Abstract
We have determined whether an adenovirus that comprises the tail and shaft domains of a serotype 5 virus and the knob domain of a serotype 3 virus expressing MDA-7/IL-24, Ad.5/3-mda-7, more effectively infects and kills renal carcinoma cells (RCCs) compared to a serotype 5 virus, Ad.5-mda-7. RCCs are a tumor cell type that generally does not express the receptor for the type 5 adenovirus; the coxsakie and adenovirus receptor (CAR). Ad.5/3-mda-7 infected RCCs to a much greater degree than Ad.5-mda-7. MDA-7/IL-24 protein secreted from Ad.5/3-mda-7-infected RCCs induced MDA-7/IL-24 expression and promoted apoptosis in uninfected "bystander" RCCs. MDA-7/IL-24 killed both infected and bystander RCCs via CD95 activation. Knockdown of intracellular MDA-7/IL-24 in uninfected RCCs blocked the lethal effects of conditioned media. Infection of RCC tumors in one flank, with Ad.5/3-mda-7, suppressed growth of infected tumors and reduced the growth rate of uninfected tumors implanted on the opposite flank. The toxicity of the serotype 5/3 recombinant adenovirus to express MDA-7/IL-24 was enhanced by combined molecular or small molecule inhibition of MEK1/2 and PI3K; inhibition of mTOR, PI3K and MEK1/2; or use of the multi-kinase inhibitor sorafenib. In RCCs, combined inhibition of cytoprotective cell signaling pathways enhanced the MDA-7/IL-24-induction of CD95 activation, with greater mitochondrial dysfunction due to loss of MCL-1 and BCL-XL expression, and tumor cell death. Treatment of RCC tumors in vivo with sorafenib also enhanced Ad.5/3-mda-7 toxicity and prolonged animal survival. Future combinations of these approaches hold promise for developing a more effective therapy for kidney cancer.
Introduction
In the United States, renal cell carcinoma (RCC) is diagnosed in ∼51,000 patients per annum. If the disease is detected at an early stage, in which a large portion or the entire kidney can be removed with the tumor, a high prolonged level of patient survival is noted (reviewed in ref. Citation1). However, if the disease has spread beyond the capsule of the kidney into the adrenal gland or surrounding fascia with nodal involvement the prognosis is poor with rapid nadir.Citation1 This occurs even under ideal circumstances where the disease is still only locally advanced and essentially all of the tumor can be surgically removed and the patients are maximally treated with palliative radiation and chemotherapy. In part, this resistance is due to the fact that RCC is characterized as frequently being highly refractory to multiple established cytotoxic chemotherapy regimens.Citation1 Clearly, there is a need to develop new approaches to treat this lethal disease.
The mda-7 gene (Interleukin 24, IL-24) was isolated from human melanoma cells induced to undergo terminal differentiation.Citation2 Protein expression of MDA-7/IL-24 decreases with increasing staging in advanced melanomas, with almost undetectable levels of the protein in metastatic tumors.Citation2–Citation14 Enforced expression of MDA-7/IL-24 inhibits the growth, kills and also radio-/chemo-sensitizes a very broad spectrum of cancer cell types without exerting toxic effects in normal human epithelial or fibroblast cells.Citation9–Citation16 In kidney cancer cells we have previously shown that GST-MDA-7 kills these cells, but not normal kidney epithelial cells, in part through activation of the death receptor CD95.Citation10 Mda-7/IL-24 has been evaluated in a Phase I clinical trial in advanced cancers that were refractory to further therapeutic intervention and this study indicated that an Ad.5-mda-7 (INGN-241) injected intra-tumorally was safe and with repeated injection a significant level of clinical activity was observed in up to ∼44% of patients.Citation10,Citation11,Citation13,Citation14,Citation17,Citation18
The apoptotic pathways by which Ad.5-mda-7 induces cell death in tumor cells are complex and seem to subtly change based on the tumor cell type being examined.Citation9–Citation16,Citation20–Citation28 In melanoma cells, MDA-7/IL-24 was shown to induce a decrease in BCL-2 and BCL-XL levels, concomitant with a modest upregulation of BAX and BAK expression; thus the ratio of protective to toxic BCL-2 family proteins in the tumor cell has been shifted towards a pro-apoptotic state.Citation10–Citation13,Citation19 The ability of Ad.5-mda-7 to induce apoptosis in DU145 prostate cancer cells, which do not produce the toxic BH3 domain protein BAX, demonstrates that MDA-7/IL-24 can mediate apoptosis in tumor cells through a BAX-independent pathway.Citation9–Citation14 More recently MDA-7/IL-24 toxicity has been strongly linked to alterations in endoplasmic reticulum (ER) stress signaling.Citation20–Citation23 MDA-7/IL-24 physically associates with the ER-localized HSP70 family chaperone protein BiP/GRP78 that inactivates this protein resulting in the activation/dimerization of its chaperone: PKR-like endoplasmic reticulum kinase (PERK).Citation10–Citation13,Citation23,Citation24 We have noted that high concentrations of GST-MDA-7 or infection of tumor cells with Ad.5-mda-7 kills tumor cells and does so in a PERK-dependent fashion that is dependent on mitochondrial dysfunction and in GBM cells, we went on to note that at least 5 toxic BH3 domain proteins facilitated MDA-7/IL-24 toxicity.Citation25–Citation28 In a wide variety of cancer cells, overexpression of multiple protective BCL-2 family proteins protects cells from MDA-7/IL-24 toxicity.Citation10–Citation13,Citation20,Citation22 In ovarian and renal carcinoma cells MDA-7/IL-24 was shown to initiate killing via the extrinsic apoptosis pathway that was dependent on ceramide generation and CD95 activation, and also upon ER stress-induced loss of MCL-1 expression.Citation10,Citation21,Citation22,Citation27
The ability of MDA-7/IL-24 to activate/inhibit the activities of many signaling pathways in transformed cells has been investigated by our laboratories and by other groups.Citation10–Citation13,Citation15–Citation32 Prior work, using bacterially synthesized GST-MDA-7, argues that in the 0.25–2.0 nM concentration range GST-MDA-7 causes growth arrest with little cell killing; effects that are due to elevated levels of ER stress signaling, whereas at ∼20-fold greater concentrations, the cytokine causes profound ER stress signals, growth arrest and tumor cell death.Citation10–Citation13,Citation24,Citation27,Citation28 Our laboratories have demonstrated that Ad.5-mda-7 kills melanoma cells and established GBM cells in part by promoting p38 MAPK activation e.g., reviewed in reference 30. In primary human GBM cells however, MDA-7/IL-24-induced activation of p38 MAPK was shown to be a protective signal at what were considered to be “toxic” doses but that at “supra-toxic” doses p38 MAPK signaling reverted to being a toxic signal; JNK signaling was toxic in GBM cells.Citation27,Citation28 In RCCs we have noted that both MDA-7/IL-24-induced JNK1/2 and p38 MAPK signaling were toxic responses.Citation10 Other groups have argued that inhibition of PI3K signaling, but not ERK1/2 signaling, modestly promotes Ad.5-mda-7 lethality in breast and lung cancer cells, whereas we have shown in GBM cells that inhibition of both PI3K and ERK1/2 signaling was required to potentiate the toxic effect of the cytokine.Citation10–Citation13,Citation21,Citation27,Citation28,Citation31,Citation32
The present studies were designed to define more effective ways of killing RCCs using mda-7/IL-24 delivered as a recombinant adenovirus. Previously we have used GST-MDA-7 as a tool to study MDA-7/IL-24 effects in RCCs; however GST-MDA-7 cannot be translated as a therapeutic for animals or for the clinic. In this manuscript we provide new data supporting the hypothesis that simultaneously inhibiting multiple cytoprotective pathways, including mTOR, PI3K and/or MEK1/2 signaling, is required to facilitate lethality of adenoviral mda-7/IL-24 delivery toward RCCs both in vitro and in vivo. In addition, we demonstrate in vitro and in vivo that the FDA approved RCC therapeutic multikinase inhibitor sorafenib, which inactivates signaling pathways and promotes ER stress signaling, enhances mda-7/IL-24 toxicity. Since delivery of therapeutic genes using Ad.Citation5 has shown significant limitations in treating RCC, because of the need for Coxsackie adenovirus receptors (CAR) on tumor cells for virus entry, we evaluated the use of a tropism-modified adenovirus that uses a serotype 3 modification of the viral knob protein to facilitate delivery of mda-7/IL-24 in a CAR-independent manner.Citation10–Citation13,Citation21,Citation29,Citation33,Citation34 This approach profoundly augmented therapeutic activity of MDA-7/IL-24 in RCCs, in vitro and in vivo, suggesting that combining both strategies, namely inhibition of cytoprotective pathways using sorafenib and virus tropism modification, may provide a means of developing an improved therapy to translate into the clinic for kidney cancer.Citation10–Citation13,Citation21,Citation29
Results
RCCs are resistant to infection by serotype 5 adenoviruses due to lack of coxsakie and adenovirus receptor (CAR) expression.Citation10,Citation24,Citation29,Citation31 To circumvent this problem, we developed a chimeric serotype 5/serotype 3 modified knob adenovirus to deliver mda-7/IL-24 to RCCs and infect these cells in a CARi-ndependent fashion: Ad.5/3-mda-7.Citation11,Citation21,Citation34,Citation35 At low multiplicities of infection, Ad.5/3-mda-7 but not Ad.5-mda-7 infected RCCs and promoted expression of MDA-7/IL-24 (). These findings were also reflected in colony formation assay data ().
