Abstract
Neuropilin-2 (NRP2) is a receptor expressed by tumor cells and endothelial cells (EC) that binds both semaphorin 3F (SEMA3F), a potent inhibitor of tumor angiogenesis and metastasis, and vascular endothelial growth factor (VEGF), a potent stimulator of tumor angiogenesis. It was found that glioblastoma and melanoma cells repressed NRP2 expression when maintained under hypoxic conditions and after treatment with the hypoxia-mimetic agent desferrioxamine (DFO), at both the mRNA and protein levels. Silencing of HIF1-α, the hypoxia-induced subunit of the hypoxia inducible factor (HIF), abrogated DFO-induced NRP2 repression. Conversely, ectopic expression of HIF1-α directly repressed NRP2 promoter activity and expression. NRP2 is the sole receptor for SEMA3F. Loss of NRP2 expression in tumor cells inhibited SEMA3F-dependent activities, such as inactivation of RhoA, depolymerization of F-actin, and inhibition of tumor cell migration. On the other hand, loss of NRP2 expression in tumor cells increased VEGF protein levels in conditioned media, with no effects on VEGF mRNA levels. This increase in VEGF protein levels promoted paracrine activation of EC, including VEGF receptor-2 phosphorylation, and activation of downstream signaling proteins such as p44/42 MAPK and p38 MAPK. In addition, the elevated VEGF levels induced EC migration and sprouting, two key steps of tumor angiogenesis in vivo. It was concluded that hypoxia regulates VEGF and SEMA3F activities through transcriptional repression of their common receptor NRP2, providing a novel mechanism by which hypoxia induces tumor angiogenesis, growth and metastasis.
Introduction
Most solid tumors become hypoxic as they grow. In order to survive in the hypoxic environment, tumor cells have developed a coordinated set of responses to increase their blood supply.Citation1–Citation3 For example, in hypoxic conditions, tumor cell expression of vascular endothelial growth factor (VEGF), a potent stimulator of angiogenesis, is induced in order to attract new blood vessels.Citation4,Citation5 A critical mediator of the hypoxic response is the hypoxia-inducible factor (HIF), a heterodimeric basic helix-loop-helix (bHLH) transcription factor composed of a HIF-α subunit (HIF1-α or HIF2-α/EPAS) and a HIF-β subunit.Citation1,Citation2,Citation6 Whereas the β-subunit is constitutively expressed, the stability and transcriptional activity of the a-subunits are precisely controlled by the intracellular oxygen concentration.Citation7 Under normoxic conditions, cells continuously synthesize, ubiquitinate and degrade the α-subunits. However, under hypoxic conditions, the degradation of the α-subunits is inhibited, resulting in accumulation of the α-subunits, dimerization with HIF1-β, binding to hypoxia response elements (HREs) within target genes and activation of transcription. For example, under hypoxic conditions, HIF-1 has been shown to bind to and activate transcription of the gene encoding VEGF.Citation4
Neuropilin-1 and -2 (NRP1 and NRP2), first described as receptors for class-3 semaphorins (SEMA3A-G) that regulate axon guidance during nervous system development, are also regulators of angiogenesis.Citation8–Citation11 In endothelial cells (EC), binding of SEMA3A to NRP1 or SEMA3F to NRP2 induced EC repulsion in vitro and inhibited tumor angiogenesis in vivo.Citation12,Citation13 NRPs also have been shown to be co-receptors of VEGF receptors (VEGFR1-3) for VEGF.Citation14–Citation16 NRP expression in EC increased VEGF binding to VEGFRs, VEGFR phosphorylation and VEGF-dependent VEGFR angiogenic activities such as EC survival and chemotaxis.Citation14,Citation17 Thus, inhibition or stimulation of tumor angiogenesis may be favored, depending on whether SEMA3F or VEGF is the ligand.
NRPs are also expressed by tumor cells and contribute to tumor progression and metastasis.Citation9–Citation11,Citation18,Citation19 Overexpression of NRP1 in prostate carcinoma, colon carcinoma and glioma cancer models induced tumor angiogenesis and promoted tumor progression.Citation20–Citation22 Similarly, NRP2 promoted tumor growth and metastasis in pancreatic adenocarcinoma and colorectal cancer models.Citation23,Citation24 In cancer patients, expression of NRP1, NRP2 or both NRPs is often upregulated and is correlated with tumor aggressiveness, advanced disease stage and poor prognosis.Citation25–Citation30 Tumor cells rarely express VEGFR-2, the major VEGFR; therefore, NRPs often represent the only VEGF receptors on tumor cells.Citation14,Citation20 Thus, in tumor cells, VEGF/NRPs interactions might transduce a signal independent of VEGFR-2. For example, exogenous expression of NRPs in tumor cells was correlated with tumor cell survival and migration in a VEGF-dependent manner.Citation20,Citation31
Tumor cells expressing NRPs bind and respond to SEMA3s. SEMA3s have been shown to inhibit tumor growth and metastasis.Citation11 A connection between SEMA3B and SEMA3F and tumor formation became apparent when it was shown that their loci mapped to a region on human chromosome 3p21.3 that is commonly deleted in lung tumors.Citation32,Citation33 Binding of SEMA3F to NRP2 inactivated RhoA, leading to F-actin cytoskeleton depolymerization and, as a result, inhibition of tumor cell migration and invasion in vitro.Citation34 A suppressive role in metastasis was based on the observations that SEMA3F expression was strongly downregulated in highly metastatic tumor cells and that SEMA3F overexpression inhibited metastasis in vivo.Citation13
Whereas much is known about how VEGF expression is regulated under hypoxic conditions, very little is known about the regulation of NRPs during hypoxia. In this report we demonstrate that tumor cells transcriptionally repress NRP2 under hypoxic conditions and after treatment with the hypoxia-mimetic agents desferrioxamine (DFO) and cobalt, in a HIF1-α-dependent manner. NRP2 repression in tumor cells affected the biological activities of its two ligands, SEMA3F and VEGF. Tumor cells no longer responded to the inhibitory activities of SEMA3F, such as inactivation of RhoA activity and inhibition of tumor cell migration. On the other hand, NRP2 repression in tumor cells increased VEGF protein levels in conditioned media, resulting in increased paracrine activation of EC. These results suggest that hypoxia-induced transcriptional repression of NRP2 in tumor cells regulates both VEGF and SEMA3F activities, providing a novel mechanism by which hypoxia induces tumor angiogenesis, growth and metastasis.
