Abstract
SU11248 is a selective inhibitor of certain protein kinases including VEGFR types 1-3 that are expressed in human breast cancer. The present study determines whether the anti-tumor activity of SU11248 results from the inhibition of angiogenesis, as well as direct anti-proliferation and anti-migration effects on breast tumors. Eight-wk old female mice (C57BL/6) were given SU11248 at 20-40 mg/kg/d in drinking (distilled) water for 4 wks. Control mice received drinking water only. In the 2nd wk, 10^6 E0771 (mouse breast cancer) cells were injected in the left fourth mammary gland. Tumor size was monitored using dial calipers. At the end, tumors were isolated for measuring tumor size and intratumoral microvessel density (IMD) using CD31 immunohistochemistry. SU11248 significantly reduced tumor weight over the control (1.220.28 vs. 3.280.31 g; n=8; P<0.01) and IMD (11110 vs. 1556 IM#/mm^2; P<0.01). RT-PCR indicated that VEGFR1 and R2 were expressed in cultured E0771 cells. VEGF (10 ng/ml) caused a 42% increase in proliferation of E0771 cells, compared to the control (P<0.01; n = 8), and there was a significant decrease in proliferation of E0771 cells treated with VEGF plus SU11248 (10 μmol/L) vs. the control (65%, P<0.01). VEGF caused a 2-fold increase in the proliferation of HUVEC vs. the control (P<0.01; n = 8), but its action was completely eradicated by SU11248. Neither VEGF nor SU11248 had any effect on the proliferation of cultured HASMC. Migration assay showed that SU11248 (10 μmol/L) significantly inhibited the migration of cultured E0771 cells. SU11248 significantly inhibited the proliferation of MCF-7 and MDA-MB-231 cells in a dose-related manner. These findings support the hypothesis that the anti-tumor activity of SU11248 on breast cancer is possibly mediated by targeting the paracrine and autocrine effects of VEGF on breast cancer to suppress tumor angiogenesis, proliferation, and migration.
Introduction
The growth and expansion of a tumor is mainly dependent on angiogenesis, the formation of new capillaries from pre-existing blood vessels. Avascular tumors are those that do not grow beyond a maximum size of 1 to 2 mm3 in the absence of neovascularization, and it may be eliminated by a normal immune system.Citation1 Angiogenesis requires stimulation of vascular endothelial cells through the release of angiogenic factors. Of these, the vascular endothelial growth factor (VEGF) is the most critical regulator in the development of the vascular system and is commonly overexpressed in a variety of human solid tumors including breast cancer.Citation2 In fact, VEGF is involved in vascular permeability, endothelial cell migration and proliferation, as well as vessel maturation. The action of VEGF is accomplished through its ligands, which specifically bind to the following three different cell membrane receptors: VEGFR1 (Flt-1), VEGFR2 (Flk/KDR) and VEGFR3 (Flt-4). The ligand-receptor interaction induces the activation of the VEGF receptor (VEGFR) tyrosine kinase domain, which in turn activates intracellular signaling transduction pathways involved in the regulation of vascular permeability, endothelial cell migration, proliferation and survival and vessel maturation. The well-established role of VEGF in promoting tumor angiogenesis in human cancers has led to the development of agents that target this pathway.
Many VEGF receptor tyrosine kinase inhibitors, such as SU5416 and SU11248, have been developed in clinical trials as an anti-tumor angiogenesis agent for some cancers including breast cancer.Citation3 These tyrosine kinase inhibitors also directly target certain tumor cells.Citation3 Recently, several studies have indicated that VEGF receptors are expressed in some human breast cancer cells and have suggested that VEGF stimulates breast cancer progression mediated by autocrine signaling.Citation4,Citation5 We have recently reported that VEGFR1 and R2 are expressed in a mouse estrogen-receptor positive breast cancer (E0771) cell line.Citation6 However, the autocrine VEGF signaling functions in breast cancer have not been explained adequately. It is critical for cancer therapy to identify specific agents that can intrinsically affect multiple targets.Citation6 We hypothesize that the paracrine effects (especially angiogenesis) and the autocrine effects (proliferation and migration) of VEGF are involved in promoting breast cancer progression. SU11248, a selective inhibitor of certain protein kinases including VEGFR types 1–3, can inhibit the paracrine and autocrine effects of VEGF by targeting both tumor vasculature and breast cancer cells, thereby suppressing the proliferation, migration, tumor angiogenesis and tumor growth of breast cancer tumors.
The present study determines the following: [1] whether oral administration of SU11248 inhibits VEGF expression, angiogenesis and growth of breast cancer (E0771) tumors in an immunocompetent female mouse model; [2] whether SU11248 directly inhibits the proliferation of cultured E0771 cells; [3] whether SU11248 directly inhibits the migration of cultured E0771 cells; [4] whether VEGF and VEGFR1 are differentially expressed in ER-positive (MCF-7) and ER-negative (MDA-MB-231) human breast cancer cells; and [5] whether SU11248 inhibits the proliferation and migration in cultured MCF-7 cells as well as MDA-MB-231 cells.
Results
Oral administration of SU11248 suppresses the progression of breast cancer tumor growth in mice.