MDA-7/IL-24 is a novel cancer-specific cytotoxic cytokine that can be secreted from cells infected with a recombinant adenovirus containing the mda-7/IL-24 gene.Citation9–Citation12,Citation37 We next determined whether direct infection of RCCs with Ad.5/3-mda-7 also permitted MDA-7/IL-24 to be secreted into the culture media and whether this also had a lethal “bystander” effect on uninfected RCCs. Infection of UOK121LN cells with Ad.5/3-mda-7, but not Ad.5-mda-7, increased expression of MDA-7/IL-24 in the culture media of cells (, upper immunoblots). Transfer of the conditioned media containing MDA-7/IL-24, but not “spent” media from vector infected cells, onto uninfected UOK121LN cells increased expression of MDA-7/IL-24. It has been argued that MDA-7/IL-24 can set up a self-stimulatory paracrine loop in tumor cells and in agreement with data in other tumor types, knockdown of MDA-7/IL-24 expression in uninfected cells blocked the conditioned media containing the cytokine from inducing MDA-7/IL-24 expression (, lower immunoblots).Citation38 The lethal effects of Ad.5/3-mda-7, in infected cells were blocked by knockdown of CD95 expression (). Transfer of media containing MDA-7/IL-24 onto uninfected RCCs promoted cell killing, an effect that was also blocked by knockdown of CD95 or knockdown of MDA-7/IL-24.
Infection of primary renal epithelial cells with Ad.5-mda-7 resulted in expression of MDA-7/IL-24 without causing significant toxicity to these non-transformed cells ( and data not shown). Forty-eight hour after infection, conditioned media from Ad.5-cmv, Ad.5-mda-7 and Ad.5-mda-7 SP- (lacking the signal peptide for cytokine secretion)-infected primary renal epithelial cells was removed and placed onto uninfected UOK121LN carcinoma cells. As a single agent, conditioned media containing from Ad.5-mda-7-infected cells, but not Ad.5-cmv or Ad.5-mda-7 SP-infected cells, suppressed the growth of RCCs as measured in an MTT assay by 38 ± 4% (). In a dose-dependent fashion conditioned media containing MDA-7/IL-24 promoted cell killing in short-term true viability assays ().
Previously we have noted that inactivation of ERK1/2 and AKT and activation of JNK1/2 and p38 MAPK were hallmarks of MDA-7/IL-24 toxicity in renal carcinoma cells.Citation21 In A498 cells molecular inhibition of PI3K, mTOR or MEK1/2 suppressed basal levels of phospho-ERK1/2 and of phospho-(S473)-AKT and in cells infected with Ad.5/3-mda-7 the dephosphorylation of ERK1/2 and of AKT was enhanced wherein kinase activity was largely abolished (). Knock down of mTOR reduced total and phospho-mTOR levels and MDA-7/IL-24 expression also reduced phospho-mTOR levels, and the combinations of Ad.5/3-mda-7 and molecular inhibition of PI3K, mTOR or MEK1/2 further reduced mTOR phosphorylation.
Expression of MDA-7/IL-24 enhanced JNK1/2 phosphorylation that was further increased by combined inhibition of signaling by: PI3K + MEK1/2; mTOR + MEK1/2; PI3K + mTOR; or PI3K + MEK1/2 + mTOR (). This correlated with increased expression of the toxic BH3 domain protein BAX and decreased expression of MCL–1. Prior studies from our group demonstrated that MDA-7/IL-24-induced PKR-like endoplasmic reticulum kinase (PERK) signaling was essential for cytokine lethality.Citation21,Citation27 MDA-7/IL-24 enhanced PERK and eIF2α phosphorylation that was strongly enhanced by combined inhibition of PI3K + MEK1/2; mTOR + MEK1/2; PI3K + mTOR; or PI3K + MEK1/2 + mTOR. These findings correlated with decreased expression of the PERK chaperone BiP/GRP78.
We next determined the impact of inhibiting signaling pathways on Ad.5/3-mda-7 toxicity in A498 RCCs. Molecular inhibition of mTOR; PI3K; MEK1/2; PI3K + MEK1/2; mTOR + MEK1/2; PI3K + mTOR; or PI3K + MEK1/2 + mTOR caused modest increases in the basal level of apoptosis in Ad.5/3-cmv infected cells (). Combined inhibition of PI3K + MEK1/2 or PI3K + mTOR modestly enhanced MDA-7/IL-24 toxicity whereas combined PI3K + MEK1/2 + mTOR inhibition strongly enhanced cell killing. The short-term viability data was also reflected in assays determining total cell numbers (). However, in long-term colony formation assays only combined PI3K + MEK1/2 + mTOR inhibition strongly enhanced MDA-7/IL-24 toxicity (). In UOK121LN RCCs only combined molecular inhibition of PI3K + MEK1/2 + mTOR signaling significantly enhanced MDA-7/IL-24 toxicity and suppression of growth in short term assays whereas molecular inhibition of any individual signaling pathway promoted MDA-7/IL-24 toxicity in long-term colony formation ().
Multiple drugs have been developed for clinical application that are designed to inhibit the PI3K/MEK1/2/mTOR signaling pathways and the multi-kinase inhibitor sorafenib which suppresses ERK1/2 pathway activity as well as inhibiting receptor tyrosine kinases is an FDA approved therapeutic for RCC. We next examined the impact of the PI3K inhibitor PX-866; the MEK1/2 inhibitor PD184352; and the mTOR inhibitors rapamycin and PI-103 on MDA-7/IL-24 lethality in RCCs. Use of kinase inhibitor drugs, as compared to transfecting plasmids to express dominant negative inhibitory proteins, resulted in a greater suppression of ERK1/2 and AKT signaling under basal conditions and promoted a greater level of PERK/eIF2α and JNK1/2 activation ().Citation37 Inhibition of MEK1/2 and PI3K significantly enhanced MDA-7/IL-24 lethality in RCCs, compared to inhibition of MEK1/2 and mTOR ( and C). Combined inhibition of mTOR, PI3K and MEK1/2 enhanced MDA-7/IL-24 toxicity to a greater extent than inhibition of MEK1/2 and PI3K (). Sorafenib enhanced MDA-7/IL-24 toxicity ().
In general agreement with data in and C; , in colony formation assays combined inhibition of mTOR, PI3K and MEK1/2 or sorafenib treatment enhanced MDA-7/IL-24 toxicity to a greater extent than inhibition of MEK1/2 and PI3K or of mTOR (). Using purified GST-MDA-7, we noted that sorafenib synergized in median dose effect colony formation assays with the cytokine to kill RCCs with combination index (CI) values of less than 1.00 ().
