Abstract
Alzheimer disease (AD) is a progressive dementia affecting a large proportion of the aging population. The histopathological changes in AD include neuronal cell death, formation of amyloid plaques and neurofibrillary tangles. There is also evidence that brain tissue in patients with AD is exposed to oxidative stress (e.g., protein oxidation, lipid oxidation, DNA oxidation and glycoxidation) during the course of the disease. Advanced glycation endproducts (AGEs) are present in amyloid plaques in AD, and its extracellular accumulation may be caused by an accelerated oxidation of glycated proteins. AGEs participate in neuronal death causing direct (chemical) and indirect (cellular) free radical production and consequently increase oxidative stress. The development of drugs for the treatment of AD that breaks the vicious cycles of oxidative stress and neurodegeneration offer new opportunities. These approaches include AGE-inhibitors, antioxidants and anti-inflammatory substances, which prevent free radical production.
Introduction
Alzheimer disease (AD) is a progressive dementia afecting a large proportion of the aging population. A lot of attention has been focused on the histopathological changes in AD, including widespread neuronal cell death, the formation of amyloid plaques and neurofibrillary tangles (NFTs). The major component of the amyloid plaques is amyloid β-peptide (Aβ). Although Aβ is toxic to neurons in cell culture, Aβ deposits formed by overexpression of the amyloid precursor protein (APP) in transgenic mice does not cause suficient neuronal death, suggesting that additional factors are necessary to promote the progression of the disease. Early signs of tangle formation in certain brain regions such as the entorhinal cortex precede the clinical diagnosis of AD. The major component of NFTs is hyperphosphorylated microtubule-associated protein tau (MAP-tau). The abnormal MAP-tau is resistant to proteolitic enzymes suggesting that glycation, disulphide bond formation, phosphorylation and/or formation of core fragments contribute to extensive cross-linking between MAP-tau monomers.
We will introduce “advance glycation end products” (AGEs) and oxidative stress as the interacting key factors, promoting the transformation of soluble proteins into insoluble proteins deposits, as well as activating the microglia through specific ligands for cell surface receptors.
Oxidative Stress and Alzheimer Disease
There is overwhelming evidence that brain tissue in AD patients is exposed to oxidative stress during the course of the disease (). Since oxidative stress is characterized by an imbalance in radical production of reactive oxygen species (ROS) and antioxidative defense, both are considered to have a major role in the process of age-related neurodegeneration and cognitive decline.Citation1–Citation9
Evidence of oxidative stress in AD is manifested through high levels of oxidised proteins, advanced glycation end products, lipid peroxidation end products, formation of toxic species, such as peroxides, alcohols, aldehydes, free carbonyles, ketones, cholestenone and oxidative modifications in nuclear and mitochondrial DNA.Citation10–Citation21
Age-related memory impairments correlate with a decrease in brain and plasma antioxidants defense mechanism.Citation22,Citation23 An important aspect of the antioxidant defense system is the low molecular weight reducing equivalent glutathione, which is responsible for the endogenous redox potential in the cell.Citation24 The most important function of glutathione is to donate electrons to ROS and by doing so to scavenge them. Intracellular glutathione (GSH) concentration decreases with age in different animal models,Citation25–Citation30 and it also decreases in aged mammalian brain regions including hippocampus.Citation31–Citation33 The decrease in GSH leads to a situation where the rate of ROS production exceeds the antioxidant ability, generating a situation that favors oxidative stress. A further reason for oxidative stress is caused by an imbalance among the radical detoxifying enzymes in AD.Citation34