Results
Hypoxia represses NRP2 expression in tumor cells in a HIF1-α-dependent manner.
Human U87MG glioblastoma and A375SM melanoma cells expressed abundant NRP2 (). These two cell lines have been used extensively by us to study SEMA3F/NRP2 interactions, signaling and biological activity. For example, we demonstrated that U87MG cells responded strongly to SEMA3F in a number of bioassays, such as inactivation of RhoA, depolymerization of F-actin, loss of stress fibers and inhibition of tumor cell migration.Citation34 In addition, overexpression of SEMA3F in A375SM cells inhibited tumor cell migration and invasion in vitro, and inhibited tumor angiogenesis, progression and metastasis in vivo.Citation13,Citation35
To study the effect of hypoxia on NRP2 expression, U87MG and A375SM cells were maintained in either normoxic (21% O2) or hypoxic (1% O2) conditions or treated with DFO, an iron chelator that inhibits the prolyl hydroxylation of HIF-α subunits and has a well-characterized hypoxia mimetic effect.Citation36 Furthermore, it has been shown that DFO was sufficient to promote angiogenesis,Citation37 providing an excellent method to study hypoxia in vitro. Compared with normoxia, hypoxia and DFO treatment suppressed NRP2 expression in both cell lines, concomitant with HIF1-α induction (). Similar results were obtained when using human U251 and SF210 glioblastoma and MMAN and WM-266-4 melanoma cells (Sup. Fig. 1). Cobalt mimics hypoxia and causes accumulation of HIF-α.Citation38 Proteasome inhibitor MG132 treatment mimics hypoxia by preventing HIF-α degradation.Citation39 Both cobalt and MG132 treatment decreased NRP2 protein levels as well (). Thus, under 4 different hypoxic conditions—hypoxia, DFO, cobalt and MG132—and in 6 different human tumor cell lines, NRP2 expression was repressed and accumulation of HIF1-α was induced ().
To determine whether HIF1-α and/or HIF2-α were necessary for hypoxia-induced NRP2 repression in tumor cells, their expression was knocked down using siRNAs. HIF1-α and HIF2-α expression in U87MG cells treated with DFO was downregulated efficiently 72 h after siRNA transfection (). DFO-induced NRP2 repression was inhibited by HIF1-α siRNA in U87MG cells (, lane 2 vs. 4), but not by HIF2-α siRNA (, lane 2 vs. 6), confirming that hypoxia-induced NRP2 repression was HIF1-α-dependent. On the other hand, ectopic expression of HIF1-α in U87MG cells resulted in downregulation of NRP2 protein levels (). Together, these results indicate that hypoxia represses tumor cell expression of NRP2 and that HIF1-α is necessary and sufficient for this hypoxia-induced NRP2 repression.
Besides NRP2, NRP1 expression was also repressed by hypoxia and DFO treatment in U87MG cells (Sup. Fig. 2A and B) in a HIF1-α-dependent manner (Sup. Fig. 2C and D). A375SM cells did not express NRP1 under normoxic conditions (data not shown).
Hypoxia represses NRP2 at the transcriptional level.
Both hypoxia and DFO treatment decreased NRP2 mRNA levels by 2-fold in U87MG cells (, top left). NRP2 mRNA levels were decreased by both hypoxia (5-fold) and DFO treatment (2-fold) in A375SM cells (, top right). On the other hand, VEGF mRNA expression was increased in both tumor cell lines when subjected to hypoxia or treated with DFO (, bottom). This increase in VEGF expression served as a positive control for the hypoxia response.
To test whether HIF1-α repressed NRP2 expression at the promoter level, we isolated a fragment extending 1,831 bp in the 5′-flanking region of the human NRP2 gene from a human genomic PAC library. Luciferase reporter assays using a NRP2 promoter reporter construct showed that ectopic expression of HIF1-α decreased NRP2 promoter activity directly in a dose-dependent manner (, top). HIF1-α is a well-known transcriptional factor that activates the transcription of target genes by binding to HRE. Thus, when an HRE-driven reporter construct was used, HIF1-α increased HRE promoter activity in a dose-dependent manner (, bottom), providing a positive control for HIF1-α overexpression. Taken together, these results show that hypoxia represses tumor cell expression of NRP2 at the transcriptional level.
Repression of NRP2 in tumor cells by hypoxia inhibits SEMA3F biological activity.
We previously reported that SEMA3F-induced depolymerization of F-actin, loss of stress fibers, inactivation of RhoA, and inhibition of tumor cell adhesion and migration are all dependent on NRP2 expression.Citation34 In those studies, NRP2 expression was repressed by either a NRP2 siRNA or an anti-NRP2 antibody. We hypothesized that hypoxia may also inhibit SEMA3F biological activity through transcriptional repression of NRP2.
Confocal microscopy showed that U87MG cells displayed abundant F-actin stress fibers, indicative of an intact F-actin cytoskeleton (). SEMA3F induced loss of F-actin stress fibers, reduced spreading and decreased cytoplasm (). However, after pretreatment with DFO, SEMA3F-induced F-actin depolymerization was inhibited ( vs. c). DFO alone had no effect on U87MG cell morphology ().