Based on our data in – we next determined the signaling and apoptosis pathways that were being modulated in RCCs. Expression of activated AKT and activated MEK1 significantly protected cells from inhibition of mTOR, PI3K and MEK1/2 to a similar extent as expression of activated AKT, activated MEK1 and activated mTOR (, p < 0.05). Inhibition of JNK1/2 signaling blunted MDA-7/IL-24 lethality and it suppressed the elevation of MDA-7/IL-24 toxicity by combined inhibition of mTOR, PI3K and MEK1/2.Citation37
Prior studies in RCCs have shown that MDA-7/IL-24 kills primarily via activation of CD95 (the extrinsic pathway), and our present studies also demonstrated that secreted MDA-7/IL-24 killed bystander RCCs in a CD95 dependent fashion (). The ability of MEK1/2 and PI3K inhibition to enhance MDA-7/IL-24 toxicity was suppressed by ∼50% when extrinsic apoptosis pathway signaling was blocked (). However, when signaling via the intrinsic pathway was blocked, MDA-7/IL-24 toxicity was abolished. As MDA-7/IL-24 activates CD95 in RCCs we determined whether modulation of signaling pathway functions altered the levels of MDA-7/IL-24-induced CD95 activation. Use of molecular tools modestly enhanced MDA-7/IL-24-induced CD95 surface localization and DISC formation whereas use of small molecule kinase inhibitors, that inhibited the signaling pathways to a greater extent, caused a much larger enhancement of CD95 surface localization, that in the case of PX-866 + PD184352 treatment or sorafenib treatment, was greater than additive (). These findings using kinase inhibitors correlated with increased association of pro-caspase 8 and FADD with CD95 (, upper blots).
Based on the data in , we wished to determine whether the in vitro toxic “bystander” effect of secreted MDA-7/IL-24 on uninfected tumor cells could be translated into an in vivo model system. A498 RCC tumors were established on two flanks of an athymic mouse. One tumor was injected every second day with Ad.5/3-cmv or with Ad.5/3-mda-7 (total two injections) and the volumes of the tumors on both sides of the animal measured using calipers. Infection of tumors with Ad.5/3-mda-7, but not Ad.5/3-cmv, caused a rapid suppression of the tumor growth rate and significantly increased animal survival ( and B (p < 0.05)). Infection of animals with Ad.5/3-mda-7, but not Ad.5/3-cmv, also suppressed the growth rate of uninfected tumors growing on the opposite flank, as previously observed in the context of prostate tumors implanted on opposite flanks of nude mice.Citation34 This correlated with caspase 3 cleavage; MDA-7/IL-24 expression and increased apoptosis in both the Ad.5/3-mda-7-infected and uninfected “bystander” RCC tumors ( and D). Of particular note, Ad.5/3-cmv virus infection promoted recruitment of CD11c and CD11b positive immune cells (dendritic and natural killer cells) into tumors, as judged by CD11c and CD11b staining in Ad.5/3-cmv infected tumors but not in the contra-lateral uninfected “bystander” tumor. In contrast to empty vector virus, Ad.5/3-mda-7 infection resulted in CD11c and CD11b staining in both the infected tumor and in the contra-lateral uninfected bystander tumor. Together with our in vitro data, these findings argue that MDA-7/IL-24 secreted from a renal carcinoma tumor in vivo has antitumor activity on an uninfected distant RCC tumor.
Finally, based on our in vitro studies we determined whether the in vivo RCC antitumor effects of MDA-7/IL-24 could be enhanced by the clinically relevant RCC therapeutic sorafenib.Citation38,Citation39 Pre-formed A498 tumors (∼150 mm3) were injected with either Ad.5/3-cmv or Ad.5/3-mda-7. A second adenoviral infusion was performed 48 h after the first infection. Twenty-four hour after the first viral infection animals were treated with vehicle or sorafenib (20 mg/kg) for the following 4 days. Infection with Ad.5/3-mda-7 or treatment with sorafenib reduced the rate of tumor growth (). However, combined exposure to Ad.5/3-mda-7 and sorafenib promoted a significantly greater suppression of tumor growth than either agent individually. Based on animal welfare guidelines, animals carrying tumors > 2.0 cm3 in volume are humanely sacrificed; treatment of animals with empty vector virus and sorafenib modestly increased animal survival whereas treatment with Ad.5/3-mda-7 significantly increased survival (). Combined treatment of animals with sorafenib and Ad.5/3-mda-7 enhanced animal survival beyond that caused by administration of Ad.5/3-mda-7 alone (p < 0.05). In isolated tumors it was noted that cleavage of pro-caspase 3 and TUNEL positivity was increased by Ad.5/3-mda-7 + sorafenib treatment and decreased Ki67 reactivity ( and D). Infection with Ad.5/3-mda-7 also promoted recruitment of CD11b and CD11c positive immune cells into the tumor; immune cells including monocytes, macrophages/dendritic cells, neutrophils, natural killer cells and B cells. In addition we found that: (a) Ad.5/3-mda-7 and sorafenib did not interact to kill primary human renal epithelial cells in vitro and; (b) no obvious normal tissue toxicity by H&E staining was noted in to be induced in either the kidneys or livers of animals exposed to Ad.5/3-MDA-7 and sorafenib (data not shown). Collectively, our data argue that MDA-7/IL-24 augments the antitumor effects of sorafenib against established renal carcinoma tumors.
Discussion
The present studies have focused on developing enhanced therapies for renal carcinoma, a tumor that frequently displays a rapid progression beyond the kidney and with no treatment providing long-term clinical benefit for metastatic disease. To achieve this objective we utilized mda-7/IL-24, which has demonstrated tumor cell-specific killing in a wide variety of tumor cell types.Citation10–Citation13,Citation14 Based on previous observations that specific signaling pathways might provide protection from mda-7/IL-24-induced toxicity, we have now explored combinatorial approaches targeting multiple specific cytoprotective signaling pathways in combination with mda-7/Il-24 on human RCCs both in vitro and in vivo. The studies in this manuscript highlight combinations of clinically relevant protein and lipid kinase inhibitors that increase MDA-7/IL-24 toxicity and we also identify a means of enhancing the therapeutic delivery of MDA-7/IL-24 for RCC using a tropism-modified Ad.5/3.
Serotype 5 human adenoviruses bind to and infect, human cells through the Coxsackie and adenovirus receptor (CAR); a protein whose expression has been shown in several studies to be significantly reduced in primary human RCCs in culture and in situ (reviewed in ref. Citation10–Citation13, Citation21 and Citation22). Thus, delivery of therapeutic genes by this method to RCCs was hampered by the relative inefficiency of this gene delivery method. Multiple laboratories are attempting to solve the relative inefficiency of serotype 5 adenovirus infections in RCCs and other malignancies by modifying specific sequences within virus capsid proteins that directly associate with CAR, i.e., they are applying targeting strategies to enhance viral infectivity via CAR-independent pathways. Initial studies from several groups modified the infective viral capsid “knob” to bind instead to surface integrin proteins whose expression is enhanced upon transformation (RGD modification) or by insertion into the knob of multiple lysine residues (pK7) which will increase viral interaction with cells by electrostatic effects on the cell's surface.
Subsequently, more subtle modifications to the proteins expressed as part of the viral capsid knob have been made with the development of viruses expressing chimeric knobs containing components of different serotype viruses. For example, in some of these viruses, the infective capsid knobs from serotype 3 adenoviruses were incorporated into the adenovirus type 5 knob and our present studies have demonstrated that modified serotype 5/3 knob adenoviruses are able to achieve enhanced gene transduction into low-CAR containing RCCs in vitro and in vivo.Citation33,Citation34
Infection of RCCs with a dose of Ad.5/3-mda-7 virus particles that caused modest levels of toxicity ∼48 h after exposure correlated with activation of the JNK1/2 and p38 MAPK pathways as well as PERK. This treatment, in parallel, suppressed ERK1/2, AKT, mTOR and p70 S6K signaling. Multiple studies using a number of toxic stimuli document that prolonged JNK1-3 and/or p38 MAPK activation in a wide variety of cell types can trigger cell death.Citation35–Citation37 It is also well established that the balance between the readouts of ERK1/2 and JNK1-3 signaling can represent a general key homeostatic mechanism that regulates cell survival versus cell death processes.Citation35–Citation37 Prior in vitro studies using RCCs treated with GST-MDA-7 have demonstrated that JNK1-3 and p38 MAPK signaling represent key pro-apoptotic signals generated by mda-7/IL-24 exposure. Inhibition of MEK1/2 and PI3K enhanced MDA-7/IL-24-induced JNK1-3 activation that was causal in promoting cell killing, but combined inhibition of MEK1/2 and PI3K and mTOR did not lead to a further activation of the JNK pathway yet caused enhanced tumor cell killing. Inhibition of the JNK pathway in vitro, however, suppressed the toxicity of MEK1/2 and PI3K and mTOR inhibition when combined with Ad.5/3-mda-7.