Protein Oxidation
ROS mediated oxidation of protein side-chains has been reviewed,Citation35 and it results in the introduction of hydroxyl groups or in the generation of protein based carbonyls. Carbonyl groups are introduced in proteins by oxidizing amino acid residue side-chain hydroxyls into ketone or aldehyde derivatives.Citation36 A variety of oxidative pathways lead to carbonylation of proteins.Citation37 Carbonyl groups can also be introduced in proteins by direct oxidation of lysine, arginine, proline and threonine residues, or from the cleavage of peptide bonds by the α-amidation pathway or by the oxidation of glutamyl residues. ROS can also react with other molecules, such as lipids (lipid oxidation), DNA (DNA oxidation) and sugars (glycoxidation), resulting in the generation of reactive carbonyl derivatives and aldehydes, which may in turn react with proteins and form protein-bound carbonyls. Measurement of protein carbonylation is thought to be a good estimation for the extent of oxidative damage of proteins associated with various conditions of oxidative stress, aging, physiological disorders and AD.Citation38–Citation40
Lipid Oxidation in AD
Aβ induces lipoperoxidation of membranes and lipid peroxidation products.Citation41 Lipids are modified by ROS and there is a strong correlation between lipid peroxides, antioxidant enzymes, amyloid plaques and NFTs in AD brains.Citation42 Several breakdown products of oxidative stress, including 4-hydroxy-2,3-nonenal (HNE), acrolein, malondialdehyde and F2-isoprostanes have been observed in AD brains compared to age-matched controls.Citation43–Citation46 HNE is able to modify proteins, resulting in a multitude of effects, including inhibition of neuronal glucose and glutamate transporters, inhibition of Na-K ATPases, activation of kinases and dysregulation of intracellular calcium signalling, that ultimately induce an apoptotic cascade mechanism.Citation47–Citation49 NFTs bear the footprints of oxidative membrane damage since they contain adducts of malondialdehyde and HNE, the most highly reactive lipid peroxidation products. Furthermore, dystrophic neurites of senile plaques that contain NFTs filaments show greater membrane damage than those that lack filaments. Evidence continues to mount that bifunctional HNE are the major cytotoxic products of lipid peroxidation. Following lipid peroxidation, a 2-pentylpyrrole modification of lysine is the only presently known “advanced” (stable end-product) adduct that forms from the modification of proteins by HNE in AD cases. These findings, together with the recent demonstration that HNE is cytotoxic to neurons and that it impairs the function of membrane proteins including the neuronal glucose transporter GLUT 3, indicate that HNE is a characteristic marker and a toxin leading to neurodegeneration in AD.Citation50
DNA Oxidation in AD
DNA bases are vulnerable to oxidative stress damage involving hydroxylation, protein carbonylation and nitration.Citation21,Citation51,Citation52 It has been observed in AD that brain ROS induces calcium influx, via glutamate receptors and triggers an excitotoxic response leading to cell death.Citation48 ROS are generated when oxygen reacts with unregulated redox-active metals.Citation53 DNA and RNA oxidation is marked by increased levels of 8-hydroxy-2-deoxyguanosine (8OHdG) and 8-hydroxyguanosine (8OHD).Citation54–Citation56 Furthermore, these markers have been localized in Aβ plaques and NFTs.Citation57 Increased levels of DNA strand breaks have been found in AD. They were first considered to be part of apoptosis, but it is now widely accepted that oxidative damage is responsible for DNA strand breaks and this is consistent with the increased free carbonyls in the nuclei of neurons and glia in AD. The induction of heme oxygenase-1, an antioxidant enzyme involved in the conversion of heme to bilirubin, is increased in AD brains and is tightly correlated with NFTs.