We used a mouse breast cancer model that mimics the human disease, in which the mouse breast adenocarcinoma (E0771) cells were injected into the pad of the fourth mammary gland of female immunocompetent mice (C57BL/6). The eight-week old female mice (n = 8) were given SU11248 10 mg/50 ml in drinking (distilled) water for four weeks, and the control group (n = 8) was given regular drinking (distilled) water only. During the second week, all mice were inoculated subcutaneously with E0771 cells. The body weight of the mice was monitored weekly. Tumor size was monitored every other day in two perpendicular dimensions parallel with the surface of the mice using dial calipers. At the conclusion of the experiment, the tumor cross section area was significantly reduced by 48% (p < 0.01) in the SU11248-treated group in contrast to the control group (), which was consistent with a reduction in tumor weight () of the SU11248-treated group compared to the control group (1.22 ± 0.28 vs. 3.28 ± 0.32 g; p < 0.01). Clearly, giving SU11248 at 20 to 40 mg/kg/day in drinking water significantly inhibited the progression of breast cancer tumor growth in female mice by decreasing the tumor size and reducing the growth curve of the breast cancer tumor. However, there was no significant difference in body, heart or kidney weight between the SU11248-treated mice and the control mice.
SU11248 treatment inhibits tumor angiogenesis of breast cancer in mice.
Growth and expansion of tumor mass is mainly dependent on angiogenesis because neovascularization contributes rapid tumor growth by providing an exchange of nutrients, oxygen and paracrine stimulus of the tumor.Citation13 Therefore, in this study, we used a morphometric analysis of immunohistochemical staining for CD31 to determine the effect of SU11248 on tumor angiogenesis of breast cancer. Representative images of CD31 staining of the breast cancer tumors showed that the SU11248-treated tumor () had fewer microvessels than the control tumor (). Morphometric analysis () indicated that SU11248 treatment caused a significant decrease in average microvessel density (AMVD, the number of microvessels per mm2 area) of breast cancer tumors when compared to the control breast cancer tumors (111 ± 10 vs. 155 ± 6 microvessels number per mm2; n = 8; p < 0.01). These results suggest that the pronounced decrease in tumor angiogenesis is associated with the decrease in tumor size found in the SU11248-treated group compared to those in the control group. These findings also indicate that the inhibition of tumor angiogenesis by SU11248 is mediated by targeting the paracrine effects of VEGF on breast cancer tumors.
VEGFR1 (Flt-1) and VEGFR2 (Flk-1) mRNA are expressed in E0771 cells.
We previously reported that Flt-1 and Flk-1 proteins were expressed in a mouse estrogen receptor positive breast cancer (E0771) cell line.Citation6 Reverse transcription-PCR analysis revealed the mRNA expression of mouse VEGF receptor-1 (Flt-1) and VEGF receptor-2 (Flk-1) in cultured E0771 cells (). These results further confirm that VEGF receptor-1 and receptor-2 are expressed in a mouse estrogen receptor positive breast cancer (E0771) cell line. It also suggests that the autocrine effects (proliferation and migration) of VEGF are involved in promoting breast cancer progression.
Effects of VEGF or VEGF plus SU11248 on the proliferation of cultured E0771, HUVEC and HASMC.
3H-thymidine incorporation assay indicated that VEGF (10 ng/ml) caused a 42% increase in the proliferation of cultured E0771 cells (), compared to the control group (p < 0.01; n = 8). There was a significant decrease of 65% in the proliferation of E0771 cells treated with VEGF plus SU11248 (10 µmol/L), as compared to the control (p < 0.01). Clearly, SU11248 blocked the actions of both exogenous and endogenous VEGF on the proliferation of E0771 cells. showed that VEGF (10 ng/ml) caused a 2-fold increase in the proliferation of HUVEC, compared to the control (p < 0.01), but its action was completely eradicated by SU11248 (10 µmol/L). Neither VEGF nor SU11248 had any effect on the proliferation of cultured HASMC that do not express VEGF receptors (). These results suggest that the inhibition of proliferation in cultured E0771 cells by SU11248 is also mediated by targeting the autocrine effects of VEGF on breast cancer tumors.
SU11248 directly inhibits the migration of E0771 cells.
We examined the inhibitory effect of SU11248 on E0771 cell migration using the BD BioCoat Matrigel Invasion Chamber. demonstrated that SU11248 at 10 µmol/L caused a 47% reduction of migrated E0771 cells compared to the control group (n = 8; p < 0.01). These results suggest that SU11248 can directly inhibit the migration of cultured E0771 cells by targeting the autocrine effects of VEGF on breast cancer.
VEGF and VEGFR1 (Flt-1) are expressed differentially in MCF-7 and MDA-MB-231 cells.
The invasive and malignant capacity of human estrogen-receptor negative (MDA-MB-231) breast cancer cells is much higher than that of human estrogen-receptor positive (MCF-7) breast cancer cells.Citation14 VEGF is involved in promoting breast cancer progression.Citation4,Citation5,Citation15 VEGF and its receptors are expressed in MCF-7 and MDA-MB-231 cells.Citation5,Citation16–Citation19 However, it has not been reported whether VEGF and its receptors are expressed differentially in MCF-7 and MDA-MB-231 cells. We examined the expression of VEGF and VEGFR1 in cultured MCF-7 and MDA-MB-231 cells using ELISA assay. shows that VEGF protein is expressed much more in MDA-MB-231 cells than MCF-7 cells (>8-fold; p < 0.01; n = 8). Also, VEGF receptor-1 (flt-1) protein is more highly expressed in MDA-MB-231 cells than MCF-7 cells (>7-fold; p < 0.01; n = 8). demonstrates that 17β-estradiol (5 nmol/L) significantly increases VEGF expression in cultured ER-positive (MCF-7) human breast cancer cells (1,762 ± 56 vs. 1,136 ± 58 pg/mg; p < 0.01; n = 8) in comparison with the control group, but not in ER-negative (MDA-MB-231) human breast cancer cells. These findings suggest that VEGF and its receptors in breast cancer could be the biological marker for breast cancer malignancy and progression. The combination of anti-VEGF and anti-estrogen therapy could be synergistic in the treatment of ER-positive breast cancer.