As previously noted using bacterially synthesized GST-MDA-7 protein, infection of RCCs with Ad.5/3-mda-7 caused activation of the CD95 death receptor that played a key role in MDA-7/IL-24 lethality. Inhibition of PI3K + MEK1/2 or use of the multi-kinase inhibitor sorafenib enhanced Ad.5/3-mda-7 lethality that correlated with increased activation of CD95 and decreased levels of MCL-1 and c-FLIP-s. Overexpression of c-FLIP-s reduced Ad.5/3-mda-7 lethality by >80% but only suppressed the enhanced killing of Ad.5/3-mda-7 cells treated with PI3K + MEK1/2 inhibitors by ∼40%. Overexpression of BCL-XL or dominant negative caspase 9 blocked not only Ad.5/3-mda-7 toxicity but also the potentiation of Ad.5/3-mda-7 toxicity by PI3K + MEK1/2 inhibitors. Thus, although PI3K + MEK1/2 inhibitors did increase MDA-7/IL-24-induced CD95 activation our data argues that the primary site of PI3K + MEK1/2 inhibitor action is by promoting further mitochondrial dysfunction due to the loss of MCL-1 and BCL-XL expression.
Activation of PERK by MDA-7/IL-24 has been linked to increased tumor cell killing and in RCCs we noted that the majority of PERK activation caused by GST-MDA-7 treatment was dependent on CD95-induced activation of PERK rather than GST-MDA-7-induced inactivation of BiP/GRP78 with the subsequent activation of the BiP/GRP78 chaperone PERK.21 Expression of MDA-7/IL-24 increased BiP/GRP78 levels, an effect that was suppressed by inhibition of multiple protective signaling pathways and the loss of BiP/GRP78 induction correlated with increased activation of PERK and eIF2α and with enhanced tumor cell killing. Overexpression of BiP/GRP78 has been linked in several systems to chemotherapy resistance and to tumor promotion.
Sorafenib (Bay 43-9006, Nexavar®; a Raf family kinase inhibitor) is a multi-kinase inhibitor that was originally developed as an inhibitor of Raf-1, a component of the ERK1/2-MEK1/2 pathway, but which was subsequently shown to inhibit multiple other kinases, including class III tyrosine kinase receptors such as platelet-derived growth factor, vascular endothelial growth factor receptors 1 and 2, c-Kit and Flt3.Citation38,Citation39 Antitumor effects of sorafenib in RCC have been ascribed to anti-angiogenic actions of this agent through inhibition of pro-angiogenic growth factor receptors.Citation40,Citation41 Several groups, including ours, have shown in vitro that sorafenib kills human tumor cells at concentrations well below the maximum achievable dose (Cmax) of 15–20 µM, through a mechanism involving downregulation of the antiapoptotic BCL-2 family member MCL-1; MCL-1 has a short half-life of ∼30 min. In these studies sorafenib-mediated MCL-1 downregulation occurred through a translational rather than a transcriptional or post-translational process that was mediated by endoplasmic reticulum (ER) stress signaling.Citation36 This effect is almost identical to the mechanism of action by which MDA-7/IL-24 suppresses MCL-1 levels.Citation42 Sorafenib enhanced Ad.5/3-mda-7 lethality and enhanced CD95 activation. Sorafenib and expression of MDA-7/IL-24 combined to abolish MCL-1 and BCL-XL expression. As sorafenib is an FDA-approved therapeutic modality for renal carcinoma patients we determined whether the drug could promote MDA-7/IL-24-induced antitumor effects in vivo. Treatment of established RCC tumors with either sorafenib or Ad.5/3-mda-7 suppressed RCC tumor growth in vivo. Combined exposure to MDA-7/IL-24 and sorafenib caused a prolonged suppression of RCC tumor growth and promoted recruitment of immune cells into the tumor.
In conclusion, the present studies demonstrate that clinically relevant protein and lipid kinase inhibitors enhance the toxicity of MDA-7/IL-24 in vitro and in vivo. Further studies will be required to determine whether MDA-7/IL-24 (Ad.5/3-mda-7) is able to enhance the tumor controlling effects of sorafenib in patients with metastatic renal carcinoma. Based on the poor prognosis of patients with advanced renal cancer, defining novel and effective therapeutic approaches is clearly a priority. Considering the safety of sorafenib and mda-7/IL-24 as single agents, future studies of this combination could prove of immense value in treating RCC patients whose disease has progressed beyond the renal capsule.Citation11,Citation17,Citation18,Citation43,Citation44
Materials and Methods
Materials.
Phospho-/total-ERK1/2, Phospho-/total-JNK1-3, Phospho (S473)-/total-AKT, Phospho-/total-p38 MAPK, Phospho-/total mTOR, Phospho-/total p70 S6K and P-PERK antibodies were purchased from both Cell Signaling Technologies (Worcester, MA) and from Santa Cruz Biotechnology (Santa Cruz, CA). Sorafenib tosylate was purchased from Eton Bioscience Inc., (San Diego, CA). The plasmid to express mTOR was from Addgene. The JNK inhibitor peptide (JNK IP), PI-103 and PX-866 were supplied by Calbiochem (San Diego, CA) as a powder, dissolved in sterile DMSO and stored frozen under light-protected conditions at −80°C. A498 cells were from the ATCC (Bethesda, MD). UOK121LN cells were supplied by Dr. W.M. Lineham (NIH, Bethesda, MD). A498 cells are re-purchased every 6–9 months. No other cell line authentication was performed by the authors. Trypsin-EDTA, DMEM and RPMI medium and penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). Other reagents were of the highest quality commercially available.Citation10–Citation13,Citation21,Citation27,Citation28,Citation35,Citation36
Methods.
Generation of Ad.5-mda-7 or Ad.5/3-mda-7. Recombinant type 5 and 5/3 adenoviruses to express MDA-7 (Ad.5-mda-7 and Ad.5/3-mda-7), control (Ad.cmv empty vector) were generated as described in reviewed in ref. Citation10–Citation13, Citation16, Citation27, Citation28 and Citation33–Citation36.
Cell culture and in vitro infection/exposure of cells to Ad.mda-7 and drugs. All cell lines were cultured at 37°C (5% (v/v CO2)) in vitro using RPMI supplemented with 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 36 h after plating were treated with MDA-7/IL-24 and/or various drugs, as indicated. In vitro small molecule inhibitor treatments were from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). For Ad infection, cells were infected 12 h after plating and the expression of the recombinant viral transgene allowed to occur for 24 h prior to any additional experimental procedure. Cells were not cultured in reduced serum media during any study.Citation10–Citation13,Citation27,Citation28
Cell treatments, SDS-PAGE and western blot analysis. Cells were treated with various drug concentrations and viral multiplicities of infection, as indicated in the Figure legends. For SDS PAGE and immunoblotting, cells were lysed in either a nondenaturing lysis buffer and prepared for immunoprecipitation as described in reference 36 or in whole-cell lysis buffer (0.5 M Tris- HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue), and the samples were boiled for 30 min. After immunoprecipitation, samples were boiled in whole cell lysis buffer. The boiled samples were loaded onto 10–14% SDS-PAGE and electrophoresis was run overnight. Proteins were electrophoretically transferred onto 0.22 µm nitrocellulose and immunoblotted with indicated primary antibodies against the different proteins.Citation10–Citation13,Citation27,Citation28
Recombinant adenoviral vectors; infection in vitro. We generated and purchased previously noted recombinant adenoviruses to express constitutively activated and dominant negative (dn) AKT and MEK1 proteins, dn caspase 9, XIAP, c-FLIP-s, CRM A and BCL-XL (Vector Biolabs, Philadelphia, PA). Note: UOK121LN cells were infected with these adenoviruses at an approximate m.o.i. of 400. A498 cells, even at 400 m.o.i. were not infected by serotype 5 adenovirus. Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.Citation10–Citation13,Citation27,Citation28
Detection of cell death by trypan blue, hoechst, TUNEL and flow cytometric assays. Cell death assays were carried out as described in references Citation10–Citation13, Citation21 and Citation25–Citation36.