Glycoxidation in AD
Advanced glycation end products (AGEs), which are formed by a non-enzymatic reaction of sugars with long lived protein deposits, are also potent neurotoxins and proinflammatory molecules (). Glycation of proteins starts as a nonenzymatic process with the spontaneous condensation of ketone or aldehyde groups of sugars with a free aminoacid group of proteins to form a labile Schiff base, consistent with the classical reaction described by Maillard in 1912.Citation13 A cascade of reactions results thereafter in the formation of AGEs, which are composed of irreversibly cross-linked heterogeneous protein aggregates. There is increasing evidence that the insolubility of Aβ plaques is caused by extensive covalent protein cross-linking.Citation58 One mechanism by which long-lived proteins can be cross-linked involves AGEs.Citation59,Citation60 Extracellular AGEs accumulation has been demostrated in senile plaques in different cortical areas in primitive plaques and coronas of classic plaques. Immunohistochemical studies demostrate that AGEs colocalize to a very high degree with ApoE.Citation61 Accumulation of extracellular AGEs in AD is caused by an accelerated oxidation of glycated proteins (“glycoxidation”).Citation62 Intracellular proteins deposits including NFTs, Lewy bodies of patients with Parkinson's disease and Hirano bodies are also cross-linked by AGEs,Citation63 which may explain their insolubility in detergents and resistance to proteases. The major component of the NFTs, the microtubuli-associated protein tau (MAP-tau) has been shown to be subject to intracellular AGEs formation. MAP-tau can be glycated in vitro, inhibiting its ability to bind to microtubules. In addition, MAP-tau isolated from brains of AD patients is glycated in the tubulin-binding region, giving rise to the formation of β-sheet fibrils.Citation64,Citation65 Some studies have shown the presence of AGEs in association with two major proteins of AD, AβCitation66 and MAP-tau.Citation13,Citation67 This observation supports the argument that AGEs are involved in the pathogenesis of AD.Citation68,Citation69 Free radicals are involved in glycation processes and clearly can foster the formation of Aβ cross-linking.Citation70
Glucose Metabolism in Alzheimer Disease
In vivo imaging of AD patients using positron emision tomography with 2-[F-18]-fluoro-2-deoxy-D-glucose demonstrates progressive reduction in brain glucose metabolism and blood flow in severe dementia. Glucose metabolism in the brain limits the synthesis of acetylcholine, glutamate, aspartate, γ-aminobutyric acid, glycine and ATP production. Whereas the cerebral energy pool is only slightly diminished during the normal aging process, glucose metabolism and cellular energy production are severely reduced in AD.Citation71,Citation72 The hypothesis that genetic or environmental factors lead to an intracellular glucose hypometabolism which might predispose for both AD and adult-onset diabetes (NIDDM) is supported by large epidemologic studies. These studies demonstrate that NIDDM significantly increases the risk to develop AD.Citation73–Citation77
The first type of evidence for a link between oxidative stress (e.g., caused by Aβ) and impaired glucose transport has been shown in cultured neurons. Aβ impairs glucose transport, which is followed by a decrease in cellular ATP levels. It has been suggested that this effect is caused by conjugation of HNE, produced by lipid peroxidation, to the neuronal glucose transport protein GLUT3.Citation78,Citation79 Lipid peroxidation caused by other sources of oxidative stress, such as activated microglia or free extracellular iron, may contribute to decreased glucose uptake and neuronal degeneration. This is consistent with histopathological findings in AD, where decreased membrane fluidity in mitochondria and increase levels of oxidized 8OHdG in mitochondrial DNA can be observed, and suggest a link between oxidative stress and glucose utilization.Citation19
Oxidative stress and energy depletion simulated by addition of chemical uncoupling agents to neuroblastoma cells leads to the appearance of NFTs; feeding a thiamine-deficient diet to rodents leads to the formation of dystrophic neurites similar to those in AD. The oxidatively-compromised animals develop AD-type neuritic dystrophy suggesting that disturbed energy metabolism and subsequent oxidative stress may be a common denominator of neuritic dystrophy.Citation80
NFTs, which are largely composed of MAP-tau protein, and senile plaques, which contain aggregates of the Aβ, are realted to disturbances in the balance between protein phosphorylation and dephosphorylation. Various studies have shown that injection of the phosphatase inhibitor okadaic acid in rat brain, results in severe memory impairment, as well as the presence of MAP-tau protein in paired helical filaments and formation of plaques containing Aβ.Citation81–Citation83
Positive Feedback Loops in the Pathogenesis of Alzheimer Disease
One of the characteristics of degenerative processes is the creation of positive feedback loops or vicious cycles. To define a vicious circle of neurodegeneration in AD, characteristic factors have to be defined which promotes the generation of ROS, amplified production of AGEs and inflammation. The “error catastrophe theory” proposes that damages to cell constituents accumulate during aging. At the same time, cellular defence mechanisms weaken, thus the accumulated damages cannot be repaired efficiently, leading to loss of function and finally cell death.