SEMA3F inactivated RhoA, a member of the Rho family of GTPases that stabilizes the F-actin cytoskeleton, within 15 min (, top). Pretreatment with DFO inhibited SEMA3F-induced inactivation of RhoA (, bottom). SEMA3F inhibited U87MG cell migration (). Pretreatment with DFO abrogated the ability of SEMA3F to inhibit cell migration (, left). Importantly, these cell migration results using DFO were very similar to those obtained by silencing NRP2 (, right). These results indicate that hypoxia induces loss of NRP2 expression in tumor cells and, as a functional consequence, inhibits NRP2-dependent SEMA3F biological activity.
Repression of NRP2 in tumor cells increases VEGF protein levels in conditioned media.
In addition to SEMA3F, VEGF also binds to NRP2. It is well established that tumor cells express VEGF and that hypoxia induces expression of VEGF in tumor cells.Citation4,Citation5 Therefore, VEGF protein levels in conditioned media (CM) were increased (, bottom), concomitant with NRP2 repression (, top), when hypoxic conditions were induced by DFO.
Tumor cells rarely express VEGFR-2; thus, they bind VEGF solely via NRPs. We hypothesized that hypoxia may inhibit VEGF/NRP2 interactions through transcriptional repression of NRP2 and consequently may increase VEGF protein levels in CM. NRP2 levels were knocked down using two single NRP2 siRNAs and a pool of four individual NRP2 siRNAs (). NRP2 protein levels in U87MG cells were diminished efficiently 48 h after siRNA transfection (, top). On the other hand, silencing of NRP2 increased VEGF protein levels in CM by 2–2.5 fold compared with U87MG cells transfected with control siRNA (, middle). VEGF mRNA levels were not affected by silencing NRP2 expression in tumor cells (, bottom). An increase in VEGF protein levels in CM was also observed when NRP2 was silenced in another human glioblastoma cell line, SF210 (Sup. Fig. 3). In another approach, VEGF binding to NRP2 was inhibited using an anti-NRP2 antibody. The anti-NRP2 antibody increased VEGF protein levels in CM as well (). It was concluded that silencing of NRP2 expression in tumor cells inhibited VEGF/NRP2 interactions and, as a consequence, there were increased levels of VEGF, not bound to tumor cells, that were released into CM.
Repression of NRP2 in tumor cells increases paracrine VEGF-induced EC function.
It was explored whether loss of NRP2 expression in tumor cells would affect VEGF/VEGFR-2 signaling in EC. VEGFR-2 phosphorylation increased in HUVEC stimulated with VEGF (5 ng/ml) (, lane 1 vs. 2). When HUVEC were stimulated with CM from U87MG cells treated with DFO, phosphorylation of VEGFR-2 was increased compared with HUVEC stimulated with CM from U87MG cells left untreated (, lane 3 vs. 4). These results were consistent with hypoxia inducing tumor cell expression and secretion of VEGF. Activation of p44/42 MAPK and p38 MAPK, two downstream proteins in VEGF/VEGFR-2 signaling, was observed in HUVEC stimulated with VEGF (5 ng/ml) (, lane 1 vs. 2). Interestingly, when HUVEC were stimulated with CM from U87MG cells transfected with a pool of 4 NRP2 siRNAs, phosphorylation of VEGFR-2, as well as phosphorylation of p44/42 MAPK and p38 MAPK, were increased compared with HUVEC stimulated with CM from U87MG cells transfected with a control siRNA (, lane 3 vs. 4).
CM from U87MG cells treated with DFO increased EC migration compared with CM from U87MG cells left untreated (, left). Importantly, silencing of NRP2 in U87MG cells increased EC migration by 2.5-fold (, right). The VEGFR-2 kinase inhibitor SU5416 totally inhibited the migration of these cells (, right), confirming that these effects are mediated by VEGF/VEGFR-2 signaling.
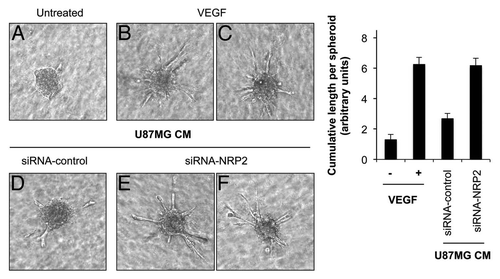
Sprouting of EC is an important property of angiogenesis. Therefore, we examined EC sprouting in a 3-dimensional in vitro angiogenesis spheroid assay. HUVEC spheroids, with defined size and cell number, were embedded in collagen gels and then treated with either VEGF or CM from U87MG cells transfected with control or a pool of four NRP2 siRNAs. Outgrowth of capillary-like structures was assessed. VEGF-treated EC spheroids elicited numerous spontaneous sprouts ( vs. B and C). CM from U87MG cells transfected with NRP2 siRNA increased EC sprouting by 3-fold compared with CM from U87MG cells transfected with control siRNA ( vs. E and F). Together, these results show that hypoxia-induced repression of NRP2 in tumor cells promotes VEGF-induced EC migration and sprouting, two key steps of angiogenesis in vivo.
Discussion
Tumor angiogenesis is mediated by a balance of angiogenesis activators, such as VEGF, and angiogenesis inhibitors, such as SEMA3F. VEGF and SEMA3F belong to two disparate families, yet they bind the same receptor, NRP2.Citation15 Here we provide evidence that hypoxia, one of the key mediators of tumor angiogenesis, causes transcriptional repression of NRP2 in tumor cells with two consequent effects that promote tumor angiogenesis and metastasis in vivo: (1) an increase in VEGF protein levels in CM that enhances VEGF-induced angiogenesis; and (2) an inhibition of the anti-tumorigenic activity of SEMA3F.