Plasmid transfection. Plasmid DNA (0.5 µg/total plasmid transfected) was diluted into 50 µl of RPMI growth media that lacked supplementation with FBS or with penicillin-streptomycin. Lipofectamine 2000 reagent (1 µl) (Invitrogen, Carlsbad, CA) was diluted into 50 µl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The two solutions were then mixed together and incubated at room temperature for 30 min. The total mix was added to each well (4-well glass slide or 12-well plate) containing 200 µl growth media that lacked supplementation with FBS or with penicillinstreptomycin. The cells were incubated for 4 h at 37°C, after which time the media was replaced with RPMI growth media containing 5% (v/v) FBS and 1X pen-strep.
Flank inoculation of A498 cells.
Athymic female NCr-nu/nu mice (NCI-Fredrick) weighing ∼20 g, were used for this study. Mice were maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, Washington, DC, the US Department of Health and Human Services, Washington, DC, and the National Institutes of Health, Bethesda, MD. A498 cells were cultured in RPMI supplemented with 5% (v/v) fetal calf serum and 100 µg/ml (1% v/v) penicillinstreptomycin. Cells were incubated in a humidified atmosphere of 5% (v/v) CO2 at 37°C. Rear flank injection of 5 × 106 A498 cells 10 µl of medium was performed over 10 min. Tumors (∼150 mm3) were permitted to form over the following month. Adenoviral vectors, Ad.5/3-mda-7 or Ad.5/3-cmv were administered ∼1 month after tumor cell implantation into animals. Viral vectors (Ad.5/3-mda-7 or Ad.cmv, 1 × 109 p.f.u.) suspended in 2 µl of PBS were delivered by slow infusion over a 6 min period.
Immunohistochemistry and staining of fixed tumor sections. Postsacrifice, tumors were fixed in OCT compound (Tissue Tek); cryostat sectioned (Leica) as 12 µm sections. Nonspecific binding was blocked with a 2% (v/v) Rat Sera, 1% (v/v)m Bovine Sera, 0.1% (v/v) Triton X100, 0.05% (v/v) Tween-20 solution then sections were stained for cell signaling pathway markers: Animals that received recombinant adenoviruses were monitored twice weekly for survival or signs of disease. Other animals were sacrificed 14–140 days as indicated after adenovirus injection. Post-sacrifice, mouse tumors were fixed in OCT compound (Tissue Tek); cryostat sectioned (Leica) as 12 µm sagittal sections. Nonspecific binding was blocked with a 2% (v/v) Rat Sera, 1% (v/v) Bovine Sera, 0.1% (v/v) Triton X100, 0.05% (v/v) Tween-20 solution then sections were stained for cell signaling pathway markers: MDA-7/IL-24; Ki67; CD11b; CD11c; cleaved caspase 3. Staining was performed as described in references Citation10–Citation13, Citation21, Citation26 and Citation36.
Data analysis. Comparison of the effects of various treatments was performed using one-way analysis of variance and a two-tailed Student's t-test. Differences with a p value of <0.05 were considered statistically significant. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Experiments shown are the means of multiple individual points from multiple experiments (±SEM).
Abbreviations
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| JNK | = | c-Jun NH2-terminal kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| MDA-7/IL-24 | = | melanoma differentiation associated gene-7/interleukin-24 |
| PERK | = | protein kinase R-like endoplasmic reticulum kinase |
| MAPK | = | mitogen activated protein kinase |
| ca | = | constitutively active |
| dn | = | dominant negative |
| EGFR | = | epidermal growth factor receptor |
| IL | = | interleukin |
| PTEN | = | phosphatase and tensin homolog on chromosome ten |
| RCC | = | renal cell carcinoma |
Figures and Tables
Figure 1A–D Infection of RCCs with Ad.5/3-mda-7 causes MDA-7/IL-24 secretion into the growth media that has a potent toxic “bystander” effect via CD95 signaling. (A) A498 or UOK121LN cells were cultured for 24 h and then infected with increasing multiplicities of infection (moi) of Ad.5-cmv; Ad.5/3-cmv; Ad.5-mda-7; Ad.5/3-mda-7. Cells were isolated for viability analyses 48 h after infection as judged in triplicate by trypan blue dye exclusion assay (±SEM, n = 3, #p < 0.05 value less than vehicle treated cells). (B) A498 or UOK121LN cells were cultured for 24 h and then infected with increasing multiplicities of infection (moi) of Ad.5-cmv; Ad.5/3-cmv; Ad.5-mda-7; Ad.5/3-mda-7. Cells were re-plated as single cells and colonies permitted for form for 14 days (±SE M, n = 3). (C) Upper blot: UOK121LN cells were infected with Ad.5-cmv; Ad.5/3-cmv; Ad.5-mda-7; Ad.5/3-mda-7 (50 moi) and the cells and the growth media isolated 48 h after infection. Lysates of infected cells and a portion (5% v/v) of the growth media were subjected to SDS PAGE and immunoblotting to determine the expression of MDA-7/IL-24 (representative, n = 3). Lower blot: UOK121LN cells were infected with Ad.5-cmv; Ad.5/3-cmv; Ad.5-mda-7; Ad.5/3-mda-7 (50 moi) and the cells and the growth media isolated 48 h after infection. Lysates of infected cells were examined for expression of MDA-7/IL-24 in virus-infected cells. Conditioned media was transferred onto UOK121LN cells that had previously been transfected with a scrambled siRNA or an siRNA to knock down MDA-7/IL-24 expression. Forty-eight hour after media transfer cells were isolated, lysed and subjected to SDS PAGE/blotting to determine MDA-7/IL-24 protein levels (representative, n = 3). (D) UOK121LN cells were transfected with either scrambled siRNA molecules (siSCR) or molecules to knockdown expression of CD95 (siCD95) or MDA-7/IL-24 (siMDA-7). Twenty-four hour after transfection cells were infected with Ad.5/3-cmv; Ad.5/3-mda-7 (50 moi) and the cells and the growth media were isolated 48 h after infection. Cell viability in infected cells was determined by trypan blue dye exclusion (n = 3, ± SEM). Conditioned media was placed onto uninfected (but transfected) cells and 48 h later their viability was determined by trypan blue dye exclusion (n = 3, ± SEM). (E) Primary human renal epithelial cells were infected with Ad.5-cmv or Ad.5-mda-7 or Ad.5-mda-7-SP- (Secretion Peptide null, SP-) (50 moi) and 48 h after infection the growth media removed. UOK121LN cells, 24 h after plating, are treated with increasing concentrations of conditioned media. Cell growth was measured by MTT assay 72 h after treatment with conditioned media (n = 2 in sextuplicate, 12 data points total ± SEM). The Inset Part shows MDA-7 expression in primary renal epithelial cells 48 h after Ad.5-mda-7 infection. (F) Conditioned media from infected primary renal epithelial cells was placed onto UOK121LN cells. Cells were isolated 48 h after treatment with conditioned media and viability determined by trypan blue dye exclusion (n = 3, ± SEM).