SU11248 inhibits the proliferation of MCF-7 and MDA-MB-231 cells.
demonstrates that 5 and 10 µmol/L of SU11248 cause a 56% and 77% decrease in the proliferation of ER-positive (MCF-7) human breast cancer cells, respectively; in contrast to the control (p < 0.01; n = 8). Also, 5 and 10 µmol/L of SU11248 cause a 36% and 75% decrease in the proliferation of ER-negative (MDA-MB-231) human breast cancer cells, respectively; in contrast to the control group (p < 0.01; n = 8). Interestingly, SU11248 at 5 µmol/L had a less effect in suppressing the proliferation of MDA-MB-231 cells than MCF-7 cells, while SU11248 at 10 µmol/L had a similar effect in suppressing the proliferation of both MDA-MB-231 and MCF-7 cells.
Discussion
The key novel findings from this study include the following: oral administration of SU11248 causes a significant decrease in tumor growth and intratumoral microvessel density of ER-positive breast cancer tumors in the wild type female mice. (2) The VEGFR1 (Flt-1) and VEGFR2 (Flk-1) mRNA are expressed in a mouse ER-positive breast cancer (E0771) cells. (3) SU11248 directly inhibits the proliferation and migration of cultured E0771 cells. (4) VEGF and VEGFR1 are more significantly expressed in ER-negative (MDA-MB-231) cells than ER-positive (MCF-7) cells. (5) SU11248 causes a dose-related inhibition in the proliferation of cultured MCF-7 and MDA-MB-231 cells.
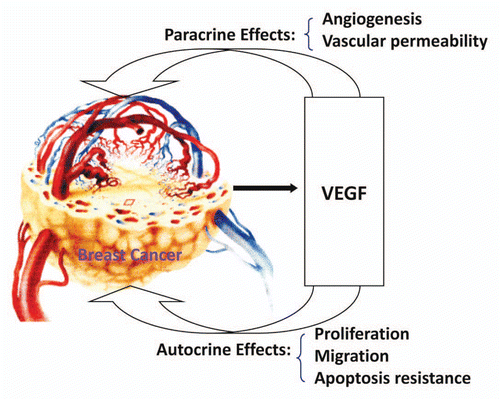
These findings support the hypothesis that the paracrine effects (especially angiogenesis) and the autocrine effects (proliferation and migration) of VEGF are involved in promoting breast cancer progression (). SU11248, a selective inhibitor of certain protein kinases including VEGFR types 1–3, can inhibit the paracrine and autocrine effects of VEGF by targeting both tumor vasculature and breast cancer cells, thereby suppressing the proliferation, migration, tumor angiogenesis and tumor growth of breast cancer tumors.
The mechanisms underlying the development of breast cancer are complex and vary among individuals.Citation20 Based on the identification of the molecular signaling pathways involved in breast cancer development and progression, specific therapy for breast cancer has been developed to target estrogen, type I growth factors (HER-2/neu and epidermal growth factor receptor), angiogenesis, cyclooxigenase-2 and farnesylation.Citation20–Citation22 VEGF is a key angiogenic factor and is upregulated in various breast cancers to promote breast cancer progression.Citation4,Citation5,Citation15 Many VEGF receptor tyrosine kinase inhibitors, such as SU5416 and SU11248, have been developed in clinical trials as an anti-tumor angiogenesis agent for breast cancer.Citation3 In the present study, we investigated the effects of oral administration of SU11248 on tumor growth and tumor angiogenesis of breast cancer in an immunocompetent wild type mouse model (C57BL/6). An important finding of this study is that SU11248 treatment at 20 to 40 mg/kg/day in drinking water significantly suppresses the progression of breast cancer tumor growth and tumor angiogenesis in mice. These results validate that SU11248 can inhibit the paracrine effects of VEGF on breast cancer tumors by suppressing tumor angiogenesis and tumor growth via targeting tumor vasculature.