Glioblastoma and melanoma cells subjected to hypoxia or treated with the hypoxia-mimetic agent DFO repressed NRP2 expression, at both the mRNA and protein levels. Cobalt and MG132, which also mimic hypoxia by accumulating HIF-α, also repressed NRP2 expression in tumor cells. Exogenous HIF1-α, the hypoxia-induced subunit of the hypoxia-inducible transcription factor HIF1, inhibited NRP2 expression as well. Furthermore, DFO-induced NRP2 repression in tumor cells was inhibited by HIF1-α siRNA, confirming that HIF1-α was necessary and sufficient to repress NRP2 expression under hypoxic conditions. Importantly, ectopic expression of HIF1-α directly repressed NRP2 promoter activity, demonstrating that hypoxia repressed NRP2 at the transcriptional level. HIF1-α activates the transcription of target genes by binding to HRE within target genes.Citation3 The presence of HRE has been demonstrated in many pro-angiogenic genes such as VEGF.Citation4 However, instead of inducing expression, both hypoxia and HIF1-α overexpression repressed NRP2 expression in tumor cells. Thus, we hypothesize that HIF1-α might repress NRP2 expression in an indirect manner by inducing a transcription factor that directly binds to the promoter region of the NRP2 gene and represses NRP2 expression.
NRPs are co-receptors of VEGFR1-3 for VEGF in EC.Citation14–Citation16 NRP expression in EC increased VEGF binding to VEGFRs, VEGFR phosphorylation and VEGF-dependent VEGFR angiogenic activities such as EC survival and chemotaxis.Citation14,Citation17 Tumor cells rarely express VEGFR-2, the major VEGFR; therefore, NRPs represent the only VEGF receptors on tumor cells.Citation14,Citation20 We found that NRP2 repression in tumor cells increased VEGF protein levels in CM, with no effects on VEGF mRNA levels. This increase in VEGF protein levels promoted paracrine VEGF-induced activation of EC VEGFR-2 and enhanced EC migration and sprouting in a 3-dimensional in vitro angiogenesis spheroid assay, two key steps of tumor angiogenesis in vivo. We suggest that, in the absence of NRP2, VEGF/NRP2 interactions are inhibited and consequently there is increased VEGF, not bound to tumor cells, that is released into CM and activates EC in a paracrine manner. Taken together, these results show that NRP2 levels in tumor cells regulate VEGF-induced angiogenic activities.
Hypoxia is one of the main activators of VEGF expression in tumor cells through direct transcriptional activation by HIFs.Citation4 However, hypoxia also upregulates VEGF at the non-transcriptional level. For example, it has been shown previously that under hypoxic conditions, VEGF mRNA was stabilized and VEGF secretion was more efficient.Citation40,Citation41 Here, we show that hypoxia increased VEGF protein levels in CM through transcriptional repression of its receptor NRP2, providing a novel mechanism by which hypoxia increases VEGF protein levels and consequently promotes VEGF-dependent angiogenic activities.
NRP2 is the sole receptor for SEMA3F. In tumor cells, binding of SEMA3F to NRP2 inactivated RhoA, resulting in depolymerization of F-actin, inhibition of tumor cell migration and invasion in vitro and inhibition of tumor growth and metastasis in vivo.Citation13,Citation34,Citation35 Hypoxia did not have any effect on SEMA3F expression (data not shown). However, hypoxia-induced transcriptional repression of NRP2 in tumor cells inhibited SEMA3F-induced biological activity. On the other hand, in EC, VEGF/NRP2 interactions induced tumor angiogenesis, whereas SEMA3F/NRP2 interactions induced EC repulsion in vitro and inhibited tumor angiogenesis in vivo.Citation12,Citation13 Thus, inhibition or stimulation of tumor angiogenesis and growth may be influenced by the relative concentrations of VEGF and SEMA3F in the tumor microenvironment. Our finding that hypoxia increases the ratio of VEGF/SEMA3F levels may be significant since the relative levels of these two proteins in a hypoxic tumor may be used as a prognostic tool. For example, in ovarian carcinoma, VEGF levels were elevated whereas SEMA3F levels remained low.Citation42
Nearly all tumor cells express NRP1, NRP2 or both. Carcinomas express NRP1, whereas neuronal tumors and melanomas predominantly express NRP2.Citation19 Our findings that hypoxia represses NRP2 expression in glioblastoma and melanoma cells are in contrast with previous studies showing that hypoxia did not affect NRP2 protein levels in either EC or human SUM-159 breast carcinoma cells.Citation43 However, we found that NRP2 expression was repressed in four different hypoxic conditions—hypoxia, DFO, cobalt and MG132—and in six human tumor cell lines, including glioblastoma and melanoma cells. Furthermore, both VEGF and HIF1-α were induced under the same hypoxic conditions, providing a positive control for the hypoxia response. The experimental approaches and conditions used in the two studies were quite different. We analyzed the effect of hypoxia on NRP2 at the transcriptional level over a 24 h time period. In contrast, Bae et al. examined the effects of hypoxia on NRP2 protein degradation over a 6 h time period, which might have been insufficient time to transcriptionally repress NRP2. Thus, further studies are needed to resolve these differences on NRP2 regulation by hypoxia. On the other hand, we found that NRP1 was transcriptionally repressed by hypoxia in U87MG cells, which was in good agreement with previous studies including Bae et al. showing that hypoxia decreased NRP1 levels in astrocytoma and breast carcinoma cells.Citation43,Citation44
In summary, our results identify NRP2, a SEMA3F and VEGF receptor, as a target of transcriptional repression by hypoxia. Loss of NRP2 expression in tumor cells inhibits the anti-tumorigenic activity of SEMA3F and increases the proangiogenic activity of VEGF, two steps that promote tumor angiogenesis and tumor growth in vivo. We conclude that hypoxia regulates VEGF and SEMA3F activities through transcriptional repression of their common receptor NRP2, providing a novel mechanism by which hypoxia induces tumor angiogenesis, growth and metastasis.
Materials and Methods
Cell culture.
Human U87MG glioblastoma and A375SM melanoma cells were cultured as described in reference Citation35. Human umbilical vein endothelial cells (HUVEC) purchased from Lonza Inc. were cultured in EBM-2, supplemented with EGM-2 SingleQuots (Lonza Inc.).
Hypoxia.
