Abstract
We have recently proposed a new model for understanding tumor metabolism, termed: "The Autophagic Tumor Stroma Model of Cancer Metabolism". In this new paradigm, catabolism (autophagy) in the tumor stroma fuels the anabolic growth of aggressive cancer cells. Mechanistically, tumor cells induce autophagy in adjacent cancer-associated fibroblasts via the loss of caveolin-1 (Cav-1), which is sufficient to promote oxidative stress in stromal fibroblasts. To further test this hypothesis, here we created human Cav-1 deficient immortalized fibroblasts using a targeted sh-RNA knock-down approach. Relative to control fibroblasts, Cav-1 deficient fibroblasts dramatically promoted tumor growth in xenograft assays employing an aggressive human breast cancer cell line, namely MDA-MB-231 cells. Co-injection of Cav-1 deficient fibroblasts, with MDA-MB-231 cells, increased both tumor mass and tumor volume by ~4-fold. Immuno-staining with CD31 indicated that this paracrine tumor promoting effect was clearly independent of angiogenesis. Mechanistically, proteomic analysis of these human Cav-1 deficient fibroblasts identified > 40 protein biomarkers that were upregulated, most of which were associated with i) myofibroblast differentiation, or ii) oxidative stress/hypoxia. In direct support of these findings, the tumor promoting effects of Cav-1 deficient fibroblasts could be functionally suppressed (nearly 2-fold) by the recombinant over-expression of SOD2 (superoxide dismutase 2), a known mitochondrial enzyme that de-activates superoxide, thereby reducing mitochondrial oxidative stress. In contrast, cytoplasmic soluble SOD1 had no effect, further highlighting a specific role for mitochondrial oxidative stress in this process. In summary, here we provide new evidence directly supporting a key role for a loss of stromal Cav-1 expression and oxidative stress in cancer-associated fibroblasts, in promoting tumor growth, which is consistent with "The Autophagic Tumor Stroma Model of Cancer". The human Cav-1 deficient fibroblasts that we have generated are a new genetically tractable model system for identifying other suppressors of the cancer-associated fibroblast phenotype, via a genetic "complementation" approach. This has important implications for understanding the pathogenesis of triple negative and basal breasts cancers, as well as tamoxifen-resistance in ER+ breast cancers, which are all associated with a Cav-1 deficient "lethal" tumor micro-environment, driving poor clinical outcome.
Introduction
The tumor micro-environment contains a diverse array of cell types, including endothelial cells, pericytes, smooth muscle cells, macrophages, mesenchymal stem cells and stromal fibroblasts, among others.Citation1,Citation2 Interestingly, in this context, it appears that the stromal fibroblasts undergo re-programming, through their interactions with cancer cells, taking on a more myo-fibroblastic phenotype.Citation3 These cells are now more commonly referred to as cancer-associated fibroblasts, and have the ability to promote tumor growth and metastasis, although the exact mechanism(s) still remain largely unknown.Citation4,Citation5 These findings have important implications for both the diagnosis and the treatment stratification of cancer patients.Citation6
In accordance with these assertions, we and others have recently identified a loss of stromal caveolin-1 (Cav-1) in the cancer associated fibroblast compartmentCitation7 as a single independent predictor of clinical outcome.Citation8–Citation12 For example, a loss of stromal Cav-1 predicts early tumor recurrence, lymph node metastasis and tamoxifen-resistance in breast cancer patients. Importantly, the predictive value of stromal Cav-1 was independent of epithelial marker status, and was effective in all the major sub-types of breast cancer, including ER+, PR+, HER2+ and triple negative (ER−, PR−, HER2−) breast cancer patients.Citation11 In triple negative (TN) patients, which is one of the most lethal types of breast cancer, stromal Cav-1 effectively distinguished between low-risk and high-risk patients.Citation10 In TN patients with high stromal Cav-1, their overall survival was >75% up to 12 years postdiagnosis. 10 In striking contrast, in TN patients with absent stromal Cav-1, their overall survival was <10% at 5 years post-diagnosis.Citation10 Thus, a loss of stromal Cav-1 is a key predictor of a “lethal” tumor micro-environment.Citation13 Similar results were also obtained in DCISCitation9 and prostate cancer patients,Citation14 indicating that a loss of stromal Cav-1 may also play a role in tumor initiation and progression, in a variety of different epithelial cancer types.