VEGF has been shown to be highly expressed in breast tumors at levels that are 7-fold higher than normal adjacent tissue.Citation23 Several studies have demonstrated that VEGF receptors are found in human breast cancer epithelial cells.Citation4,Citation5,Citation23–Citation25 We have reported that VEGF and its receptor (Flt-1 and Flk-1) proteins are expressed in a mouse estrogen-receptor positive breast cancer (E0771) cell line.Citation6 The present study confirms that the mRNA of VEGF receptor-1 (Flt-1) and VEGF receptor-2 (Flk-1) are expressed in cultured E0771 cells. In fact, VEGF enhanced the proliferation and migration of breast cancer cells.Citation26,Citation27 These findings suggest that the autocrine effects of VEGF are involved in promoting breast cancer progression. On the other hand, a study that sought to distinguish autocrine and paracine mechanisms of VEGF on breast cancer cells suggested that in 22–24% of cases, VEGF acted in an autocrine manner, whereas in 38–40% of the cases, it acted in a paracrine manner.Citation29 It has also been reported that VEGF is not able to stimulate T-47D breast cancer cell proliferation.Citation24 However, the autocrine effects of VEGF on breast cancer progression have not been rigorously studied. In this study, we demonstrate for the first time that SU11248, a selective inhibitor of certain protein kinases including VEGFR types 1–3, exerts a profound inhibition that suppresses the autocrine effects of VEGF on the proliferation and migration of mouse breast cancer (E0771) cells. We believe that VEGF and its receptors could be the biological marker for breast cancer malignancy and progression. Of significance, SU11248 targets breast cancer tumor vasculature as well as the tumor epithelial cells directly.VEGF and its receptors are expressed in MCF-7 and MDA-MB-231 cells.Citation5,Citation16–Citation19 The invasive and malignant capacity of human estrogen-receptor negative (MDA-MB-231) breast cancer cells is much higher than that of human estrogen-receptor positive (MCF-7) breast cancer cells.Citation14 However, it has not been reported that VEGF and its receptors are more highly expressed in MDA-MB-231 cells than MCF-7 cells. Interestingly, in the present study, we have demonstrated that the protein levels of VEGF and VEGF receptor-1 (Flt-1) in MDA-MB-231 cells are 7-fold higher than those in MCF-7 cells. These results may explain the amplified invasive and malignant capacity of MDA-MB-231 cells. However, the mechanisms of upregulating VEGF and its receptor in MDA-MB-231 cells are not clear. A recent study showed a more than 10-fold increase in nuclear factor-κB (NFκB) activation in MDA-MB-231 cells when compared to MCF-7 cells.Citation28 NFκB is a redox-regulated transcription factor and a sensor for oxidative stress.Citation29 It is highly expressed in numerous malignancies including breast cancer, and it is critically involved in cancer development processes.Citation30 It was reported that NFκB stimulated VEGF expression.Citation31 We previously reported that pyrrolidine dithiocarbamate (PDTC) suppresses VEGF expression, tumor angiogenesis, growth and migration of breast cancer via inhibiting NFκB activation.Citation6 It is possible that MDA-MB-231 cells are under greater oxidative stress than MCF-7 cells, leading to the activation of NFκB and the upregulation of VEGF. In future studies, we will examine the combination of PDTC and VEGF receptor inhibitor SU11248 and its possible capability to synergistically inhibit breast cancer progression.
In the present study, SU11248 at 5 µmol/L has a less effect in suppressing proliferation of MDA-MB-231 cells than MCF-7 cells, while SU11248 at 10 µmol/L has a similar effect in suppressing the proliferation of both MDA-MB-231 and MCF-7 cells. Also, VEGF and its receptors are more highly expressed in MDA-MB-231 cells than in MCF-7 cells. These findings support the hypothesis that higher doses of SU11248 may be necessary to treat ER-negative breast cancers such as MDA-MB-231. Future studies should investigate this phenomena in a wider spectrum using other ER (−) and (+) cell lines. Since 17β-estradiol significantly increased VEGF expression in ER-positive (MCF-7) human breast cancer cells, it will be crucial in the future to investigate the combination of anti-VEGF and anti-estrogen therapy and its synergistic effect in the treatment of ER-positive breast cancer.
In conclusion, our results indicate that oral administration of SU11248, a selective inhibitor of certain protein kinases including VEGFR types 1–3, significantly inhibits tumor growth and tumor angiogenesis of breast cancer in an immunocompetent mouse model using mouse breast cancer (E0771) cells that express estrogen receptor-α (ERα), VEGF receptor-1 (Flt-1) and VEGF receptor-2 (Flk-1). SU11248 directly suppresses the proliferation and migration of cultured mouse breast cancer cells. VEGF and VEGFR1 are more highly expressed in ER-negative (MDA-MB-231) cells than ER-positive (MCF-7) cells. These finding suggest that VEGF and its receptors are important biological markers for breast cancer malignancy and progression. SU11248 effectively inhibits the paracrine and autocrine effects of VEGF by targeting both tumor vasculature and breast cancer cells, thereby suppressing the proliferation, migration, tumor angiogenesis and tumor growth of breast cancer tumors. This work will have important implications for translating anti-VEGF therapy to human breast cancer treatment.
Future studies will address how combination therapies, such as SU11248 plus PDTC and SU11248 plus tamoxifen, may improve breast cancer treatment options.
Materials and Methods
Chemicals and cell lines.
SU11248, Sunitinib, was purchased from LC Laboratories (Woburn, MA). Human VEGF protein was obtained from PROSPECT-TANY TECHNOGENE LTD., (Israel). The hormone, 17-beta-estradiol was purchased from Sigma (St. Louis, MO). The mouse breast cancer cells (E0771), which were originally isolated from an immunocompetant C57BL/6 mouse, were provided by Dr. Sirotnak FM at Memorial Sloan Kettering Cancer Center, New York, NY.Citation7 Human umbilical vein endothelial cells (HUVEC), human aortic smooth muscle cells (HASMC), human estrogen-receptor positive breast cancer (MCF-7) cells and human estrogen-receptor negative breast cancer (MDA-MB-231) cells were purchased from the American Type Culture Collection (Rockville, MD).
Animal protocols.