Hypoxia was created by placing the cells in a hypoxia workstation (1% O2, 5% CO2, 37°C) for 24 h. Duplicate plates were maintained in “normoxia,” a standard tissue culture incubator maintained at 37°C with 21% O2 and 5% CO2. In some experiments, cells were treated with desferrioxamine (DFO) (250 µM, Sigma-Aldrich Corp.), MG132 (5 µM, Calbiochem) or cobalt (250 µM, Sigma-Aldrich Corp.) for 24 h.
Transfections.
The full-length human HIF1-α construct cloned in the pCEP4 vector was purchased from ATCC. The pCEP4-HIF1-α and the pcDNA3 (Invitrogen Corp.) vectors were digested with Not I and Kpn I. The HIF1-α insert and the digested pcDNA3 vector were ligated together with the DNA ligation kit (Takara Bio Inc.). Cells were transfected using Fugene6 reagent (Roche Applied Science).
siRNA knock down.
HIF1-α and HIF2-α/EPAS siRNAs were purchased from Santa Cruz Biotechnology, Inc. siRNAs of NRP2 (SMARTpool M-017721-01, D-017721-02 and D-017721-05) were purchased from Thermo Fisher Scientific. As a control, a siRNA duplex with an irrelevant sequence (Ambion Inc.) was used. Cells were transfected with 20 nM siRNA using SilentFect reagent (Bio-Rad Laboratories).
Anti-NRP2.
Goat anti-human anti-NRP2 antibody (R&D Systems) (20 µg/ml) and normal goat immunoglobulin as a control were added to U87MG cells for 24 h.
Immunoblot.
Cells were lysed and immunoblotted as described previously in reference Citation35. Specific proteins were detected after incubation with anti-NRP1, -NRP2, -HIF1-α, -HIF2-α/EPAS (Santa Cruz Biotechnology, Inc.) and -β-actin (Sigma-Aldrich Corp.) antibodies.
RNA isolation and analysis.
Total RNA was isolated from the cells using the RNeasy kit (Qiagen). cDNA was prepared using Superscript II enzyme (Invitrogen Corp.) and 1 µg total RNA. For real-time RT-PCR analysis, the DyNAmo Sybr-Green-based system (New England BioLabs, Inc.) was used. Oligonucleotide primers are listed in Supplemental Table 1. Reactions were run on a LightCycler (Roche Applied Science). Each experiment was done in duplicate and repeated three times.
Promoter luciferase constructs.
The human NRP2 promoter region was cloned from a human genomic PAC library as previously described in reference Citation45. A fragment spanning −2,037 to −206 relative to the start codon at position +1 was obtained using the following primers: forward 5′-TCA AGT CAG AGA TCT GGT AGT CAG GTG TGT GTT TCT C-3′; reverse 5′-CAG TGC TGC AAG CTT ATC AGC AAA GAG GGA ACA AGC C-3′. The PCR product was digested with Bgl II and Hind III and ligated into the Bgl II/Hind III sites of the pGL3 basic luciferase reporter vector (Promega Corp.) with the DNA ligation kit (Takara Bio Inc.). An HRE-driven luciferase construct generated to contain 3 HRE in a row was kindly provided by Dr. Jorge Ruas (Dana-Farber Cancer Institute, Boston, MA).
Luciferase reporter assay.
U87MG cells were transfected with Fugene6 (Roche Applied Science). Briefly, U87MG cells were transfected with 500 ng of the NRP2 or HRE promoter-luciferase constructs or pGL3 empty vector, 10 ng of a Renilla luciferase vector (used as a transfection efficiency control) (Promega Corp.) and either 0.5–1 µg of pcDNA3-HIF1-α plasmid or pcDNA3 control vector. After 36 h, cells were lysed and luciferase activities were measured with the dual-Luciferase reporter Assay System (Promega Corp.), with the reporter activities normalized to Renilla luciferase activity. Each transfection was done in duplicate and repeated at least three times.
Stress fibers.
SEMA3F was purified as in reference Citation34. U87MG cells were seeded on glass coverslips into 6-well plates the day before the treatment. U87MG cells were treated with DFO for 24 h followed by a 30 min incubation with 20 ng/ml SEMA3F. Cells were fixed and stained for F-actin as described earlier in reference Citation34.
RhoA activity.
U87MG cells were treated with DFO and SEMA3F as described above. At the end of the experiment, Rho activity assay was performed and quantified using the Rho activation assay kit (rhotekin pull-down) as described previously in reference Citation34.
Secretion of VEGF.
CM from U87MG cells were collected and centrifuged for 20 min at 4°C. VEGF protein levels in CM were measured by a sandwich enzyme immunoassay using a VEGF Quantitative ELISA kit from R&D Systems, Inc.
Cell migration.
Migration assays were performed in Transwell chambers (Corning Inc.) as described earlier in reference Citation34. Cells that migrated through the filter after 16 h were stained and counted by phase microscopy. The experiment was repeated three times in duplicate. The results represent the average of the three experiments. VEGF was provided by the National Cancer Institute.
Migration of U87MG cells. U87MG cells in MEM containing 0.5% FBS were treated with DFO for 24 h or transfected with either control or NRP2 siRNAs for 72 h. U87MG cells (2 × 104) in serum-free MEM medium were added to upper wells. MEM media containing 1% fetal bovine serum (FBS) and 320 ng/ml SEMA3F were added to the lower wells.
Migration of HUVEC. CM from U87MG cells were collected and filtered through a 0.45 µm filter. HUVEC (5 × 104) in MEM media containing 0.5% FBS were added to upper wells. MEM media (600 µl) containing 0.5% FBS, 5 ng/ml VEGF and 1 µg/ml heparin were added to the lower wells. For experiments using CM from U87MG cells, MEM media containing 0.5% FBS (500 µl) plus CM isolated from U87MG (100 µl) and 1 µg/ml heparin were added to the lower wells. Where indicated, 1 µM SU5416 was added to the lower wells.