Our most recent results indicate that a loss of stromal Cav-1 induces oxidative stress and autophagy in the tumor microenvironment, which then drives the local production of recycled high-energy nutrients that cancer cells can use to “fuel” their anabolic growth.Citation15–Citation17 We have termed this new paradigm “The Autophagic Tumor Stroma Model of Cancer.”Citation18,Citation19 In this context, cancer cells use oxidative stress as a “weapon” to extract nutrients from adjacent stromal cells (via autophagy), reflecting a true host-parasite relationship.Citation20 Thus, it would be predicted that alleviation of oxidative stress in the tumor micro-environment could inhibit or reduce tumor growth.Citation21 We have now tested this hypothesis experimentally.
Here, we have developed a new xenograft system for modeling the lethality of a loss of stromal Cav-1. More specifically, we see that a loss of stromal Cav-1 in human cancer associated fibroblasts dramatically promotes the growth of triple negative breast cancer cells (MDA-MB-231), increasing both tumor mass and tumor volume by ∼4-fold, without any increase in angiogenesis. We also show that we can significantly revert this phenotype by reducing oxidative stress in the tumor micro-environment. This was achieved via the recombinant overexpression of mitochondrially-targeted super-oxide dismutase (SOD2), in Cav-1 deficient cancer associated fibroblasts. As such, this new xenograft model provides a genetically tractable system for dissecting the key factors that govern the lethality of a Cav-1 deficient “pro-oxidative” tumor micro-environment.
Results
A new cellular model to mimic a lethal tumor microenvironment.
A loss of stromal Cav-1 is associated with early tumor recurrence, metastasis and tamoxifen-resistance, resulting in poor clinical outcome in human breast cancer patients.Citation8–Citation11,Citation22 Thus, we sought to develop a new model system to better study the lethality of a Cav-1 negative tumor micro-environment.
For this purpose, we stably-transduced an immortalized human fibroblast cell line (hTERT-BJ1 cells) with two different sh-RNA vectors targeting Cav-1. shows that both of these vectors successfully decreased the expression of the Cav-1 protein product by >95%. Importantly, the control sh-RNA vector did not affect Cav-1 protein expression.
A loss of stromal Cav-1 is sufficient to dramatically increase tumor growth.
We have previously demonstrated that a loss of stromal Cav-1 is particularly lethal in triple negative breast cancers.Citation10,Citation11 For example, in triple negative patients with a loss of stromal Cav-1, their 5 year survival rate is less than 10%.Citation10,Citation11 In striking contrast, in triple negative patients with high stromal Cav-1, their 12 year survival rate is greater than 75%.Citation10,Citation11
As a consequence, we used a triple negative basal breast cancer cell line, namely MDA-MB-231 cells, to evaluate the effects of a loss of stromal Cav-1 in vivo in a murine xenograft model. Control and Cav-1-deficient fibroblasts were co-injected into the flanks of immuno-deficient (nude) mice, along with equal amounts of MDA-MB-231 cells. After 4.5 weeks, the tumors were harvested, and subjected to a detailed analysis.
Remarkably, a loss of Cav-1 in human fibroblasts is sufficient to dramatically promote the growth of MDA-MB-231 cells. shows that targeted downregulation of Cav-1 drives an ∼4-fold increase in tumor mass and an ∼4-fold increase in tumor volume.
Thus, a loss of Cav-1 in the tumor stromal compartment is indeed sufficient to promote the growth of aggressive human breast cancer derived tumor cells.
The tumor-promoting effects of a loss of stromal Cav-1 are independent of tumor angiogenesis.
One possible explanation for the tumor promoting effects of a loss of stromal Cav-1 might be that they efficiently stimulate an increase in tumor angiogenesis. To address this issue, frozen tissue sections derived from xenografted tumors were subjected to immuno-staining with a well-established vascular marker, namely CD31.
shows that a loss of stromal Cav-1 does not have a significant effect on tumor vascularization. The number of vessels per field (vascular density) was virtually identical in both tumors grown with control and Cav-1-deficient fibroblasts. Representative images are shown in .
