De Strooper B. Aph-1, Pen-2 and nicastrin with presenilin generate an active γ-secretase complex. Neuron 2003; 38:9 - 12
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of γ-secretase activity. Nat Cell Biol 2003; 5:486 - 488
- Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature 2006; 440:1208 - 1212
- Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem 2004; 279:24601 - 24611
- Von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, et al. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci 2004; 117:4435 - 4448
- Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, et al. Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem 2002; 82:283 - 294
- Lu DC, Soriano S, Bredesen DE, Koo EH. Caspase cleavage of the amyloid precursor protein modulates amyloid β-protein toxicity. J Neurochem 2003; 87:733 - 741
- Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, et al. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem 2006; 281:34277 - 34287
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 2005; 120:545 - 555
- Ertekin-Taner N. Genetics of Alzheimer's disease: a centennial review. Neurol Clin 2007; 25:611 - 667
- Kowalska A. Amyloid precursor protein gene mutations responsible for early-onset autosomal dominant Alzheimer's disease. Folia Neuropathol 2003; 41:35 - 40
- Czech C, Tremp G, Pradier L. Presenilins and Alzheimer's disease: biological functions and pathogenic mechanisms. Prog Neurobiol 2000; 60:363 - 384
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 1996; 383:710 - 713
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 1997; 3:67 - 72
- Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci 2004; 23:189 - 204
- Poirier J. Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer's disease. Neurobiol Aging 2005; 26:355 - 361
- Sambamurti K, Granholm AC, Kindy MS, Bhat NR, Greig NH, Lahiri DK, et al. Cholesterol and Alzheimer's disease: clinical and experimental models suggest interactions of different genetic, dietary and environmental risk factors. Curr Drug Targets 2004; 5:517 - 528
- Sjogren M, Mielke M, Gustafson D, Zandi P, Skoog I. Cholesterol and Alzheimer's disease- is there a relation?. Mech Ageing Dev 2006; 127:138 - 147
- Whitfield JF. Can statins put the brakes on Alzheimer's disease?. Expert Opin Investig Drugs 2006; 15:1479 - 1485
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 2007; 39:168 - 177
- Tang BL, Liou YC. Novel modulators of amyloid-β precursor protein processing. J Neurochem 2007; 100:314 - 323
- He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat Med 2004; 10:959 - 965
- Murayama KS, Kametani F, Saito S, Kume H, Akiyama H, Araki W. Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid β-protein. Eur J Neurosci 2006; 24:1237 - 1244
- Park JH, Gimbel DA, GrandPre T, Lee JK, Kim JE, Li W, et al. Alzheimer precursor protein interaction with the Nogo-66 receptor reduces amyloid-β plaque deposition. J Neurosci 2006; 26:1386 - 1395
- Park JH, Widi GA, Gimbel DA, Harel NY, Lee DH, Strittmatter SM. Subcutaneous Nogo receptor removes brain amyloid-β and improves spatial memory in Alzheimer's transgenic mice. J Neurosci 2006; 26:13279 - 13286
- Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X, et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Aβ42 by inhibiting Rho. Science 2003; 302:1215 - 1217
- Pedrini S, Carter TL, Prendergast G, Petanceska S, Ehrlich ME, Gandy S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med 2005; 2:18
- Gianni D, Zambrano N, Bimonte M, Minopoli G, Mercken L, Talamo F, et al. Platelet-derived growth factor induces the beta-gamma-secretase-mediated cleavage of Alzheimer's amyloid precursor protein through a Src-Rac-dependent pathway. J Biol Chem 2003; 278:9290 - 9297
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 2006; 440:528 - 534
- Bishop GM, Robinson SR. The amyloid paradox: amyloid-beta-metal complexes can be neurotoxic and neuroprotective. Brain Pathol 2004; 14:448 - 452
- Adlard PA, Bush AI. Metals and Alzheimer's disease. J Alzheimer's Dis 2006; 10:145 - 163
- Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer's disease amyloid beta peptide. Biochim Biophys Acta 2007; 1768:1976 - 1990
- Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer's disease. J Neurochem 2006; 96:1 - 13
- Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 2007; 64:2202 - 2210
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 2006; 52:15 - 31
- Pasternak SH, Callahan JW, Mahuran DJ. The role of the endosomal/lysosomal system in amyloid-β production and the pathophysiology of Alzheimer's disease: reexamining the spatial paradox from a lysosomal perspective. J Alzheimer's Dis 2004; 6:53 - 65
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, et al. BACE1 overexpression alters the subcellular processing of APP and inhibits Aβ deposition in vivo. J Cell Biol 2005; 168:291 - 302