VEGF signaling.
CM from U87MG cells were collected and filtered through a 0.45 µm filter. VEGF-induced VEGFR-2, p44/42 MAPK and p38 MAPK phosphorylation was analyzed as in in reference Citation46. Briefly, HUVEC were plated on 6-well plates (400,000 cells/well). After two days, cells were starved overnight in MEM media containing 0.5% FBS. The following day, HUVEC were either left untreated, treated with VEGF (5 ng/ml) or incubated with 1 ml of MEM media containing 0.5% FBS and 1 ml of CM from U87MG cells and 1 µg/ml heparin. After a 10 min stimulation at 37°C, cells were washed with PBS/pervanadate and lysed in RIPA buffer supplemented with a protease inhibitor cocktail tablet (Roche) and pervanadate. Equal amounts of lysates were boiled in SDS sample buffer for 10 min at 95°C and analyzed by SDS-PAGE. Then, proteins were transferred onto polyvinylidene fluoride (PVDF) membranes and immunoblotted with anti-phospho-VEGFR-2, -p44/42 MAPK and -p38 MAPK antibodies (Cell Signaling Technology, Inc.). Total proteins were detected after stripping the PVDF membranes and re-blotting with anti-VEGFR-2, -p44/42 MAPK and -p38 MAPK (Cell Signaling) or anti-β-actin antibodies as described above.
Spheroid-based angiogenesis assay.
Early passage HUVEC were suspended and aggregated overnight to form cellular spheroids (500 cells/spheroid). HUVEC spheroids were embedded into collagen gels and either left untreated or treated for 16 h with 5 ng/ml VEGF or CM from U87MG cells. In vitro angiogenesis was quantified by measuring the number of sprouts and the cumulative length of sprouts that had grown out of each spheroid using NIH ImageJ software. Ten to fifteen spheroids per experiment group were analyzed.
Abbreviations
| CM | = | conditioned media |
| DFO | = | desferrioxamine |
| EC | = | endothelial cells |
| HIF | = | hypoxia inducible factor |
| HRE | = | hypoxia responsive elements |
| HUVEC | = | human umbilical vein endothelial cells |
| NRP | = | neuropilin |
| SEMA3 | = | class-3 semaphorins |
| VEGF | = | vascular endothelial growth factor |
| VEGFR | = | VEGF receptor |
Figures and Tables
Figure 1 Hypoxia represses NRP2 expression in tumor cells in a HIF1-α-dependent manner. (A) NRP2, HIF1-α and β-actin protein levels in U87MG glioblastoma and A375SM melanoma cells maintained in either normoxic (21% O2) or hypoxic (1% O2) condtions or treated with either DFO, cobalt or MG132 for 24 h. (B) HIF1-α (top) and HIF2-α (bottom) mRNA levels in U87MG cells transfected with either control, HIF1-α (top) or HIF2-α (bottom) siRNAs and either left untreated or treated with DFO for 24 h. (C) NRP2, HIF1-α, HIF2-α and β-actin protein levels in U87MG cells transfected with either control, HIF1-α or HIF2-α siRNAs and either left untreated or treated with DFO for 24 h. Note that there are two HIF1-α bands on the gel. The upper band was specific since it was induced by DFO treatment (lanes 1 vs. 2) and blocked by HIF1-α siRNA (lanes 2 vs. 4). However, the lower band appeared to be non-specific since it was present under normoxia in both control and HIF1-α siRNA transfections (lanes 1 and 3). (D) NRP2, HIF1-α and β-actin protein levels in U87MG cells transfected with either control or pcDNA3-HIF1-α vectors.
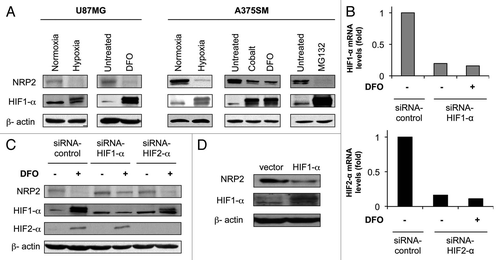
Figure 2 Hypoxia represses NRP2 at the transcriptional level. (A) U87MG glioblastoma and A375SM melanoma cells were maintained in either normoxic or hypoxic conditions or treated with DFO for 24 h. NRP2 (top) and VEGF (bottom) mRNA levels were measured by quantitative PCR and normalized to B2M mRNA. (B) A NRP2 luciferase promoter vector (top) or a HRE-driven reporter construct (bottom) were co-transfected with either control or pcDNA3-HIF1-α vectors. Luciferase activities were measured after 32 h.
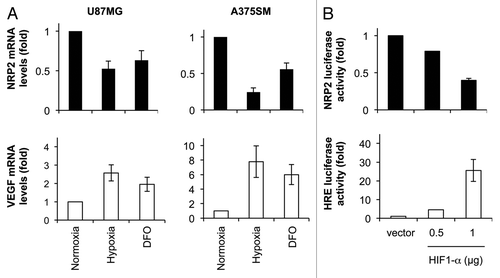
Figure 3 Repression of NRP2 in tumor cells by hypoxia inhibits SEMA3F biological activity. (A) Confocal microscopy images of U87MG cells either left untreated (a) or treated with SE MA3F (b), DFO and SE MA3F (c) or DFO alone (d). F-actin and nuclei were visualized using Alexa Fluor 488 phalloidin (green) and Hoescht (blue), respectively. (B) U87MG cells, either left untreated (top) or treated with DFO for 24 h (bottom), were given SEMA3F for 0–15 min. GTP-bound RhoA (active RhoA) and total RhoA levels in lysates were analyzed. (C) Transwell cell migration of U87MG cells either left untreated or treated with DFO for 24 h (left) and U87MG cells transfected with control or NRP2 siRNAs for 72 h (right). SE MA3F was added to the lower wells.