Proteomic analysis of Cav-1 deficient fibroblasts provides evidence for the onset of a myofibroblast phenotype and mitochondrial oxidative stress.
In order to mechanistically dissect the tumor promoting activity associated with human Cav-1 knockdown fibroblasts, they were subjected to an extensive unbiased proteomic analysis, as detailed in the Materials and Methods section.
Interestingly, Cav-1 deficient fibroblasts showed the upregulation of 15 gene products associated with the myo-fibroblast phenotype, including numerous proteins associated with collagen synthesis and processing (COL1A1, COL6A1/2, P4HA1/2, HSP47, PLOD1), muscle-specific proteins (VIM, MYL6, TPM1/4), and other components of the extracellular matrix (LGALS1/3, CRTAP) ().
In addition, in Cav-1 deficient fibroblasts, we observed the upregulation of 21 gene products associated with oxidative stress and hypoxia, including glycolytic enzymes (LDHA, GAPDH), mitochondrial components involved in ROS production (NDUFA5, NDUFS3, UQCRFS1, ETFB), enzymes that function as anti-oxidants (PRDX1, PRDX4, TXNDC5) or that fuel the production of reduced glutathione (ANPEP, IDH2, GLUD1), as well as factors that are involved in oxidative-stress induced DNA repair (XRCC6BP1) (). In accordance with the idea that oxidative stress induces autophagy/mitophagy, we observed the upregulation of factors associated with the autophagic destruction of mitochondria (DNM1L), and lipid/protein catabolism (DMGDH, PSME1). The upregulation of CDC42 is consistent with the activation of ROS production via NADPH oxidase.
The overexpression of DMGDH (dimethylglycine dehydrogenase) is also consistent with oxidative stress. DMGDH functions in the catabolism of choline, catalyzing the oxidative demethylation of dimethylglycine to form sarcosine. Arul Chinnaiyan and colleagues recently identified sarcosine as a metabolic biomarker for lethal prostate cancer.Citation23 Interestingly, re-analysis of their data sets directly shows that the increase in sarcosine directly parallels a loss of reduced glutathione, indicative of an oxidative tumor micro-environment. Thus, increased sarcosine is a biomarker for oxidative stress, and is associated with advanced prostate cancer and the development of metastatic disease.
We also see that four calcium-binding proteins are upregulated (CALU, CALR, RCN1, S100A13), suggesting that there may be dys-regulation of calcium homeostasis, in Cav-1 deficient fibroblasts (). Importantly, oxidative stress is known to be associated with the disruption of calcium homeostasis.Citation24,Citation25 Again, this may be due to ROS-related mitochondrial dysfunction, as mitochondria play a key role in maintaining calcium homeostasis.Citation26–Citation28
The upregulation of both myofibroblast and oxidative stress markers is consistent with many previous studies showing that oxidative stress is indeed sufficient and/or required for the induction of the myofibroblast phenotype.Citation6,Citation29–Citation33 In other words, oxidative stress is sufficient to induce the myofibroblast differentiation program.Citation34
Importantly, many of the proteins that were induced by Cav-1 knock-down in fibroblasts are highly expressed in the tumor stroma of human breast cancer patients, and are associated with tumor recurrence or metastasis (see ).
Proteomic analysis of eNOS transfected fibroblasts identifies SOD2 as a potential stromal tumor suppressor.
To further study the functional effects of oxidative stress on the phenotypic behavior of human fibroblasts, we previously generated hTERT-BJ1 fibroblasts stably-overexpressing eNOS.Citation15 The overexpression of eNOS phenocopies many of the effects we see due to loss of Cav-1, as Cav-1 normally functions as an inhibitor of NOS, thereby preventing NO (nitric oxide) production.Citation15 In this model, NO over-production is toxic for mitochondria, resulting in mitochodrial oxidative stress.Citation15 Thus, these eNOS-fibroblasts were also subjected to proteomic analysis.