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003; 9:3 - 4
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, et al. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci USA 2004; 101:3632 - 3637
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE1 knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet 2001; 10:1317 - 1324
- Vassar R. Beta-secretase (BACE1) as a drug target for Alzheimer's disease. Adv Drug Deliv Rev 2002; 54:1589 - 1602
- Durham TB, Shepherd TA. Progress toward the discovery and development of efficacious BACE1 inhibitors. Curr Opin Drug Discov Devel 2006; 9:776 - 791
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 1994; 269:17386 - 17389
- Tate BA, Mathews PM. Targeting the role of the endosome in the pathophysiology of Alzheimer's disease: a strategy for treatment. Sci Aging Knowledge Environ 2006; 2006:2
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, et al. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J Biol Chem 1999; 274:18851 - 18856
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, et al. Rab5-stimulated upregulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J Biol Chem 2003; 278:31261 - 31268
- Carey RM, Balcz BA, Lopez-Coviella I, Slack BE. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid β protein. BMC Cell Biol 2005; 6:30
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme, the Alzheimer's disease β-secretase. J Biol Chem 2000; 275:33729 - 33737
- Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE1 interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci 2003; 116:3339 - 3346
- Ermolieff J, Loy JA, Koelsch G, Tang J. Proteolytic activation of recombinant promemapsin 2 (pro-β-secretase) studied with new fluorogenic substrates. Biochemistry 2000; 39:12450 - 12456
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391:387 - 390
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000; 405:689 - 694
- Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, et al. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med 1996; 2:224 - 229
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, et al. Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol 1999; 147:277 - 294
- Kaether C, Haass C, Steiner H. Assembly, trafficking and function of γ-secretase. Neurodegener Dis 2006; 3:275 - 283
- Kim J, Kleizen B, Choy R, Thinakaran G, Sisodia SS, Schekman RW. Biogenesis of γ-secretase early in the secretory pathway. J Cell Biol 2007; 179:951 - 963
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, et al. Nicastrin, presenilin, APH-1, and PEN-2 form active γ-secretase complexes in mitochondria. J Biol Chem 2004; 279:51654 - 51660
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic 2006; 7:408 - 415
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 1998; 21:1213 - 1221
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of β-amyloid precursor protein. J Biol Chem 2003; 278:3446 - 3454
- Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, et al. Presenilin-1 uses phospholipase D1 as a negative regulator of β-amyloid formation. Proc Natl Acad Sci USA 2006; 103:1941 - 1946
- Cai D, Zhong M, Wang R, Netzer WJ, Shields D, Zheng H, et al. Phospholipase D1 corrects impaired βAPP trafficking and neurite outgrowth in familial Alzheimer's disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci USA 2006; 103:1936 - 1940
- Zhang M, Haapasalo A, Kim DY, Ingano LA, Pettingell WH, Kovacs DM. Presenilin/γ-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J 2006; 20:1176 - 1178
- Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V. Neuronal and glial β-secretase (BACE1) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res 2001; 64:437 - 446
- Tang BL. Emerging aspects of membrane traffic in neuronal dendrite growth. Biochim Biophys Acta 2008; 1783:169 - 176
- Tang BL. Alzheimer's disease: channeling APP to non-amyloidogenic processing. Biochem Biophys Res Commun 2005; 331:375 - 378
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, et al. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 1999; 96:3922 - 3927
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in the trans-Golgi network. J Biol Chem 2000; 275:2568 - 2575
- Haass C, Koo EH, Teplow DB, Selkoe DJ. Polarized secretion of β-amyloid precursor protein and amyloid β-peptide in MDCK cells. Proc Natl Acad Sci USA 1994; 91:1564 - 1568
- Capell A, Meyn L, Fluhrer R, Teplow DB, Walter J, Haass C. Apical sorting of β-secretase limits amyloid β-peptide production. J Biol Chem 2002; 277:5637 - 5643
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, et al. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 1990; 87:1561 - 1565
- Simons M, Ikonen E, Tienari PJ, Cid-Arregui A, Mönning U, Beyreuther K, et al. Intracellular routing of human amyloid protein precursor: axonal delivery followed by transport to the dendrites. J Neurosci Res 1995; 41:121 - 128