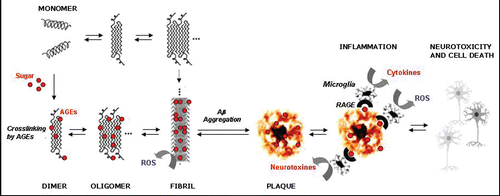
We propose that AGEs are one of these factors, which participate dynamicly in neuronal death and exert multiple detrimental effects on cells.Citation84,Citation85 Elucidation of the effects of AGEs on cells, particularly their interaction with cell surface receptors (RAGE) and downstream signal transduction, might help to solve certain aspects of the etiopathogenesis of AD. For example, glycation and AGEs cause ():
Direct radical production (chemical): Glycated proteins produce nearly 50 fold more radicals than non-glycated proteins. This process commences with the production of superoxide radicals by the transition metal-catalyzed oxidation of protein-bound Amadori products, followed by the dismutation of superoxide to hydrogen peroxide, and the generation of hydroxyl radicals by the Fenton reaction. | |||||
Indirect radical production (cellular): Interaction of AGEs with cells increases oxidative stress. It is not clear whether this is initiated by binding of AGEs to the cell surface and subsequent diffusion of chemically produced free radicals across the membrane or by receptor mediated signalling pathway, in which a radical producing enzyme (e.g., NADPH-oxidase) and AGEs specific receptor (RAGE), is involved. Yan et al.Citation86 showed that RAGE is also a receptor for Aβ. This discovery supports the idea of a relation between AGEs and AD as well as the production of free radicals. The combination of AGEs and RAGE can cause oxidative stress, as shown in the production of thiobarbituric acid-reactive substances, secretion of cytokines, TNFα, heme oxygenase-1, and the activation of nuclear transcription factor κB (NFκB). Aβ binding with RAGE also elicits the macrophage colony stimulation factor which introduces an inflammatory pathway, according to a procedure linking oxidative processes and inflammation in AD.Citation87 | |||||
Pharmacological Interference with Age Formation or Signalling as a Novel Treatment Strategy
The development of drugs for the treatment of AD remains at a very unsatisfying state. However, pharmacological approaches which break the vicious cycle of oxidative stress and neurodegeneration offer new opportunities for the treatment of AD. These approaches include AGE-inhibitors (aminoguanidine, pyridoxamine), antioxidants (thioctic acid, vitamin E, vitamin C, β-carotin) and nonsteroidal antiinflammatory substances, which do not only scavenge radicals passively but interfere with signal transduction pathways, thereby preventing radical production.
AGE inhibitors might be able to stop formation of AGE-modified Aβ deposits or modify their structure with subsequent loss of AGEs binding to RAGE.Citation88,Citation89 Antioxidants are likely to scavenge intracellular and extracellular superoxide radicals and hydrogen peroxide before these radicals damage cell constituents or activate microglia through their action as intracellular second messengers.Citation90–Citation94 Antiinflammatory drugs act similarly, attenuating microglial radical and cytokine production.Citation95–Citation97
With our growing understanding of the molecular basis of the clinical symptoms of dementia, particularly positive feedback loops involving oxidative stress, it is hoped that elucidation of the etiopathogenesis of AD will help to develop novel “neuroprotective” treatment strategies able to interrupt the vicious cycle of oxidative stress and neurodegeneration.