Interestingly, eNOS-fibroblasts show a similar proteomic profile () as we observe here with Cav-1 knock-down fibroblasts (), with the simultaneous upregulation of both myofibroblast markers and proteins associated with oxidative stress/hypoxia. The eNOS proteomic data is also consistent with our previous analyses of several other Cav-1 deficient fibroblastic cell lines (murine Cav-1 [-/-] knock-out fibroblasts and murine mesenchymal stem cells; or HIF-alpha and IKBKE-transfected hTERT-BJ1 human fibroblasts, which also lack Cav-1).Citation20,Citation35,Citation36 Thus, the induction of mitochondrial oxidative stress with eNOS is indeed sufficient to induce a proteomic profile similar to the one we observed due to a loss of Cav-1. This observation fits nicely with the well-established idea that a loss of Cav-1 leads to increased NO production.Citation37,Citation38
However, unlike Cav-1 deficient fibroblasts (), eNOS overexpressing fibroblasts did not promote tumor growth (). Thus, we hypothesized that eNOS overexpressing fibroblasts may have upregulated a “tumor suppressor protein” to allow them to adjust or adapt to a very high-level of mitochondrial oxidative stress, thereby “repressing” their tumor promoting activity.
Interestingly, one candidate stromal tumor suppressor gene is mitochondrial SOD2, which is upregulated in eNOS fibroblasts (). SOD2 localizes to mitochondria and detoxifies super-oxide, resulting in decreased oxidative stress. SOD2 is also transcriptionally upregulated in human breast cancer tumor stroma (), but it is not associated with either tumor recurrence or metastasis, consistent with a potential stromal tumor suppressor role.
The tumor-promoting effects of a loss of stromal Cav-1 are specifically related to mitochondrial oxidative stress in the tumor micro-environment.
We have shown that cancer cells induce oxidative stress in adjacent fibroblasts, resulting in the activation of an autophagic program in the tumor micro-environment, which results in the lysosomal degradation of Cav-1 and the production of recycled nutrients to literally “feed” cancer cells.Citation15–Citation17,Citation39 Also, we have previously demonstrated that a loss of stromal Cav-1 in fibroblasts is sufficient to induce ROS production and oxidative stress, indicating that a loss of Cav-1 provides a feed-forward mechanism for promoting oxidative stress and the autophagic program.Citation15–Citation17,Citation20 We have attributed this increase in ROS production to mitochondrial disfunction.Citation15–Citation19,Citation35,Citation40 Consistent with these findings, our proteomic and functional analysis of eNOS overexpressing fibroblasts identified SOD2 as a candidate stromal tumor suppressor gene ( and and ), due to its ability to combat mitochondrial oxidative stress. SOD2 (super oxide dismutase 2) is an enzyme that is specifically localized to mitochondria and detoxifies super-oxide, resulting in decreased oxidative stress.
Thus, here we investigated whether a genetic reduction in oxidative stress, via SOD2 overexpression, could revert the tumor-promoting phenotype of Cav-1 deficient fibroblasts. For this purpose, we used Cav-1 deficient fibroblasts as a genetically tractable system, and stably transduced them with SOD2 (). Note that SOD2 transduced fibroblasts show an ∼2-fold increase in SOD2 protein expression.
Importantly, shows that recombinant expression of mitochondrially-targeted SOD2 is sufficient to significantly revert the tumor promoting effects of a Cav-1 deficient tumor micro-environment, resulting in a near 2-fold reduction in tumor volume. In contrast, recombinant expression of cytoplasmic soluble SOD1 is not sufficient to revert the tumor promoting effects of a loss of Cav-1, further highlighting the specific role of mitochondrial oxidative stress in this process ().
Thus, these studies provide proof-of-principal that Cav-1 deficient hTERT fibroblasts can be used successfully as a genetically tractable system to identify the key factors that govern the tumor-promoting “lethal” effects of a Cav-1 deficient tumor micro-environment.