- Back S, Haas P, Tschäpe JA, Gruebl T, Kirsch J, Müller U, et al. β-amyloid precursor protein can be transported independent of any sorting signal to the axonal and dendritic compartment. J Neurosci Res 2007; 85:2580 - 2590
- Beher D, Elle C, Underwood J, Davis JB, Ward R, Karran E, et al. Proteolytic fragments of Alzheimer's disease-associated presenilin 1 are present in synaptic organelles and growth cone membranes of rat brain. J Neurochem 1999; 72:1564 - 1573
- Icking A, Amaddii M, Ruonala M, Höning S, Tikkanen R. Polarized transport of Alzheimer amyloid precursor protein is mediated by adaptor protein complex AP1-1B. Traffic 2007; 8:285 - 296
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol 2004; 5:23 - 32
- Von Arnim CA, Spoelgen R, Peltan ID, Deng M, Courchesne S, Koker M, et al. GGA1 acts as a spatial switch altering amyloid precursor protein trafficking and processing. J Neurosci 2006; 26:9913 - 9922
- Wahle T, Thal DR, Sastre M, Rentmeister A, Bogdanovic N, Famulok M. GGA1 is expressed in the human brain and affects the generation of amyloid β-peptide. J Neurosci 2006; 26:12838 - 12846
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, et al. Depletion of GGA3 stabilizes BACE1 and enhances beta-secretase activity. Neuron 2007; 54:721 - 737
- Seman MN. Recycle your receptors with retromer. Trends Cell Biol 2005; 15:68 - 75
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE1). J Biol Chem 2005; 280:11696 - 11703
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, et al. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol 2005; 58:909 - 919
- Riemenschneider M, Schoepfer-Wendels A, Friedrich P, Konta L, Laws SM, Mueller JC, et al. No association of vacuolar protein sorting 26 polymorphisms with Alzheimer's disease. Neurobiol Aging 2007; 28:883 - 884
- Dugan JM, DeWit C, McConlogue L, Maltese WA. The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J Biol Chem 1995; 270:10982 - 10989
- McConlogue L, Castellano F, DeWit C, Schenk D, Maltese WA. Differential effects of a Rab6 mutant on secretory versus amyloidogenic processing of Alzheimer's beta-amyloid precursor protein. J Biol Chem 1996; 271:1343 - 1348
- Wang Y, Tang BL. SNAREs in neurons—beyond synaptic vesicle exocytosis. Mol Membr Biol 2006; 23:377 - 384
- Kawasumi M, Matsuda S, Matsuoka M, Nishimoto I. Cytoplasmic tail adaptors of Alzheimer's amyloid-β protein precursor. Mol Neurobiol 2004; 30:185 - 200
- Russo C, Venezia V, Repetto E, Nizzari M, Violani E, Carlo P, Schettini G. The amyloid precursor protein and its network of interacting proteins: physiological and pathological implications. Brain Res Rev 2005; 48:257 - 264
- Miller CC, McLoughlin DM, Lau KF, Tennant ME, Rogelj B. The X11 proteins, Aβ production and Alzheimer's disease. Trends Neurosci 2006; 29:280 - 285
- He X, Cooley K, Chung CH, Dashti N, Tang J. Apolipoprotein receptor 2 and X11 α/β mediate apolipoprotein E-induced endocytosis of amyloid-β precursor protein and β-secretase, leading to amyloid-beta production. J Neurosci 2007; 27:4052 - 4060
- Shrivastava-Ranjan P, Faundez V, Fang G, Rees H, Lah JJ, Levey AI. Mint3/X11γ is an ADP-ribosylation factor-dependent adaptor that regulates the traffic of the Alzheimer's precursor protein from the Trans-Golgi network. Mol Biol Cell 2008; 19:51 - 64
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem 2006; 281:35176 - 35185
- Parisiadou L, Efthimiopoulos S. Expression of mDab1 promotes the stability and processing of amyloid precursor protein and this effect is counteracted by X11α. Neurobiol Aging 2007; 28:377 - 388
- McLoughlin DM, Miller CC. The Fe65 proteins and Alzheimer's disease. J Neurosci Res 2008; 86:744 - 754
- Hoe HS, Magill LA, Guenette S, Fu Z, Vicini S, Rebeck GW. Fe65 interaction with the ApoE receptor ApoEr2. J Biol Chem 2006; 281:24521 - 24530
- Wiley JC, Smith EA, Hudson MP, Ladiges WC, Bothwell M. Fe65 stimulates proteolytic liberation of the β-amyloid precursor protein intracellular domain. J Biol Chem 2007; 282:33313 - 33325
- Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci 2001; 21:6597 - 6607
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, et al. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem 2003; 278:22946 - 22955
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol 2007; 17:1313 - 1317
- Harris-White ME, Frautschy SA. Low density lipoprotein receptor-related proteins (LRPs), Alzheimer's and cognition. Curr Drug Targets CNS Neurol Disord 2005; 4:469 - 480
- Waldron E, Jaeger S, Pietrzik CU. Functional role of the low-density lipoprotein receptor-related protein in Alzheimer's disease. Neurodegener Dis 2006; 3:233 - 238
- Cam JA, Bu G. Modulation of β-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol Neurodegener 2006; 18:1 - 8