Abbreviations
| AGEs | = | advance glycation end products |
| AD | = | Alzheimer disease |
| Aβ | = | amyloid β peptide |
| APP | = | amyloid precursor protein |
| GSH | = | glutathione |
| HNE | = | 4-hydroxy-2,3-nonenal |
| 8OHdG | = | 8-hydroxy-2-deoxyguanosine |
| 8OHD | = | 8-hydroxyguanosine |
| MAP-tau | = | hyperphosphorylated microtubule-associated protein tau |
| NFTs | = | neurofibrillary tangles |
| NFκB | = | nuclear transcription factor κB |
| ROS | = | reactive oxygen species |
Figures and Tables
Figure 1 Sources and effects of oxidative stress on a molecular and cellular level.
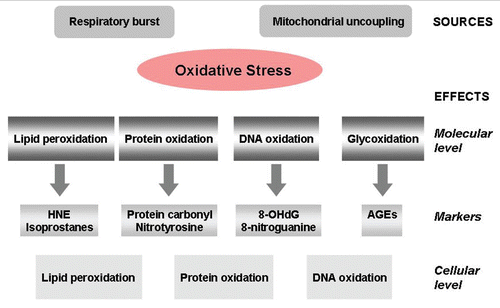
Figure 2 Chemical reactions leading to the formation of advanced glycation endproducts.
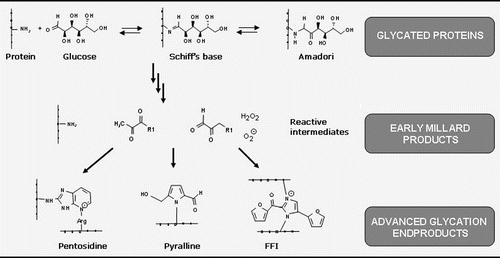
Figure 3 Direct and indirect effects of advanced glycation endproducts through crosslinking of Aβ peptide.
References
- Gsell W, Strein I, Riederer P. The neurochemistry of Alzheimer type, vascular type and mixed type dementias compared. J Neural Transm 1996; 47:73 - 101
- Gsell W, Strein I, Krause U, Riederer P. Neurochemical abnormalities in Alzheimer's disease and Parkinson's disease—a comparative review. J Neural Transm 1997; 51:145 - 159
- Retz W, Gsell W, Münch G, Rosler M, Riederer P. Free radicals in Alzheimer's disease. J Neural Transm 1998; 54:221 - 236
- Rösler M, Retz W, Thome J, Riederer P. Free radicals in Alzheimer's dementia: currently available therapeutic strategies. J Neural Transm 1998; 54:211 - 219
- Durany N, Münch G, Michel T, Riederer P. Investigations on oxidative stress and therapeutical implications in dementia. Eur Arch Psychiatry Clin Neurosci 1999; 249:68 - 73
- Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr 2000; 71:621 - 629
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004; 3:205 - 214
- Sayre LM, Moreira PI, Smith MA, Perry G. Metal ions and oxidative protein modification in neurological disease. Ann Ist Super Sanita 2005; 41:143 - 164
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol 2008; 21:172 - 188
- Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci USA 1996; 93:4765 - 4769
- Schippling S, Kontush A, Arlt S, Buhmann C, Stürenburg HJ, Mann U, et al. Increased lipoprotein oxidation in Alzheimer's disease. Free Radic Biol Med 2000; 28:351 - 360
- Mohsen MMAE, Iravani MM, Spencer JPE, Rose S, Fahim AT, Motawi TMK, et al. Age-associated changes in protein oxidation and proteasome activities in rat brain: Modulation by anti-oxidants. Biochemical Biophysical Res Commun 2005; 336:386 - 391
- Smith MA, Kutty RK, Rickey PL, Yan SD, Stern D, Chader GJ, et al. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol 1994; 145:42 - 47
- Gupta A, Hasan M, Chander R, Kapoor K. Age-related elevation of lipid peroxidation products: diminution of superoxide dismutase activity in the central nervous system of rats. Gerontology 1991; 37:305 - 309
- Cini M, Moretti A. Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol Aging 1995; 16:53 - 57
- Ramassamy C, Krzywkowski P, Averill D, Lussier-Cacan S, Theroux L, Christen Y, et al. Impact of ApoE deficiency on oxidative insults and antioxidant levels in the brain. Brain Res Mol Brain Res 2001; 86:76 - 83
- Picklo MJ, Montine TJ, Amarnath V, Neely MD. Carbonyl toxicology and Alzheimer's disease. Toxicol Appl Pharmacol 2002; 184:187 - 197
- Bassett CN, Montine TJ. Lipoproteins and lipid peroxidation in Alzheimer's disease. J Nutr Health Aging 2003; 7:24 - 29
- Mecocci P, Beal MF, Cecchetti R, Polidori MC, Cherubini A, Chionne F, et al. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and human brain. Mol Chemi Neuropathol 1997; 31:53 - 64
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, et al. Does oxidative damage to DNA increase with age?. Proc. Natl Acad. Sci USA 2001; 98:10469 - 10474
- Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res 2007; 35:7497 - 7504
- Perrig WJ, Perrig P, Stähelin HB. The relation between antioxidants and memory performance in the old and very old. J Am Geriatr Soc 1997; 45:718 - 724
- Berr C. Cognitive impairment and oxidative stress in the elderly: results of epidemiological studies. Biofactors 2000; 13:205 - 209
- Thornalley PJ. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system—involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interactions 1998; 112:137 - 151
- Chen TS, Richie JP, Lang CA. The effect of aging on glutathione and cysteine levels in different regions of the mouse brain. Proc Soc Exp Biol Med 1989; 190:399 - 402
- Iantomasi T, Favilli F, Marraccini P, Stio M, Treves C, Quatrone A, et al. Age and GSH metabolism in rat cerebral cortex, as related to oxidative and energy parameters. Mech Ageing Dev 1993; 70:65 - 82
- Sasaki T, Senda M, Kim S-N, Kojima S, Kubodera A. Age-related changes of glutathione content, glucose transport and metabolism, and mitochondrial electron transfer function in mouse brain. Nucl Med Biol 2001; 28:25 - 31
- Liu RM. Downregulation of g-glutamil cysteine synthetase regulatory subunit gene expression in rat brain tissue during aging. J Neurosci Res 2002; 68:344 - 351
- Sandhu SK, Kaur G. Alterations in oxidative stress scavenger system in aging rat brain and lymphocytes. Biogerontology 2002; 3:161 - 173
- Wang H, Liu H, Liu R. Gender difference in glutathione metabolism during aging in mice. Exp Gerontol 2003; 38:507 - 517
- Calabrese V, Scapagnini G, Ravagna A, Colombrita C, Spadaro F, Butterfield DA, et al. Increased expression of heat shock proteins in rat brain during aging: relationship with mitochondrial function and glutathione redox state. Mech Ageing Dev 2004; 125:325 - 335
- Donahue AN, Aschner M, Lash LH, Syversen T, Sonntag WE. Growth hormone administration to aged animals reduces disulfide glutathione levels in hippocampus. Mech Ageing Dev 2006; 127:57 - 63
- Zhu Y, Carvey PM, Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res 2006; 1090:35 - 44
- Gsell W, Conrad R, Hickethier M, Sofic E, Frölich L, Wichart I, et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of Alzheimer type. J Neurochem 1995; 64:1216 - 1223
- Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta 2005; 1703:93 - 109
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease and oxidative stress. J Biol Chem 1997; 272:20313 - 20316
- Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction and disease progression. J Cell Mol Med 2006; 10:389 - 406
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol 1994; 233:346 - 357
- Smith MA, Sayre LM, Anderson VE, Harris PL, Beal MF, Kowall N, et al. Cytochemical demonstration of oxidative damage in Alzheimer's disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem 1998; 46:731 - 735
- Korolainen MA, Nyman TA, Nyyssönen P, Hartikainen ES, Pirttilä T. Multiplexed proteomic analysis of oxidation and concentrations of cerebrospinal fluid proteins in Alzheimer's disease. Clin Chem 2007; 53:657 - 665
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4 Hydroxynonenalderived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem 1997; 68:2092 - 2097
- Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology 1995; 45:1594 - 1601
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid b-peptide. J Neurochem 1997; 68:255 - 264
- Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging 1998; 19:33 - 36
- Arlt S, Beisiegel U, Kontush A. Lipid peroxidation in neurodegeneration: new insights into Alzheimer's disease. Curr Opin Lipidol 2002; 13:289 - 294