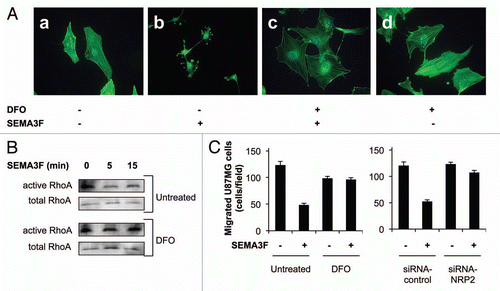
Figure 4 Repression of NRP2 in tumor cells increases VEGF protein levels in conditioned media. (A) NRP2 and β-actin protein levels in U87MG cells either left untreated or treated with DFO for 24 h. VEGF protein levels in CM are shown in the part below. (B) NRP2 and β-actin protein levels in U87MG cells transfected with either control, a pool of 4 NRP2 siRNAs or 2 different NRP2 siRNAs for 48 h. VEGF mRNA and protein levels in CM are shown in the parts below. (C) VEGF protein levels in CM from U87MG cells incubated with anti-NRP2 antibody for 24 h.
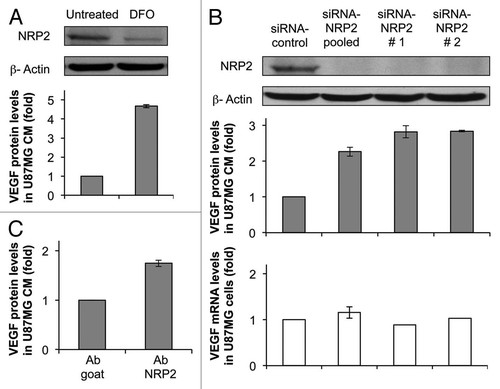
Figure 5 Repression of NRP2 in tumor cells increases EC VEGFR-2 activation and migration. (A) Western blot showing phosphorylation of VEGFR-2 (Tyr1175) and total VEGFR-2 in HUVEC stimulated with either VEGF or CM form U87MG cells either left untreated or treated with DFO for 24 h. (B) Western blot showing phosphorylation of VEGFR-2 (Tyr1175), p44/42 MAPK (Thr202/Tyr204), p38 MAPK (Thr180/Tyr182) and total VEGFR-2, p44/42 MAPK and p38 MAPK in HUVEC stimulated with either VEGF or CM from U87MG cells transfected with control or a pool of 4 NRP2 siRNAs for 48 h. (C) Transwell cell migration of HUVEC. Media containing VEGF or CM isolated from U87MG cells either left untreated or treated with DFO for 24 h (left) or CM from U87MG cells transfected with either control or a pool of 4 NRP2 siRNAs for 48 h (right) were added to the lower wells. When indicated, SU5416 was added to the lower wells.
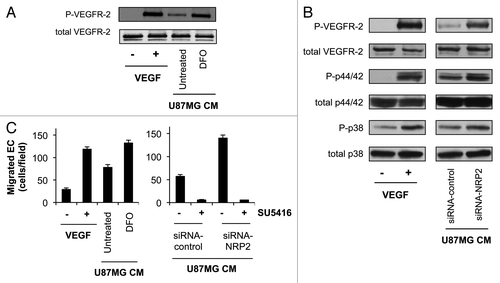
Figure 6 Repression of NRP2 in tumor cells induces in vitro angiogenesis. Representative images (left) and statistical summary (right) of 3-dimensional spheroid-based in vitro angiogenesis assays, with HUVEC spheroids either left untreated or treated with VEGF (5 ng/ml) or CM from U87MG cells transfected with either control or a pool of four NRP2 siRNAs. Extensive endothelial outgrowth can be observed in spheroids treated either with VEGF and CM from U87MG cells transfected with NRP2 siRNA.
Additional material
Download Zip (880.3 KB)Acknowledgments
We thank Dr. Andrew Dudley, Dr. Elena Geretti and Marc Allard-Ratick for valuable discussions and for critical reading of the manuscript. We thank Sandy Smith and Dr. Andreas Stahl for technical assistance with the VEGF ELISA and the spheroid sprouting assay, respectively. We thank Dr. Alex Mitsialis for assistance with the hypoxia workstation and Dr. Mireille Rossignol for cloning the human NRP2 gene. We thank Melissa Herman and Kristin Johnson for preparation of the manuscript. This study was supported by grants from the National Institutes of Health (NIH)—CA37392 and CA45548.