The protocols were carried out according to the guidelines for the care and use of laboratory animals implemented by the National Institutes of Health and the Guidelines of the Animal Welfare Act, and were approved by the University of Mississippi Medical Center's Institutional Animal Care and Use Committee. Seven week-old female C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, Maine). The mice were allowed to acclimate for 1 week with standard chow diet (Teklad, Harlan Sprague Dawley; Indianapolis, IN) and tap water before beginning the experiment. The eight week old female mice (n = 8) were given SU11248 10 mg/50 ml in drinking (distilled) water for 4 weeks, and the control group (n = 8) was given regular drinking (distilled) water only. Each mouse (20 g) drank 2 to 4 ml of water per day. Therefore, the mice consumed 20 to 40 mg/kg/day of SU11248. During the 2nd week, all mice were inoculated subcutaneously on the left pad of the fourth mammary gland with 100 µl of 106 E0771 cells suspended in phosphate-buffered saline, using a 23-gauge needle. The body weight of the mice was monitored weekly. Tumor size was monitored every other day in two perpendicular dimensions parallel with the surface of the mice using dial calipers. At the end of the experiment, the tumors were removed and weighted for analysis. Then, they were placed into either liquid nitrogen for total protein extraction and nuclear protein extraction or 10% neutral formalin for immunohistological study.
Morphometric analysis of tumor angiogenesis.
The quantification of blood vessels in tumor tissues was determined by our previously reported methods.Citation6,Citation9 Consecutive thin cryosections (5 µm) of OCT compound (Sakura Finetek, Torrance, CA) embedded tumor tissues were fixed in acetone at 4°C for 10 min. After washing in phosphate-buffered saline (PBS), the sections were treated with 3% H2O2 for 10 minutes to block endogenous peroxidase activity, and then blocked with normal rabbit serum. Then, the sections were washed in PBS and incubated with rat anti-mouse CD31 (PECAM-1) monoclonal antibody (BD Pharmingen, San Diego, CA) at a 1:200 dilution overnight at 4°C. Negative controls were incubated with rat serum IgG at the same dilution. All sections were washed in PBS containing 0.05% Tween-20, and then incubated with a 2nd antibody, mouse anti-rat IgG (Vector laboratories, Burlingame, CA), at a 1:200 dilution for 1 hour at room temperature, again followed by washing with PBS containing 0.05% Tween-20. The sections were incubated in a 1:400 dilution of Extravadin Peroxidase (Sigma, St. Louis, MO) for 30 min. After washing in PBS containing 0.05% Tween-20, the sections were incubated in peroxidase substrate (Vector laboratories, Burlingame, CA) for 5 min. The sections were washed in PBS containing 0.05% Tween-20 and were counterstained with hematoxylin. A positive reaction was indicated by brown staining. The microvascular vessels were quantified by manual counting under light microscopy. A microscopic field (0.7884 mm2) was defined by a grid laced in the eye-piece. At least 20 microscopic fields were randomly taken from each tumor for analysis. Any endothelial cell or cell cluster showing antibody staining and clearly separated from an adjacent cluster was considered to be a single, countable microvessel. The value of average microvascular density (AMVD) was determined by calculating the mean of the vascular counts per mm2 obtained in the microscopic fields for each tumor sample.
Reverse transcription polymerase chain reaction.
The total RNA isolation from cultured E0771 cells was performed as previously described.Citation9 The VEGFR1 mRNA and VEGFR2 mRNA of E0771 cells were determined by RT-PCR as previously described.Citation10 Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed with an iScript cDNA synthesis kit from Bio-Rad following the manufacturer's instructions: 2 µg total RNA, 4 µl 5X iScript reaction mix, 1 µl iScript reverse transcriptase in a total volume of 20 µl. The RT mixture was incubated at 25°C for 5 min, 42°C for 30 min, and 4°C for 5 min in a programmable thermal cycler (Bio-Rad). The PCR mixture included 5 µl of the RT mixture, 2 µl sense primer, 2 µl antisense primer and 25 µl Taq mix in a total volume of 50 µl. The amplifications were performed as follows: 40 cycles at 94°C for 5 min/94°C, 0.5 min; 60°C for 0.5 min; and 72°C for 1 min/72°C, 7 min, then 4°C. The following primer sequences were used: VEGFR1 sense 5′-GAG GAG GAT GAG GGT GTC TAT AGG T-3′ and antisense 5′-GTG ATC AGC TCC AGG TTT GAC TT-3′ (115 bp); VEGFR2 sense 5′-GCC CTG CTG TGG TCT CAC TAC-3′ and antisense 5′-CAA AGC ATT GCC CAT TCG AT-3′ (115 bp). The VEGFR1 mRNA and VEGFR2 mRNA were observed by running the RT-PCR products on 2% agarose gel with TAE buffer.
Proliferation assay.
E0771 cells, MCF-7 cells and MDA-MB-231 cells were maintained as monolayer cultures in RPMI Medium 1640 (GIBCO) supplemented with 10% FBS (HyClone), 100 U/ml penicillin, 100 µg/ml streptomycin and 0.25 µg/ml amphotericin B and incubated at 37°C in a humidified 5% CO2/air injected atmosphere. The cell lines of human umbilical vein endothelial cells (HUVEC) and human aortic smooth muscle cells (HASMC) were cultured using M199 media (GIBCO) supplemented with 10% FBS (HyClone), 100 U/ml penicillin, 100 µg/ml streptomycin and 0.25 µg/ml amphotericin B and incubated at 37°C in a humidified 5% CO2/air injected atmosphere. When the monolayer reached approximately 80% confluence, the cells were washed with PBS and incubated with fresh media with 10% FBS in the absence (vehicle control) and presence of VEGF (10 ng/ml), VEGF plus SU11248 (10 µmol/L) or SU11248 (5 and 10 µmol/L) for 18 hrs. 3H-thymidine incorporation assay was used to determine the cell proliferation during the last 6 hours of incubation as previously described.Citation11
Migration assay.