- Selley ML, Close DR, Stern SE. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer's disease. Neurobiol Aging 2002; 23:383 - 388
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, et al. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide—role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 1997; 69:273 - 284
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium 2003; 34:385 - 397
- Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, et al. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating Jnos and p38MAPK. Exp Neurol 2003; 180:144 - 155
- Brucekeller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, et al. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J Neuropath Exp Neur 1998; 57:257 - 267
- Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem 1998; 71:2034 - 2040
- Collins AR, Dusinská M, Gedik CM, Stetina R. Oxidative damage to DNA: do we have a reliable biomarker?. Environ Health Perspect 1996; 104:465 - 469
- White AR, Barnham KJ, Bush AI. Metal homeostasis in Alzheimer's disease. Expert Rev Neurother 2006; 6:711 - 722
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neuroscience 1999; 19:1959 - 1964
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer's disease. J Neuropathol Exp Neurol 2001; 60:759 - 767
- Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreassed levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem 1999; 72:771 - 776
- Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol 1994; 36:747 - 751
- Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, et al. Oxidative damage in Alzheimer's. Nature 1996; 382:120 - 121
- Ortwerth BJ, Olesen PR. Ascorbic acid-induced crosslinking of lens proteins: evidence supporting a Maillard reaction. Biochim Biophys Acta 1988; 956:10 - 22
- Prabhakaram M, Ortwerth BJ. Determination of glycation crosslinking by the sugar-dependent incorporation of [14C]lysine into protein. Anal Biochem 1994; 216:305 - 312
- Li YM, Dickson DW. Enhanced binding of advanced glycation endproducts (age) by the apoe4 isoform links the mechanism of plaque deposition in Alzheimer's disease. Neurosci Lett 1997; 226:155 - 158
- Münch G, Cunningham AM, Riederer P, Braak E. Advanced glycation endproducts are associated with Hirano bodies in Alzheimer's disease. Brain Res 1998; 796:307 - 310
- Loske C, Gerdemann A, Schepl W, Wycislo M, Schinzel R, Palm D, et al. Transition metal-mediated glycoxidation accelerates cross-linking of beta-amyloid peptide. Eur J Biochem 2000; 267:4171 - 4178
- González C, Farías G, Maccioni RB. Modification of tau to an Alzheimer's type protein interferes with its interaction with microtubules. Cell Mol Biol 1998; 44:1117 - 1127
- Ledesma MD, Pérez M, Colaco C, Avila J. Tau glycation is involved in aggregation of the protein but not in the formation of filaments. Cell Mol Biol 1998; 44:1111 - 1116
- Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, et al. Advanced glycation end products contribute to amyloidosis in Alzheimer's disease. Proc Natl Acad Sci USA 1994; 91:4766 - 4770
- Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, et al. Glycated tau protein in Alzheimer's disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA 1994; 91:7787 - 7791
- Colaco CA, Harrington CR. Glycation: a pathological modification in neuropathies?: a hypothesis. Neuroreport 1994; 5:859 - 861
- Smith MA, Sayre LM, Monnier VM, Perry G. Radical AGEing in Alzheimer's disease. Trends Neurosci 1995; 18:172 - 176
- Mattson MP, Carney JW, Butterfield DA. A tombstone in Alzheimer's?. Nature 1995; 373:481
- Blum-Degen D, Frölich L, Hoyer S, Riederer P. Altered regulation of brain glucose metabolism as a cause of neurodegenerative disorders?. J Neural Transm 1995; 46:139 - 147
- Hoyer S. Is sporadic Alzheimer disease the brain type of non-insulin dependent diabetes mellitus? A challenging hypothesis. J Neural Transm 1998; 105:415 - 422