References
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677 - 684
- Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol 2006; 59:15 - 26
- Fong GH. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis 2008; 11:121 - 140
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996; 16:4604 - 4613
- Ryan HE, Lo J, Johnson RS. HIF-1alpha is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17:3005 - 3015
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230 - 1237
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 1996; 271:1172 - 1180
- Klagsbrun M, Eichmann A. A role for axon guidance receptors and ligands in blood vessel development and tumor angiogenesis. Cytokine Growth Factor Rev 2005; 16:535 - 548
- Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther 2006; 5:1099 - 1107
- Guttmann-Raviv N, Kessler O, Shraga-Heled N, Lange T, Herzog Y, Neufeld G. The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett 2006; 231:1 - 11
- Gaur P, Bielenberg DR, Samuel S, Bose D, Zhou Y, Gray MJ, et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin Cancer Res 2009; 15:6763 - 6770
- Kessler O, Shraga-Heled N, Lange T, Gutmann-Raviv N, Sabo E, Baruch L, et al. Semaphorin-3F is an inhibitor of tumor angiogenesis. Cancer Res 2004; 64:1008 - 1015
- Bielenberg DR, Hida Y, Shimizu A, Kaipainen A, Kreuter M, Kim CC, et al. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest 2004; 114:1260 - 1271
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998; 92:735 - 745
- Geretti E, Shimizu A, Klagsbrun M. Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 2008; 11:31 - 39
- Kawamura H, Li X, Goishi K, van Meeteren LA, Jakobsson L, Cebe-Suarez S. Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood 2008; 112:3638 - 3649
- Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006; 108:1243 - 1250
- Klagsbrun M, Takashima S, Mamluk R. The role of neuropilin in vascular and tumor biology. Adv Exp Med Biol 2002; 515:33 - 48
- Bielenberg DR, Pettaway CA, Takashima S, Klagsbrun M. Neuropilins in neoplasms: expression, regulation and function. Exp Cell Res 2006; 312:584 - 593
- Miao HQ, Lee P, Lin H, Soker S, Klagsbrun M. Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. FASEB J 2000; 14:2532 - 2539
- Parikh AA, Fan F, Liu WB, Ahmad SA, Stoeltzing O, Reinmuth N, et al. Neuropilin-1 in human colon cancer: expression, regulation and role in induction of angiogenesis. Am J Pathol 2004; 164:2139 - 2151
- Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, et al. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene 2007; 26:5577 - 5586
- Dallas NA, Gray MJ, Xia L, Fan F, van Buren G 2nd, Gaur P, et al. Neuropilin-2-mediated tumor growth and angiogenesis in pancreatic adenocarcinoma. Clin Cancer Res 2008; 14:8052 - 8060
- Gray MJ, Van Buren G, Dallas NA, Xia L, Wang X, Yang AD, et al. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J Natl Cancer Inst 2008; 100:109 - 120
- Handa A, Tokunaga T, Tsuchida T, Lee YH, Kijima H, Yamazaki H, et al. Neuropilin-2 expression affects the increased vascularization and is a prognostic factor in osteosarcoma. Int J Oncol 2000; 17:291 - 295
- Hansel DE, Wilentz RE, Yeo CJ, Schulick RD, Montgomery E, Maitra A. Expression of neuropilin-1 in high-grade dysplasia, invasive cancer and metastases of the human gastrointestinal tract. Am J Surg Pathol 2004; 28:347 - 356
- Kawakami T, Tokunaga T, Hatanaka H, Kijima H, Yamazaki H, Abe Y, et al. Neuropilin 1 and neuropilin 2 co-expression is significantly correlated with increased vascularity and poor prognosis in nonsmall cell lung carcinoma. Cancer 2002; 95:2196 - 2201
- Kreuter M, Woelke K, Bieker R, Schliemann C, Steins M, Buechner T, et al. Correlation of neuropilin-1 overexpression to survival in acute myeloid leukemia. Leukemia 2006; 20:1950 - 1954
- Lantuejoul S, Constantin B, Drabkin H, Brambilla C, Roche J, Brambilla E. Expression of VEGF, semaphorin SEMA3F, and their common receptors neuropilins NP1 and NP2 in preinvasive bronchial lesions, lung tumours and cell lines. J Pathol 2003; 200:336 - 347
- Latil A, Bieche I, Pesche S, Valeri A, Fournier G, Cussenot O, et al. VEGF overexpression in clinically localized prostate tumors and neuropilin-1 overexpression in metastatic forms. Int J Cancer 2000; 89:167 - 171
- Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, et al. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res 2001; 61:5736 - 5740
- Roche J, Boldog F, Robinson M, Robinson L, Varella-Garcia M, Swanton M, et al. Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene 1996; 12:1289 - 1297
- Sekido Y, Bader S, Latif F, Chen JY, Duh FM, Wei MH, et al. Human semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc Natl Acad Sci USA 1996; 93:4120 - 4125
- Shimizu A, Mammoto A, Italiano JE Jr, Pravda E, Dudley AC, Ingber DE, et al. ABL2/ARG tyrosine kinase mediates SEMA3F-induced RhoA inactivation and cytoskeleton collapse in human glioma cells. J Biol Chem 2008; 283:27230 - 27238
- Coma S, Amin DN, Shimizu A, Lasorella A, Iavarone A, Klagsbrun M. Id2 promotes tumor cell migration and invasion through transcriptional repression of semaphorin 3F. Cancer Res 2010; 70:3823 - 3832
- Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood 1993; 82:3610 - 3615
- Veschini L, Belloni D, Foglieni C, Cangi MG, Ferrarini M, Caligaris-Cappio F, et al. Hypoxia-inducible transcription factor-1alpha determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood 2007; 109:2565 - 2570
- Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem 2004; 279:40337 - 40344
- Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 2003; 302:1975 - 1978
- Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol 1998; 18:3112 - 3119
- Ozawa K, Kondo T, Hori O, Kitao Y, Stern DM, Eisenmenger W, et al. Expression of the oxygen-regulated protein ORP150 accelerates wound healing by modulating intracellular VEGF transport. J Clin Invest 2001; 108:41 - 50
- Osada R, Horiuchi A, Kikuchi N, Ohira S, Ota M, Katsuyama Y, et al. Expression of semaphorins, vascular endothelial growth factor, and their common receptor neuropilins and alleic loss of semaphorin locus in epithelial ovarian neoplasms: increased ratio of vascular endothelial growth factor to semaphorin is a poor prognostic factor in ovarian carcinomas. Hum Pathol 2006; 37:1414 - 1425
- Bae D, Lu S, Taglienti CA, Mercurio AM. Metabolic stress induces the lysosomal degradation of neuropilin-1 but not neuropilin-2. J Biol Chem 2008; 283:28074 - 28080
- Ding H, Wu X, Roncari L, Lau N, Shannon P, Nagy A, et al. Expression and regulation of neuropilin-1 in human astrocytomas. Int J Cancer 2000; 88:584 - 592
- Rossignol M, Gagnon ML, Klagsbrun M. Genomic organization of human neuropilin-1 and neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics 2000; 70:211 - 222
- Geretti E, van Meeteren LA, Shimizu A, Dudley AC, Claesson-Welsh L, Klagsbrun M. A mutated soluble neuropilin-2 B domain antagonizes vascular endothelial growth factor bioactivity and inhibits tumor progression.. Mol Cancer Res 2010; 8:1063 - 1073