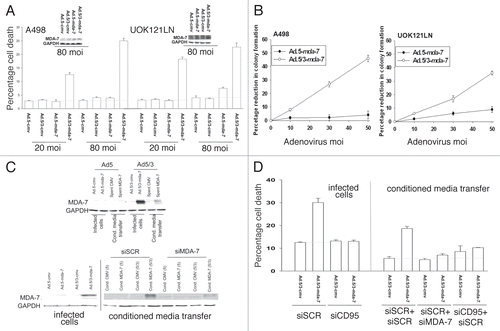
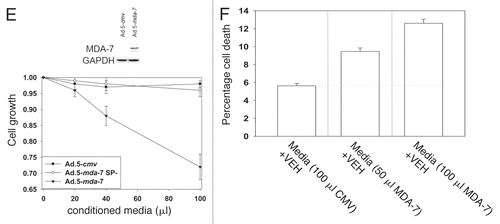
Figure 2 Ad.5/3-mda-7 lethality is enhanced by combined molecular inhibition of PI3K/MEK/mTOR pathways in A498 cells. (A) A498 cells were infected with empty vector control virus (Ad.5/3-cmv) or with viruses expressing MDA-7/IL-24 (Ad.5/3-mda-7); and in parallel transfected with empty vector control plasmid CMV or plasmids to express dominant negative forms of MEK1 or the p85 subunit of PI3K; or scrambled siRNA to siRNA to knockdown mTOR. Twenty-four hours after infection cells were isolated and the phosphorylation/expression of the indicated proteins was determined by immunoblotting (a representative blot from 3 studies is shown). (B and C) A498 cells were infected with empty vector control virus (Ad.5/3-cmv) or with viruses expressing MDA-7/IL-24 (Ad.5/3-mda-7); and in parallel transfected with empty vector control plasmid CMV or plasmids to express dominant negative forms of MEK1 or the p85 subunit of PI3K; or scrambled siRNA to siRNA to knock down mTOR. Forty-eight hours after infection cells were isolated and in (B) cell viability was determined by trypan blue exclusion assay and in (C) total cell numbers were counted in parallel and expressed in proportion to control cell growth (± SEM, n = 3). (D) A498 cells were infected with empty vector control virus (Ad.5/3-cmv) or with viruses expressing MDA-7/IL-24 (Ad.5/3-mda-7); and in parallel transfected with empty vector control plasmid CMV or plasmids to express dominant negative forms of MEK1 or the p85 subunit of PI3K; or scrambled siRNA to siRNA to knock down mTOR. Twelve hours after infection/transfection cells were re-plated as single cells (250–1,000 cells per 60 mm dish) and colonies of >50 cells/colony permitted to form over the following ∼14 days. Data are the means of four plates per study from two independent studies (± SEM).
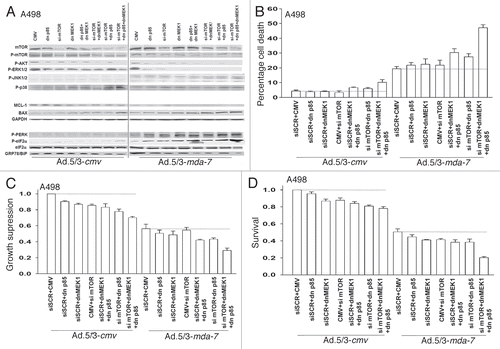
Figure 3 Ad.5/3-mda-7 lethality is enhanced by combined molecular inhibition of PI3K/MEK/mTOR pathways in UOK121LN cells. (A and B) UOK121LN cells were infected with empty vector control virus (Ad.5/3-cmv) or with viruses expressing MDA-7/IL-24 (Ad.5/3-mda-7); and in parallel transfected with empty vector control plasmid CMV or plasmids to express dominant negative forms of MEK1 or the p85 subunit of PI3K; or scrambled siRNA to siRNA to knock down mTOR. Forty-eight hours after infection cells were isolated and in (A) cell viability was determined by trypan blue exclusion assay and in (B) total cell numbers were counted in parallel and expressed in proportion to control cell growth (± SEM, n = 3). (C) UOK121LN cells were infected with empty vector control virus (Ad.5/3-cmv) or with viruses expressing MDA-7/IL-24 (Ad.5/3-mda-7); and in parallel transfected with empty vector control plasmid CMV or plasmids to express dominant negative forms of MEK1 or the p85 subunit of PI3K; or scrambled siRNA to siRNA to knock down mTOR. Twelve hours after infection/transfection cells were re-plated as single cells (250–1,000 cells per 60 mm dish) and colonies of > 50 cells/colony permitted to form over the following ∼14 days. Data are the means of four plates per study from two independent studies (± SEM).
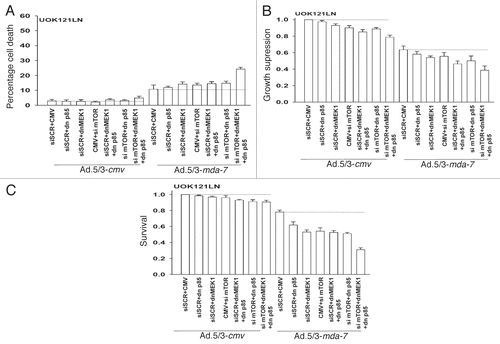
Figure 4 Inhibition of mTOR, PI3K and MEK1/2 signaling using clinically relevant small molecule kinase inhibitors enhances Ad.5/3-mda-7 lethality. (A) A498 cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus vector expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 24 h after infection treated with vehicle (DMSO), PD184352 + PX866 (PD + P X, 1 µM + 100 nM), Rapamycin (Rap, 100 nM) or all three drugs combined. Six hours after drug treatment, cells were isolated and the phosphorylation/expression of the indicated proteins was determined by immunoblotting (a representative blot from 3 studies is shown). (B) A498 cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus vector expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 24 h after infection treated with vehicle (DMSO), PD184352 + PX866 (PD + PX, 1 µM + 100 nM), Rapamycin (Rap, 100 nM) or all three drugs combined. Forty eight hours after infection cells were isolated and cell viability was determined by trypan blue exclusion assay (± SEM, n = 3). (C) UOK121LN cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus vector expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 24 h after infection treated with vehicle (DMSO), PD184352 (1 µM), PX-866 (100 nM), Rapamycin (Rap, 100 nM); PI-103 (1 µM); or drugs combined as indicated. Forty eight hours after infection cells were isolated and cell viability was determined by trypan blue exclusion assay (± SE M, n = 3). (D) A498 and UOK121LN cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus vector expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 24 h after infection treated with vehicle (DMSO), PD184352 (1 µM), PX-866 (100 nM), Rapamycin (Rap, 100 nM); or drugs combined as indicated. Forty eight hour after infection cells were isolated and cell viability was determined by trypan blue exclusion assay (± SE M, n = 3). (E) A498 and UOK121LN cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 24 h after infection treated with vehicle (DMSO) or sorafenib (3 µM). Forty eight hours after infection cells were isolated and cell viability was determined by trypan blue exclusion assay (± SE M, n = 3). Upper Inset: Immunoblots of cells isolated 24 h after addition of sorafenib to virus infected cells (n = 2). (F) A498 and UOK121LN cells were infected with empty adenovirus vector (Ad.5/3-cmv) or an adenovirus expressing MDA-7/IL-24 (Ad.5/3-mda-7) and 12 h after infection treated with vehicle (DMSO) or PD184352 (1 µM), PX-866 (100 nM), Rapamycin (Rap, 100 nM); or drugs combined as indicated or sorafenib (3 µM). Forty eight hours after infection cells were washed free of drugs and colonies permitted to form for 10–14 days (± SE M, n = 3).
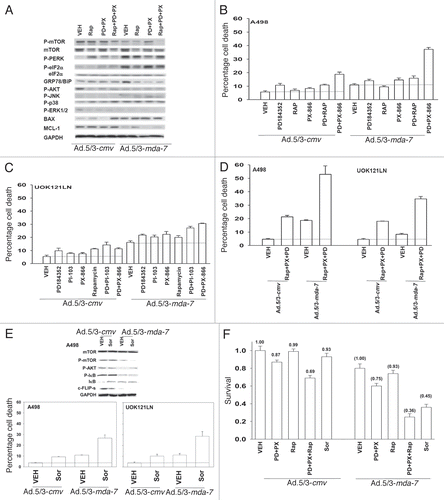
Figure 5 Inhibition of signaling pathways enhances Ad.5-mda-7-induced CD95 activation and facilitates killing independently of CD95 activation. (A) A498 cells were infected with Ad.5/3-cmv or Ad.5/3-mda-7 and in parallel transfected with plasmids to express activated AKT (caAKT); activated MEK1 (caMEK1); activated mTOR (camTOR) and 12 h after infection/transfection treated with vehicle (DMSO), PD184352 (PD, 1 µM) + PX866 (100 nM) + rapamycin (Rap, 100 nM) (PD + PX + Rap) or with JNK inhibitory peptide (JNK-IP, 10 µM). Cells were isolated 48 h after infection and cell viability was determined by trypan blue dye exclusion assay (± SE M, n = 3). (B) UOK121LN cells were infected with Ad.5/3-cmv or Ad.5/3-mda-7 and in parallel infected with type 5 viruses (400 moi) to express c-FLIP-s, CRM A, BCL-XL or dominant negative caspase 9. Twelve hours after infection cells were treated with vehicle (DMSO) or PD184352 (PD, 1 µM) + PX866 (100 nM) (PD + PX). Cells were isolated 48 h after infection and cell viability was determined by trypan blue dye exclusion assay (± SE M, n = 3). (C) A498 cells were infected with Ad.5/3-cmv or Ad.5/3-mda-7 and 24 h after infection cells were treated with vehicle (DMSO) or PD184352 (PD, 1 µM) + PX866 (100 nM) (PD + PX) or sorafenib (Sor, 3 µM). Cells were fixed 6 h after drug exposure and surface plasma membrane CD95 levels determined (± SE M, n = 3). Upper inset: 6 h after drug treatment cells were isolated, CD95 immunoprecipitated and the association of caspase 8 and FADD with CD95 (DISC formation) determined.