Migration was determined using BD BioCoat Matrigel Invasion Chamber (BD Bioscience Discovery Labware, Sedford, MA) as described in a previous study, in which only invasive cells digested the matrix and moved through the insert membrane.Citation12 1 × 105 E0771 cells per well in 0.5 ml medium (RPMI Medium 1640) were seeded in the matrigel-coated upper compartment (insert) of a Transwell (24-well format, 8 µm pore) in the absence of SU11248 (control) and presence of SU11248 (10 µmol/L) and the medium with 10% FBS was added to the lower part of the well. After overnight incubation at 37°C and 5% CO2, cells on the upper surface of the insert were removed using a cotton wool swab. Migrated cells on the lower surface of the insert were stained using DiffQuit (Dada Behring, Düdinen, Switzerland). The images of migrated cells were taken and the number of migrated cells was counted using a microscope (Leica, Germany) in a 20x objective.
Measurements of protein levels of VEGF and VEGFR1 by ELISA.
Protein levels of VEGF and VEGF receptor-1 in cultured MCF-7 and MDA-MB-231 cells were determined using mouse VEGF and VEGF Flt-1 ELISA kits (R&D Systems, Minneapolis, MN), according to the manufacturer's instructions. The total proteins of cultured MCF-7 or MDA-MB-231 cells were extracted using NE-PER Cytoplasmic Extraction Reagents (Pierce, Rockford, IL), according to the manufacturer's protocol. Protein levels of VEGF and VEGFR1 (Flt-1) in cultured MCF-7 and MDA-MB-231 cells were determined in the absence (vehicle control) and presence of 17beta-estradiol (5 nmol/L) for 18 hrs. The total protein concentration of cultured MCF-7 and MDA-MB-231 cells was determined using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). The protein concentrations of VEGF and VEGFR1 were normalized and expressed as pictograms per milligram of total cellular protein.
Statistical analyses.
All determinations were performed in duplicated sets. Where indicated, data is presented as mean ± SE. Statistically significant differences in mean values between the two groups were tested by an unpaired Student's t-test. ANOVA was used to analyze the differences between two groups with multiple comparisons. A value of p < 0.05 was considered statistically significant. All statistical calculations were performed using SPSS software (SPSS Inc., Chicago, IL).
Figures and Tables
Figure 1 The inhibition of the progression of breast cancer growth by SU11248 in the immunocompetent female mice (C57BL/6) allografted with mouse breast cancer (E0771) cells. SU11248 treatment at 20 to 40 mg/kg/d in drinking (distilled) water significantly reduced a growth curve of breast cancer monitored by the tumor cross section area (A, p < 0.01; n = 8), tumor weight (B, p < 0.01) and tumor size (C, p < 0.01), compared to the control group.
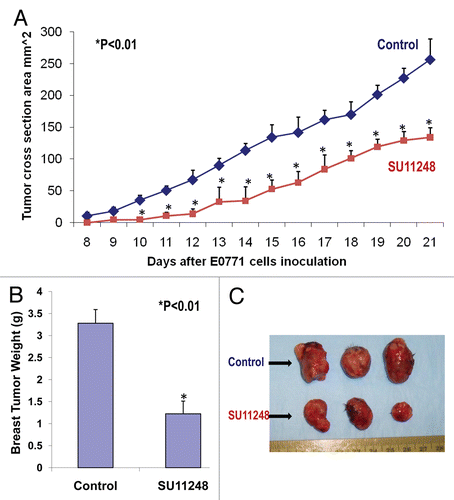
Figure 2 The digital images of CD31 immunohistochemistry staining in OCT-embedded cryosections of mouse breast cancer tumors obtained from the control and SU11248-treated mice (A). Sections were incubated with rat anti-mouse CD31 antibody followed by mouse antirat IgG (Vector laboratories, Burlingame, CA), Extravadin Peroxidase (Sigma, St. Louis, MO) and peroxidase substrate (Vector laboratories, Burlingame, CA). Sections were counterstained with hemotoxylin. The brown staining indicated microvascular vessels. Morphometric analysis (B) indicated that SU11248 treatment caused a significant decrease in average microvessel density (AMVD, the number of microvessels per mm2 area) of breast cancer tumors compared to the control breast cancer tumors (155 ± 6 vs. 111 ± 10 microvessels number per mm2; n = 8; p < 0.01).
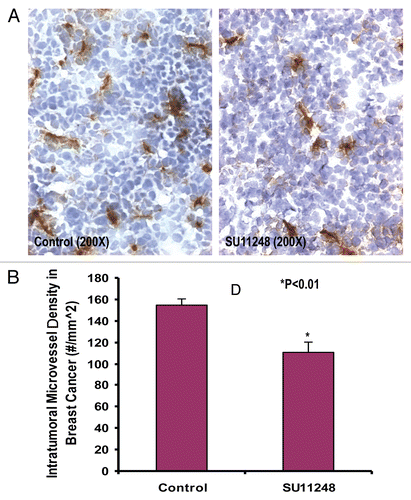
Figure 3 The mRNA expression of mouse VEGF receptor-1 (Flt-1) and VEGF receptor-2 (Flk-1) in cultured E0771 cells by reverse transcriptase-PCR analysis. (A) shows Flt-1 bands (115 bp). (B) shows Flk-1 bands (115 bp).