Discussion
Here, we have developed a new genetically tractable model system for identifying the genetic factors that govern the tumor promoting effects of cancer-associated fibroblasts. We have previously identified a loss of stromal Cav-1 in the cancer-associated fibroblast compartmentCitation7 as a new biomarker of a lethal tumor microenvironment.Citation8–Citation11,Citation18 To better understand the lethality associated with a loss of stromal Cav-1, we created human immortalized fibroblast cell lines specifically lacking Cav-1, by using a targeted sh-RNA approach. By using MDA-MB-231 cell xenografts, as a model for human triple negative breast cancer, we show that a loss of stromal Cav-1 increases both tumor mass and tumor volume by ∼4-fold, without any increases in tumor angiogenesis. This is consistent with our previous hypothesis that oxidative stress and autophagy in cancer associated fibroblasts produces recycled high-energy nutrients to directly feed cancer cells, without the need for blood vessels/vascularization.Citation21 We have termed this new idea “The Autophagic Tumor Stroma Model of Cancer Metabolism”Citation21 or “Battery-Operated Tumor Growth”.Citation18 In this sense, oxidative stress and autophagy in the tumor micro-environment function as a “battery” or “fuel source” to promote tumor growth.Citation18
A key finding related to these studies is that oxidative stress in cancer associated fibroblasts leads to mitochondrial dysfunction, ROS production and the autophagic destruction of mitochondria, in stromal fibroblasts.Citation15–Citation17,Citation20 This also then drives the “Reverse Warburg Effect”, whereby autophagic stromal fibroblasts undergo aerobic glycolysis under normoxic conditions, producing energy-rich metabolites (such as lactate, pyruvate and ketones) to feed cancer cells.Citation18,Citation19,Citation35,Citation40 Based on these findings, it would be predicted that alleviation of mitochondrial oxidative stress in Cav-1 deficient stromal fibroblasts could potentially prevent tumor growth, by cutting off the fuel supply to adjacent cancer cells.Citation21
In support of this hypothesis, here we directly overexpressed SOD2 in Cav-1 deficient fibroblasts. Importantly, our results indicate that recombinant expression of mitochondrially targeted SOD2 is indeed sufficient to significantly revert the tumor promoting phenotype of Cav-1 deficient fibroblasts. These findings provide further experimental evidence for the idea that mitochondrial oxidative stress in the tumor micro-environment can play a critical positive role in promoting tumor growth.
Undoubtedly, this new genetically tractable model system for cancer associated fibroblasts will help identify other key genetic “suppressors” that can block their tumor promoting properties.
Our current findings are consistent with many previous studies that identified SOD2 as a candidate tumor suppressor gene.Citation41–Citation53 However, all these previous studies focused exclusively on the expression of SOD2 in epithelial cancer cells and not on SOD2 expression in the tumor micro-environment. Thus, SOD2 may also function as a stromal tumor suppressor gene, by lowering oxidative stress in the tumor micro-environment.
Interestingly, in Cav-1 deficient fibroblasts, we observed the upregulation of collagen VI protein expression (COL6A1, COL6A2) (); it was among the highest upregulated protein products (up to 3.3-fold). A previous analysis of collagen gene expression in the stroma of human breast cancer patients showed that collagen VI (COL6A1) is the only collagen gene that is upregulated specifically in breast cancer patients that are prone to lymph node metastasis.Citation40 Thus, collagen VI may also be implicated in the lethal tumor micro-environment created by a loss of stromal Cav-1 expression.
Consistent with our current findings, other studies have implicated collagen VI accumulation in metabolic dys-regulation (associated with diabetes/insulin resistance), and during adipose tissue associated hypoxia, fibrosis and inflammation.Citation54–Citation56 Conversely, genetic deletion of collagen VI gene(s), in the MMTV-PyMT mouse model, severely reduces both the formation of mammary dysplastic lesions, as well as frank mammary tumors.Citation57,Citation58 In this context, it appears that collagen VI activates WNT/β-catenin signaling, via the stabilization of β-catenin.Citation57,Citation58 This provides another paracrine mechanism by which a loss of stromal Cav-1 could promote tumor growth, via the upregulation of collagen VI and other extracellular matrix components, in the tumor/stromal micro-environment.
Materials and Methods
Cell cultures.