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, et al. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007; 56:66 - 78
- Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem 2004; 279:29639 - 29646
- Dodson SE, Gearing M, Lippa CF, Montine TJ, Levey AI, Lah JJ. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol 2006; 65:866 - 872
- Spoelgen R, Von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, et al. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase beta-site APP-cleaving enzyme. J Neurosci 2006; 26:418 - 428
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA 2005; 102:13461 - 13466
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI. The lipoprotein receptor LR11 regulates amyloid β production and amyloid precursor protein traffic in endosomal compartments. J Neurosci 2006; 26:1596 - 1603
- Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, et al. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem 2007; 282:32956 - 32964
- Sager KL, Wuu J, Leurgans SE, Rees HD, Gearing M, Mufson EJ, et al. Neuronal LR11/sorLA expression is reduced in mild cognitive impairment. Ann Neurol 2007; 62:640 - 647
- Ma QL, Teter B, Ubeda OJ, Morihara T, Dhoot D, Nyby MD, et al. Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer's disease (AD): relevance to AD prevention. J Neurosci 2007; 27:14299 - 14307
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, et al. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med 2005; 352:884 - 894
- Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, Jones PB, et al. Ubiquilin 1 modulates amyloid precursor protein trafficking and Aβ secretion. J Biol Chem 2006; 281:32240 - 32253
- Zheng P, Eastman J, Vande Pol S, Pimplikar SW. PAT1, a microtubule-interacting protein, recognizes the basolateral sorting signal of amyloid precursor protein. Proc Natl Acad Sci USA 1998; 95:14745 - 14750
- Kuan YH, Gruebl T, Soba P, Eggert S, Nesic I, Back S, et al. PAT1a modulates intracellular transport and processing of amyloid precursor protein (APP), APLP1, and APLP2. J Biol Chem 2006; 281:40114 - 40123
- Vetrivel KS, Gong P, Bowen JW, Cheng H, Chen Y, Carter M, et al. Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol Neurodegener 2007; 2:4
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 2005; 6:201 - 214
- Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol 2005; 109:5 - 13
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta 2006; 1762:1094 - 1108
- El-Kadi AM, Soura V, Hafezparast M. Defective axonal transport in motor neuron disease. J Neurosci Res 2007; 85:2557 - 2566
- Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004; 63:724 - 726
- Trushina E, Dyer RB, Badger JD 2nd, Ure D, Eide L, Tran DD, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 2004; 24:8195 - 8209
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1β. Cell 2001; 105:587 - 597
- Meggouh F, Bienfait HM, Weterman MA, De Visser M, Baas F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology 2006; 67:1476 - 1478
- Brownlees J, Ackerley S, Grierson AJ, Jacobsen NJ, Shea K, Anderton BH, et al. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum Mol Genet 2002; 11:2837 - 2844
- Dickson TC, Vickers JC. The morphological phenotype of β-amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience 2001; 105:99 - 107
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci 2004; 15:267 - 278
- Munoz DG, Wang D. Tangle-associated neuritic clusters. A new lesion in Alzheimer's disease and aging suggests that aggregates of dystrophic neurites are not necessarily associated with β/A4. Am J Pathol 1992; 140:1167 - 1178
- Tabaton M, Mandybur TI, Perry G, Onorato M, Autilio-Gambetti L, Gambetti P. The widespread alteration of neurites in Alzheimer's disease may be unrelated to amyloid deposition. Ann Neurol 1989; 26:771 - 778
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 2005; 307:1282 - 1288
- Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, et al. High β-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: implications for the treatment of Alzheimer disease. J Biol Chem 2005; 280:32957 - 32967
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, et al. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci 2007; 27:7011 - 7020
- Wirths O, Weis J, Szczygielski J, Multhaup G, Bayer TA. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer's disease. Acta Neuropathol 2006; 111:312 - 319
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 1999; 155:2153 - 2165
- Leroy K, Bretteville A, Schindowski K, Gilissen E, Authelet M, De Decker R, et al. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol 2007; 171:976 - 992
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A, et al. Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol 2000; 157:1495 - 1510