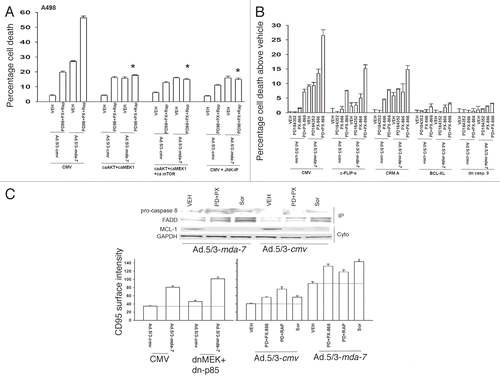
Figure 6 Ad.5/3-mda-7 infection of RCC tumors in vivo exhibits a toxic bystander effect on uninfected RCC tumors. (A) A498 cells were injected into the rear right and left flanks of athymic mice. Tumors of ∼150 mm3 grew over the following 29 days. Animals were segregated into tumor volumes of approximate equivalent mean tumor size and standard error. The tumor on the right flank of the animal was injected with 2 µl (1 × 109 infectious particles) of either Ad.5/3-cmv or Ad.5/3-mda-7. Tumor volumes are measured every two to three days. Two days after the first virus infection, tumors are infected in an identical fashion with adenovirus. The mean volumes of the tumors on each flank are presented as a -Fold increase over the pre-infected volume (defined as 1.00) (n = 2, ± SE M; 6–7 mice per group total). Data are presented for each condition for each animal; animals whose tumors grew to > 2.0 cm3 and had to be humanely sacrificed are indicated in the following Figure. (B) A498 cells were injected into the rear right and left flanks of athymic mice. Tumors of ∼150 mm3 grew over the following 29 days. Animals were segregated into tumor volumes of approximate equivalent mean tumor size and standard error. The tumor on the right flank of the animal was injected with 2 µl (1 × 109 infectious particles) of either Ad.5/3-cmv or Ad.5/3-mda-7. The survival of animals in both groups was plotted as a Kaplan Meir curve (n = 2, ± SE M; 6–7 mice per group total). (C) A498 tumors were isolated 2 days after the second virus infection. Sections (10 microns) were taken and stained for caspase 3 cleavage; Ki67 reactivity (proliferation); MDA-7/IL-24 expression; and Dapi for nuclei. Data are from representative images from multiple tumors shown at X20 magnification. (D) A498 tumors were isolated 2 days after the second virus infection. Sections (10 microns) were taken and stained for CD11c; H&E (morphology); TUNEL (apoptosis); and Dapi for nuclei. Data are from representative images from multiple tumors shown at X20 magnification.

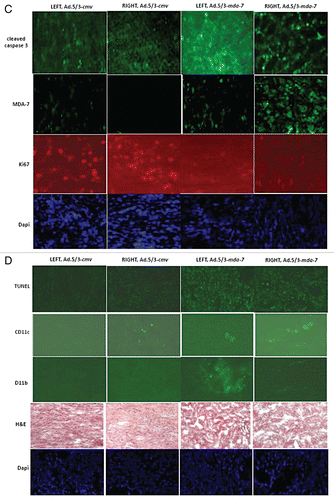
Figure 7 Sorafenib enhances the lethal effects of Ad.5/3-mda-7 in vivo. (A) A498 tumors formed over 29 days in the flank of athymic mice (∼150 mm3). Animals were separated into tumor volumes of approximate equivalent mean tumor size and standard error. Tumors were injected with 2 µl (1 × 109 infectious particles) of either Ad.5/3-cmv or Ad.5/3-mda-7. Animals were treated with sorafenib (20 mg/kg) by oral gavage 24 h after the first virus administration and subsequently for the following 4 days. Tumor volume was noted every 2–3 days (± SE M, n = 5 animals per condition from two experiments). (B) A498 tumors from one experiment (6–8 animals per condition) were treated with virus and sorafenib in an identical fashion as in (A). Tumor volumes were measured every 2–3 days and animals were humanely sacrificed when their tumor volume was ≥ 2.0 cm2. (C) A498 tumors were isolated 2 days after the second virus infection. Sections (10 microns) were prepared and stained for caspase 3 cleavage; MDA-7/IL-24 expression; CD11c; CD11b and Dapi for nuclei. Data are from representative images from multiple tumors shown at x20 magnification. (D) A498 tumors were isolated 2 days after the second virus infection. Sections (10 microns) were taken and stained for Ki67 reactivity (proliferation); H&E (morphology); TUNEL (apoptosis); and Dapi for nuclei. Data are from representative images from multiple tumors shown at x20 magnification.

Table 1 GST-MDA-7 and sorafenib synergize to kill RCCs
Acknowledgements
Support for the present study was provided: to P.D. from PHS grants (P01-CA104177, R01-CA108520, R01-DK52825; R01-CA141703; R01-CA150214), The Jim Valvano “V” foundation and Department of Defense Award (W81XWH-10-1-0009); to P.B.F. from PHS grants (P01-CA104177, R01-CA097318; R01-CA127641; R01-CA134721; R01-CA108520), the Samuel Waxman Cancer Research Foundation (SWCRF) and the National Foundation for Cancer Research (NFCR); to D.T.C. from PHS grant (P01-CA104177). P.D. is The Universal Inc., Professor in Signal Transduction Research. PBF holds the Thelma Newmeyer Corman Chair in Cancer Research at the VCU Massey Cancer Center, VCU, School of Medicine and is a SWCRF Investigator.