Human immortalized fibroblasts (hTERT-BJ1) and human breast cancer cells (MDA-MB-231-GFP) were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum in a 37°C, 5% CO2 incubator. MDA-MB-231 cells were the generous gift of Dr. A. Fatatis (Drexel University, Philadelphia, PA). hTERT-BJ1 fibroblasts stably-expressing eNOS, and the corresponding vector alone control, were as we previously described in reference Citation15.
sh-RNA silencing and retroviral infection.
sh-RNA control and two pre-designed sh-RNAs targeting nucleotides 383–403 (5′-GCT GAG CGA GAA GCA AGT GTA-3′) or 660-680 (5′-TGG GCA GTT GTA CCA TGC ATT-3′) of the CAV1 mRNA (NM_001753.3) were obtained from Invitrogen and were subcloned into the pQCXIP-GFP retroviral vector (Clontech, Inc.). The sh-RNA negative control contains an insert that forms a hairpin structure that is predicted not to target any known vertebrate gene (Invitrogen, Inc.). For retroviral infection, vectors were transiently transfected into the amphotropic Phoenix packaging cell line, using a modified calcium phosphate method. Forty-eight hours post-transfection, the viral supernatant was collected, 0.45 µm sterile filtered, and added to the target cells. Two infection cycles were carried out (every 12 hours) with hTERT-BJ1 cells. Effective knockdown of CAV1 was determined by western blot analysis of FACS-sorted GFP-positive cells.
Recombinant expression of SOD2.
hTERT-BJ1 Cav-1 knock-down cells were transduced with a lenti-viral vector encoding SOD2 (Human superoxide dismutase 2, mitochondrial; Accession #'s Y00985.1/NM_000636.2), with puromycin resistance (pReceiver-I0569-Lv105) or with the vector alone (pReceiver-Lv105), as a critical negative control (GeneCopoeia, Inc.). The same strategy was also used to overexpress SOD1 (Human superoxide dismutase 1, soluble; Accession # X02317), with the construct pReceiver-K2710-Lv105 (GeneCopoeia, Inc.).
Human tumor xenograft assay.
Athymic Ncr-nu/nu mice (7–9 weeks old) were purchased from Taconic. For each injection, 106 MDA-MB-231 cells and 300,000 hTERT-BJ1 fibroblasts in 100 µl of sterile PBS were injected subcutaneously into the flanks of the nude mice. Two flank injections were performed per mouse. Tumors weights and volumes were then measured at 4.5 weeks post-injection, essentially as we previously described in reference 20, 36, 39 and 59.
Quantitation of tumor angiogenesis.
Immunohistochemical staining for CD31 was performed on frozen tumor sections using a 3-step biotin-streptavidin-horseradish peroxidase method. Frozen tissue sections (6 µm) were fixed in 2% paraformaldehyde in PBS for 10 min and washed with PBS. After blocking with 10% rabbit serum the sections were incubated overnight at 4°C with rat anti-mouse CD31 antibody (BS Biosciences) at a dilution of 1:200, followed by biotinylated rabbit anti-rat IgG (1:200) antibody and streptavidin-HRP. Immunoreactivity was revealed with 3, 3′-diaminobenzidine. For quantitation of vessels, CD31-positive vessels were enumerated in 4–6 fields within the central area of each tumor using a x20 objective lens and an ocular grid (0.25 mm2 per field). The total number of vessels per unit area was calculated using Image J and the data was represented graphically.
Proteomic analysis.
2-D DIGE (two-dimensional difference gel electrophoresis)Citation60 and mass spectrometry protein identification were run by Applied Biomics (Hayward, CA). Image scans were carried out immediately following the SDS-PAGE using Typhoon TRIO (Amersham BioSciences) following the protocols provided. The scanned images were then analyzed by Image QuantTL software (GE-Healthcare), and then subjected to in-gel analysis and cross-gel analysis using DeCyder software version 6.5 (GE-Healthcare). The ratio of protein differential expression was obtained from in-gel DeCyder software analysis. The selected spots were picked by an Ettan Spot Picker (GE-Healthcare) following the DeCyder software analysis and spot picking design. The selected protein spots were subjected to in-gel trypsin digestion, peptides extraction, desalting and followed by MALDI-TOF/TOF (Applied Biosystems) analysis to determine the protein identity.