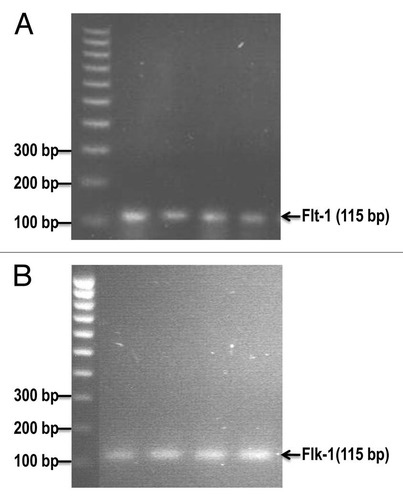
Figure 4 Effects of VEGF or VEGF + SU11248 on the proliferation of cultured mouse breast cancer (E0771) cells, human umbilical vein endothelial cells (HUVEC) and human aortic smooth muscle cells (HAS MC). VEGF (10 ng/ml) caused a 42% increase in the proliferation of E0771 cells, compared to the control (p < 0.01; n = 8) and there was a significant decrease in the proliferation of E0771 cells treated with VEGF plus SU11248 vs. the control (65%, p < 0.01) (A). VEGF caused a 2-fold increase in the proliferation of HUVEC vs. the control (p < 0.01; n = 8), but its action was completely abolished by SU11248 (B). Neither VEGF nor SU11248 exerted any effect on the proliferation of cultured HAS MC (1) that do not express VEGF receptors (C).
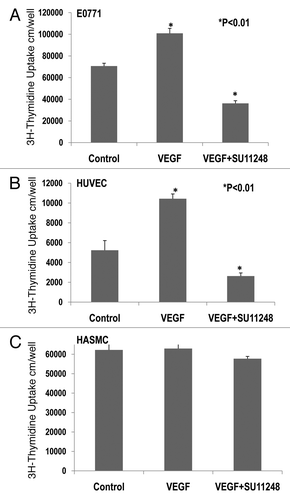
Figure 5 The light microscope images of the invaded E0771 cells on the outside of Matrigel inserts (A: Control; B: SU11248). VEGF receptor inhibitor, SU11248 at 10 µmol/L, significantly inhibited the migration of mouse breast cancer cells (E0771) in Matrigel by 47%, compared to the control group (61 ± 3 vs. 32 ± 2 invaded cell numbers/mm2; p < 0.01; n = 8).
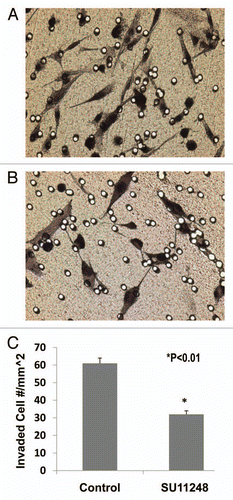
Figure 6 The expression of VEGF and VEGFR1 in cultured ER-positive (MCF-7) and ERnegative (MDA-MB-231) human breast cancer cells. ELISA assay showed that VEGF protein was expressed much more in ER-negative (MDA-MB-231) human breast cancer cells than in ER-positive (MCF-7) human breast cancer cells (>8-fold; p < 0.01; n = 8). Also, VEGF receptor-1 (flt-1) protein was more highly expressed in ER-negative (MDA-MB-231) human breast cancer cells than ER-positive (MCF-7) human breast cancer cells (>7-fold; p < 0.01; n = 8).
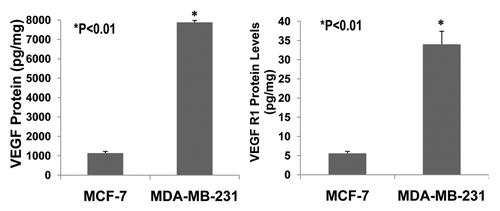
Figure 7 Effects of 17β-estradiol on the expression of VEGF protein in cultured ER-positive (MCF-7) and ER-negative (MDA-MB-231) human breast cancer cells. ELISA assay showed that 17 β-estradiol (5 nmol/L) significantly increased VEGF expression in cultured ER-positive (MCF-7) human breast cancer cells (1,762 ± 56 vs. 1,136 ± 58 pg/mg; p < 0.01; n = 8) compared to the control group, but not in ER-negative (MDA-MB-231) human breast cancer cells.
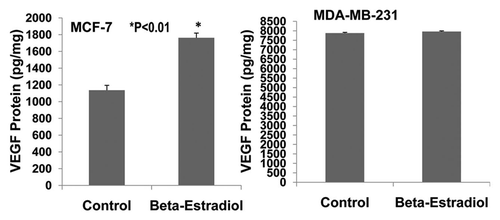
Figure 8 Effects of SU11248 on the proliferation of cultured ER-positive (MCF-7) and ER-negative (MDA-MB-231) human breast cancer cells. 3H-thymidine incorporation showed that 5 and 10 µmol/L of SU11248 caused a 56% and 77% decrease in the proliferations of ER-positive (MCF-7) human breast cancer cells, respectively; in contrast to the control (p < 0.01; n = 8). 5 and 10 µmol/L of SU11248 caused a 36% and 75% decrease in the proliferations of ER-negative (MDA-MB-231) human breast cancer cells, respectively; in contrast to the control (p < 0.01; n = 8).
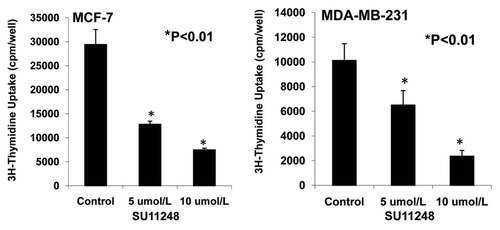
Figure 9 Mechanisms of VEGF in promoting breast cancer progression.