- Breteler MM, Bots ML, Ott A, Hofman A. Risk factors for vascular disease and dementia. Haemostasis 1998; 28:167 - 173
- Curb JD, Rodriguez BL, Abbott RD, Petrovitch H, Ross GW, Masaki KH, et al. Longitudinal association of vascular and Alzheimer's dementias, diabetes and glucose tolerance. Neurology 1999; 52:971 - 975
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, et al. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann N Y Acad Sci 1997; 826:422 - 427
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53:1937 - 1942
- Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med 1999; 16:93 - 112
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, et al. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 1997; 69:273 - 284
- Luo Y, Bond JD, Ingram VM. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci USA 1997; 94:9705 - 9710
- Yen SH, Liu WK, Hall FL, Yan SD, Stern D, Dickson DW. Alzheimer neurofibrillary lesions: molecular nature and potential roles of different components. Neurobiol Aging 1995; 16:381 - 387
- Arendt T, Hanisch F, Holzer M, Brückner MK. In vivo phosphorylation in the rat basal nucleus induces PHF-like and APP immunoreactivity. Neuroreport 1994; 5:1397 - 1400
- Arendt T, Holzer M, Fruth R, Brückner MK, Gärtner U. Paired helical filament-like phosphorylation of tau, deposition of beta/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience 1995; 69:691 - 698
- Janke C, Gärtner U, Holzer M, Arendt T. Reversible in vivo phosphorylation of tau induced by okadaic acid and by unspecific brain lesion in rat. J Hirnforsch 1998; 39:143 - 153
- Münch G, Gerlach M, Sian J, Wong A, Riederer P. Advanced glycation end products in neurodegeneration: more than early markers of oxidative stress?. Ann Neurol 1998; 44:85 - 88
- Thome J, Kornhuber J, Münch G, Schinzel R, Taneli Y, Zielke B, et al. New hypothesis on etiopathogenesis of Alzheimer syndrome. Advanced glycation end products (AGEs) Nervenarzt 1996; 67:924 - 929
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 1996; 382:685 - 691
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 2001; 108:949 - 955
- Münch G, Taneli Y, Schraven E, Schindler U, Schinzel R, Palm D, et al. The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect 1994; 8:193 - 1208
- Shoda H, Miyata S, Liu BF, Yamada H, Ohara T, Suzuki K, et al. Inhibitory effects of tenilseton the Maillard reaction. Endocrinology 1997; 138:1886 - 1892
- Staehelin HB. Micronutrients and Alzheimer's disease. Proc Nutr Soc 2005; 64:565 - 570
- Ueda Y, Miyata T, Hashimoto T, Yamada H, Izuhara Y, Sakai H, et al. Implication of altered redox regulation by antioxidant enzymes in the increased plasma pentosidine, an advanced glycation end product, in uremia. Biochem Biophys Res Commun 1998; 245:785 - 790
- Wells-Knecht KJ, Zyzak DV, Litchfield JE, Thorpe SR, Baynes JW. Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry 1995; 34:3702 - 3709
- Wells-Knecht KJ, Brinkmann E, Wells-Knecht MC, Litchfield JE, Ahmed MU, Reddy S, et al. New biomarkers of Maillard reaction damage to proteins. Nephrol Dial Transplant 1996; 11:41 - 47
- Grundman M. Vitamin E and Alzheimer's disease: the basis for additional clinical trials. Am J Clin Nutr 2000; 71:630 - 636
- Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology 1995; 45:51 - 55
- Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, et al. Delayed onset of Alzheimer's disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging 1995; 16:523 - 530
- Fiebich BL, Lieb K, Kammerer N, Hüll M. Synergistic inhibitory effect of ascorbic acid and acetylsalicylic acid on prostaglandin E2 release in primary rat microglia. J Neurochem 2003; 86:173 - 178