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol 1999; 9:489 - 492
- Medina D, DeToledo-Morrell L, Urresta F, Gabrieli JD, Moseley M, Fleischman D, et al. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiol Aging 27:663 - 672
- Van Es AC, Van der Flier WM, Admiraal-Behloul F, Olofsen H, Bollen EL, Middelkoop HA, et al. Magnetization transfer imaging of gray and white matter in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging 2006; 27:1757 - 1762
- Kins S, Lauther N, Szodorai A, Beyreuther K. Subcellular trafficking of the amyloid precursor protein gene family and its pathogenic role in Alzheimer's disease. Neurodegener Dis 2006; 3:218 - 226
- Walter J. Control of amyloid-β-peptide generation by subcellular trafficking of the betaamyloid precursor protein and β-secretase. Neurodegener Dis 2006; 3:247 - 254
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released betaamyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 2002; 22:9785 - 9793
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000; 28:449 - 459
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 2001; 414:643 - 648
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci 2005; 25:2386 - 2395
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol 2004; 165:357 - 371
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 2007; 208:185 - 192
- Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK, Povlishock JT, et al. Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol 2004; 190:59 - 69
- Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med 2003; 4:59 - 72
- Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP. Molecular motors implicated in the axonal transport of tau and α-synuclein. J Cell Sci 2005; 118:4645 - 4654
- Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci 2004; 117:5721 - 5729
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 2002; 156:1051 - 1063
- Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 2003; 24:1079 - 1085
- Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, et al. Interaction of tau protein with the dynactin complex. EMBO J 26:4546 - 4554
- Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid β peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 2002; 115:201 - 211
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol Aging 2008; 29:1334 - 1347
- Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein and neurodegeneration in Alzheimer's disease. Neuromolecular Med 2002; 2:151 - 165
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004; 43:321 - 332
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med 2004; 10:452 - 458
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid β in vivo. J Neurosci 2006; 26:10536 - 10541
- Stokin GB, Goldstein LS. Axonal transport and Alzheimer's disease. Annu Rev Biochem 2006; 75:607 - 627
- Leuner K, Hauptmann S, Abdel-Kader R, Scherping I, Keil U, Strosznajder JB, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer's disease?. Antioxid Redox Signal 2007; 9:1659 - 1675
- Coleman M. Axon degeneration mechanisms: Commonality amid diversity. Nat Rev Neurosci 2005; 6:889 - 898
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Aβ oligomerinduced aberrations in synapse composition, shape and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 2007; 27:796 - 807
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S. Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol Cell Neurosci 2007; 35:183 - 193
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, et al. Amyloid beta oligomers (Aβ(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 2008; 28:788 - 797
- Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience 2007; 147:60 - 70
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer's disease amyloid β-peptide-induced dysfunction. Biochem Soc Trans 2007; 35:1219 - 1223
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci USA 2007; 104:353 - 358
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res 2000; 60:65 - 72
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535 - 539
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 2007; 23:613 - 643
- Greger IH, Esteban JA. AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol 2007; 17:289 - 297
- Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, et al. Amyloid beta-peptide Aβ(1–42) but not Aβ(1–40) attenuates synaptic AMPA receptor function. Synapse 2007; 61:367 - 374
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006; 52:831 - 843
- Kamenatz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron 2003; 37:925 - 937
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27:2866 - 2875
- Pérez-Otaño I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci 2005; 28:229 - 238
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci 2005; 8:1051 - 1058
- Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci 2005; 8:977 - 979