References
- Sunela KL, Kataja MJ, Lehtinen ET, Salminen TK, Kujala PM, Virman JP, et al. Prognostic factors and long-term survival in renal cell cancer patients. Scand J Urol Nephrol 2009; 43:454 - 460
- Jiang H, Lin JJ, Su ZZ, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene 1995; 11:2477 - 2486
- Ekmekcioglu S, Ellerhorst J, Mhashilkar AM, Sahin AA, Read CM, Prieto VG, et al. Downregulated melanoma differentiation associated gene (mda-7) expression in human melanomas. Int J Cancer 2001; 94:54 - 59
- Ellerhorst JA, Prieto VG, Ekmekcioglu S, Broemeling L, Yekell S, Chada S, et al. Loss of MDA-7 expression with progression of melanoma. J Clin Oncol 2002; 20:1069 - 1074
- Huang EY, Madireddi MT, Gopalkrishnan RV, Leszczyniecka M, Su Z, Lebedeva IV, et al. Genomic structure, chromosomal localization and expression profile of a novel melanoma differentiation associated (mda-7) gene with cancer specific growth suppressing and apoptosis inducing properties. Oncogene 2001; 20:7051 - 7063
- Parrish-Novak J, Xu W, Brender T, Yao L, Jones C, West J, et al. Interleukins 19, 20 and 24 Signal through two distinct receptor complexes. Differences in receptor-ligand interactions mediate unique biological functions. J Biol Chem 2002; 277:47517 - 47523
- Caudell EG, Mumm JB, Poindexter N, Ekmekcioglu S, Mhashilkar AM, Yang XH, et al. The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol 2002; 168:6041 - 6046
- Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol 2004; 22:929 - 979
- Gupta P, Su ZZ, Lebedeva IV, Ekmekcioglu S, Mhashilkar AM, Yang XH, et al. mda-7/IL-24: multifunctional cancer-specific apoptosis-inducing cytokine. Pharmacol Ther 2006; 111:596 - 628
- Park MA, Walker T, Martin AP, Allegood J, Vozhilla N, Emdad L, et al. MDA-7/IL-24-induced cell killing in malignant renal carcinoma cells occurs by a ceramide/CD95/PERK-dependent mechanism. Mol Cancer Ther 2009; 8:1280 - 1291
- Hamed HA, Yacoub A, Park MA, Eulitt PJ, Dash R, Sarkar D, et al. Inhibition of multiple protective signaling pathways and Ad.5/3 delivery enhances mda-7/IL-24 therapy of malignant glioma. Mol Ther 2010; 18:1130 - 1142
- Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res 2010; 70:1120 - 1129
- Lebedeva IV, Emdad L, Su ZZ, Gupta P, Sauane M, Sarkar D, et al. mda-7/IL-24, novel anticancer cytokine: focus on bystander antitumor, radiosensitization and antiangiogenic properties and overview of the phase I clinical experience. Intl J Oncol 2007; 31:985 - 1007
- Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, et al. Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer Biol Ther 2009; 8:391 - 400
- Su ZZ, Lebedeva IV, Gopalkrishnan RV, Goldstein NI, Stein CA, Reed JC, et al. A combinatorial approach for selectively inducing programmed cell death in human pancreatic cancer cells. Proc Natl Acad Sci USA 2001; 98:10332 - 10337
- Su ZZ, Madireddi MT, Lin JJ, Young CS, Kitada S, Reed JC, et al. The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proc Natl Acad Sci USA 1998; 95:14400 - 14405
- Cunningham CC, Chada S, Merritt JA, Tong A, Senzer N, Zhang Y, et al. Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: a phase I study. Mol Ther 2005; 11:149 - 159
- Fisher PB, Sarkar D, Lebedeva IV, Emdad L, Gupta P, Sauane M, et al. Melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24): novel gene therapeutic for metastatic melanoma. Toxicol Appl Pharmacol 2007; 224:300 - 307
- Lebedeva IV, Su ZZ, Chang Y, Kitada S, Reed JC, Fisher PB. The cancer growth suppressing gene mda-7 induces apoptosis selectively in human melanoma cells. Oncogene 2002; 21:708 - 718
- Su ZZ, Lebedeva IV, Sarkar D, Emdad L, Gupta P, Kitada S, et al. Ionizing radiation enhances therapeutic activity of mda-7/IL-24: overcoming radiation- and mda-7/IL-24-resistance in prostate cancer cells overexpressing the antiapoptotic proteins bcl-xL or bcl-2. Oncogene 2006; 25:2339 - 2348
- Hamed HA, Yacoub A, Park MA, Eulitt P, Sarkar D, Dimitriev IP, et al. OSU-03012 enhances Ad.mda-7-induced GBM cell killing via ER stress and autophagy and by decreasing expression of mitochondrial protective proteins. Cancer Biol Ther 2010; 9:526 - 536
- Yacoub A, Liu R, Park MA, Hamed HA, Dash R, Schramm DN, et al. Cisplatin enhances protein kinase R-like endoplasmic reticulum kinase- and CD95-dependent melanoma differentiation-associated gene-7/interleukin-24-induced killing in ovarian carcinoma cells. Mol Pharmacol 2010; 77:298 - 310
- Gupta P, Walter MR, Su ZZ, Lebedeva IV, Emdad L, Randolph A, et al. BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer Res 2006; 66:8182 - 8191
- Sauane M, Gopalkrishnan RV, Choo Tt, Gupta P, Lebedeva IV, Yacoub A, et al. Mechanistic aspects of mda-7/IL-24 cancer cell selectivity analyzed via a bacterial fusion protein. Oncogene 2004; 23:7679 - 7690
- Yacoub A, Mitchell C, Hong Y, Gopalkrishnan RV, Su ZZ, Gupta P, et al. MDA-7 regulates cell growth and radiosensitivity in vitro of primary (non-established) human glioma cells. Cancer Biol Ther 2004; 3:739 - 751
- Yacoub A, Hamed H, Emdad L, Dos Santos W, Gupta P, Broaddus WC, et al. MDA-7/IL-24 plus radiation enhance survival in animals with intracranial primary human GBM tumors. Cancer Biol Ther 2008; 7:917 - 933
- Yacoub A, Park MA, Gupta P, Rahmani M, Zhang G, Hamed H, et al. Caspase-, cathepsin- and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells. Mol Cancer Ther 2008; 7:297 - 313
- Yacoub A, Gupta P, Park MA, Rhamani M, Hamed H, Hanna D, et al. Regulation of GST-MDA-7 toxicity in human glioblastoma cells by ERBB1, ERK1/2, PI3K and JNK1-3 pathway signaling. Mol Cancer Ther 2008; 7:314 - 329
- Yacoub A, Mitchell C, Brannon J, et al. MDA-7 (interleukin-24) inhibits the proliferation of renal carcinoma cells and interacts with free radicals to promote cell death and loss of reproductive capacity. Mol Cancer Ther 2003; 2:623 - 632
- Sarkar D, Su ZZ, Lebedeva IV, Sauane M, Gopalkrishnan RV, Valerie K, et al. mda-7 (IL-24) Mediates selective apoptosis in human melanoma cells by inducing the coordinated overexpression of the GADD family of genes by means of p38 MAPK. Proc Natl Acad Sci USA 2002; 99:10054 - 10059
- Mhashilkar AM, Stewart AL, Sieger K, Yang HY, Khimani AH, Ito I, et al. MDA-7 negatively regulates the beta-catenin and PI3K signaling pathways in breast and lung tumor cells. Mol Ther 2003; 8:207 - 219
- Chada S, Bocangel D, Ramesh R, Grimm EA, Mumm JB, Mhashilkar AM, et al. mda-7/IL24 kills pancreatic cancer cells by inhibition of the Wnt/PI3K signaling pathways: identification of IL-20 receptor-mediated bystander activity against pancreatic cancer. Mol Ther 2005; 11:724 - 733
- Dash R, Dmitriev IP, Su ZZ, Bhutia SK, Azab B, Rahmani M, et al. Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) improves therapeutic efficacy in low CAR prostate cancer cells. Cancer Gene Ther 2010; 17:447 - 456
- Murakami M, Ugai H, Belousova N, Pereboev A, Dent P, Fisher PB, et al. Chimeric adenoviral vectors incorporating A fiber of human adenovirus 3 efficiently mediate transgene expression into prostate cancer cells. Prostate 2010; 70:362 - 376
- Zhang G, Park MA, Mitchell C, Walker T, Hamed H, Studer E, et al. Multiple cyclin kinase inhibitors promote bile acid-induced apoptosis and autophagy in primary hepatocytes via p53-CD95-dependent signaling. J Biol Chem 2008; 283:24343 - 24358
- Park MA, Zhang G, Martin AP, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther 2008; 7:1648 - 1662
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270:1326 - 1331
- Li N, Batt D, Warmuth M. B-Raf kinase inhibitors for cancer treatment. Curr Opin Investig Drugs 2007; 8:452 - 456
- Flaherty KT. Sorafenib: delivering a targeted drug to the right targets. Expert Rev Anticancer Ther 2007; 7:617 - 626
- Gollob JA. Sorafenib: scientific rationales for single-agent and combination therapy in clear-cell renal cell carcinoma. Clin Genitourin Cancer 2005; 4:167 - 174
- Strumberg D. Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 2005; 41:773 - 784
- Dash R, Richards JE, Su Zz, et al. Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10 related-cytokine. Cancer Res 2010; 70:5034 - 5045
- Sun M, Lughezzani G, Perrotte P, Karakiewicz PI. Treatment of metastatic renal cell carcinoma. Nature Rev Urol 2010; 7:327 - 338
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 2009; 27:3312 - 3318