Acknowledgements
This work was supported by the National Institute on Alcohol Abuse and Alcoholism Grant AA-013821 and the National Heart, Lung and Blood Institute Grant HL-51971.
References
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285:1182
- Ferrara N, Gerber HP, LeCouter J. The biological properties of VEGF and its recepters. Nat Med 2003; 9:669 - 676
- Morabito A, De Maio E, Di Mio M, Normanno N, Perrone F. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist 2006; 11:753 - 764
- Brown LF, Berse B, Jackman RW, Tognazzi K, Guiddi AJ, Dvorak HF, et al. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in breast cancer. Hum Pathol 1995; 26:86 - 91
- Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, et al. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT-1. PLOS Med 2007; 4:1101 - 1115
- Gu JW, Young E, Busby B, Covington J, Tan W, Johnson JW. Oral administration of pyrrolidine dithiocarbamate (PDTC) inhibits VEGF expression, tumor angiogenesis and growth of breast cancer in female mice. Cancer Biol Ther 2009; 8:514 - 521
- Sirotnak FM, Otter GM, Schmid FA. Markedly improved efficacy of edatrexate compared to methotrexate in a high-dose regimen with leucovorin rescue against metastatic murine solid tumor. Cancer Res 1993; 53:587 - 591
- Maione P, Gridelli C, Troiani T, Ciardiello F. Combining targeted therapies and drugs with multiple targets in the treatment of NSCLC. Oncologist 2006; 11:274 - 284
- Tan W, Bailey AP, Shparago M, Busby B, Covington J, Gu JW, et al. Chronic alcohol consumption stimulates VEGF expression, tumor angiogenesis and progression of melanoma in mice. Cancer Biol Ther 2007; 6:1211 - 1217
- Shih SC, Robinson GS, Perruzzi CA, Calvo A, Desai K, Green JE, et al. Molecular profiling of angiogenesis markers. Am J Pathol 2002; 161:35 - 42
- Gu JW, Bailey AP, Sartin A, Makey I, Brady AL. Ethanol stimulates tumor progression and expression of vascular endothelial growth factor in chick embryos. Cancer 2005; 103:422 - 431
- Fife RS, Sledge GW Jr. Effects of doxycycline on in vitro growth, migration and gelatinase activity of breast carcinoma cells. J Lab Clin Med 1995; 125:407 - 411
- Folkman J. Angiogenesis and breast cancer. J Clin Oncol 1994; 12:441
- Zuo Y, Shields SK, Chakraborty C. Enhanced intrinsic migration of aggressive breast cancer cells by inhibition of Rac1 GTPase. Biochem Biophys Res Commun 2006; 15:361 - 367
- Hu Z, Fan C, Livasy C, He X, Oh DS, Ewend MG, et al. A compact VEGF signature associated with distant metastases and poor outcome. BMC Med 2009; 7:9
- Ruohola JK, Valve EM, Karkkainen MJ, Joukov V, Alitalo K, Harkonen PL. Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol Cell Endocrinol 1999; 149:29 - 40
- Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, et al. Vascular endothelial growth factor is an autocrine survival factor for neuroplin-expressing breast carcinoma cells. Cancer Res 2001; 61:5736 - 5740
- Buteau-Lozano H, Ancelin M, Lardeux B, Malanini J, Perrot-Applanat M. Transcriptional regulation of vascular endothelial growth factor by estradiol and tamoxifen in breast cancer cells: a complex interplay between estrogen receptors α and β. Cancer Res 2002; 62:4977 - 4984
- Wang S, Liu Q, Zhang Y, Liu K, Yu P, Liu K, et al. Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol Cancer 2009; 8:81
- Gasparini G, Longo R, Torino F, Morabito A. Therapy of breast cancer with molecular targeting agents. Ann Oncol 2005; 16:26 - 28
- Rosen LS, Louise H, Chap L. Targeting signal transduction pathways in metastatic breast cancer: a comprehensive review. Oncologist 2010; 15:216 - 235
- Anderson WF, Chatterjee N, Ershler WB, Brawley OW. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology and End Results database. Breast Cancer Res 2002; 76:27 - 36
- Yoshiji H, Gomez DE, Shibuya M, Thorgeirsson UP. Expression of vascular endothelial growth factor, its receptor and other angiogenic factors in human breast cancer. Cancer Res 1996; 56:2013 - 2016
- Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H. Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ 2001; 12:129 - 135
- Kallergi G, Markomanolaki H, Giannoukaraki V, Papadaki MA, Strati A, Lianidou ES, et al. Hypoxia-inducible factor-1α and vascular endothelial growth factor expression in circulating tumor cells of breast cancer patients. Breast Cancer Res 2009; 11:84
- Liang Y, Brekken PA, Hyder SM. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of antihormones. Endocr Relat Cancer 2006; 13:905 - 919
- Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res 2002; 62:7203 - 7206
- Yan D, Qin N, Zhang H, Liu T, Yu M, Jiang X, et al. Expression of TNFα leader sequence renders MCF-7 tumor cells resistant to the cytotoxicity of soluble TNFα. Breast Cancer Res Treat 2009; 116:91 - 102
- Li N, Karin M. Is NFkappaB the sensor of oxidative stress?. FASEB J 1999; 13:1137 - 1143
- Bharti A, Aggarwal BB. Nuclear factor-kappaB and cancer: its role in prevention and therapy. Biochem Pharmacol 2002; 64:883 - 888
- Karin M. Nuclear factor-kappaB in cancer development and progression Nature 2006; 441:431 - 436