Intersection of fibroblast proteomics with the transcriptome of human breast cancer tumor stroma.
We speculated that the proteomic profiles obtained from Cav-1 deficient and eNOS-transfected fibroblasts might overlap with the transcriptional stromal profiles obtained from human breast caners. To test this hypothesis, we obtained the transcriptional profiles of a large data set of human breast cancer patientsCitation61 whose tumors were subjected to laser-capture micro-dissection, to selectively isolate the tumor stroma. Based on this data set,Citation61 we then generated three human breast cancer stromal genes lists:Citation40
Tumor stroma vs. normal stroma list. Compares the transcriptional profiles of tumor stroma obtained from 53 patients to normal stroma obtained from 38 patients. Gene transcripts that were consistently upregulated in tumor stroma were selected and assigned a p value, with a cut-off of p < 0.05 (contains 6,777 genes) (discussed in ref. Citation40).
Recurrence stroma list. Compares the transcriptional profiles of tumor stroma obtained from 11 patients with tumor recurrence to the tumor stroma of 42 patients without tumor recurrence. Gene transcripts that were consistently upregulated in the tumor stroma of patients with recurrence were selected and assigned a p value, with a cut-off of p < 0.05 (contains 3,354 genes) (discussed in ref. Citation40).
Lymph-node (LN) metastasis stroma list. Compares the transcriptional profiles of tumor stroma obtained from 25 patients with LN metastasis to the tumor stroma of 25 patients without LN metastasis. Gene transcripts that were consistently upregulated in the tumor stroma of patients with LN metastasis were selected and assigned a p value, with a cutoff of p < 0.05 (contains 1,182 genes) (discussed in ref. Citation40).
These three gene lists were then individually intersected with the proteomic profiles of Cav-1 deficient and eNOS-transfected fibroblasts.
Figures and Tables
Figure 1 Targeted knock-down of Cav-1 protein expression in hTERT-fibroblasts. To dissect the role of Cav-1 in promoting the growth of triple negative breast cancers, we have created a matched set of hTERT-immortalized human fibroblast cell lines (from parental hTERT-BJ1), either expressing an shRNA targeting Cav-1 or a control shRNA. This retroviral vector also contains GFP, so transduced cells were recovered by FACS sorting. Successful knock-down of Cav-1 was verified by western blot analysis. The expression of beta-actin is shown as a control for equal protein loading. Ctl, control sh-RNA; sh-Cav1, harboring sh-RNA targeting Cav-1.
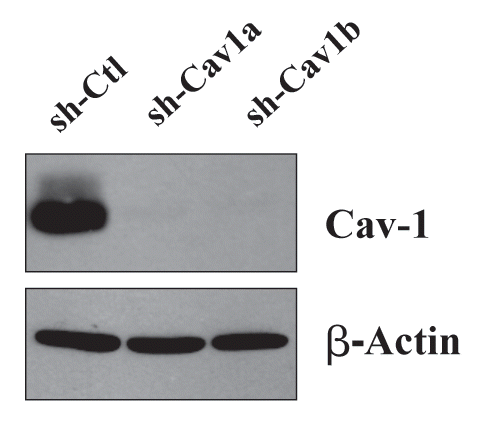
Figure 2 Targeted knock-down of Cav-1 in stromal fibroblasts dramatically promotes breast cancer tumor growth. Control or Cav-1 knock-down fibroblasts (300,000 cells) were co-injected with MDA-MB-321 cells (1 million cells) in the flanks of nude mice. After 4.5 weeks post-injection, the tumors were harvested. Note that relative to control fibroblasts, Cav-1 knock-down fibroblasts increased tumor mass by ∼4-fold (A) and increased tumor volume by ∼4-fold (B). An asterisk indicates that p ≤ 0.01. Fibroblasts injected alone did not form tumors. MDA-MB-231 cells injected alone, behaved as MDA-231 cells injected with control fibroblasts. N = 20 flank injections for each experimental group. Ctl, control sh-RNA; sh-Cav1, harboring sh-RNA targeting Cav-1.
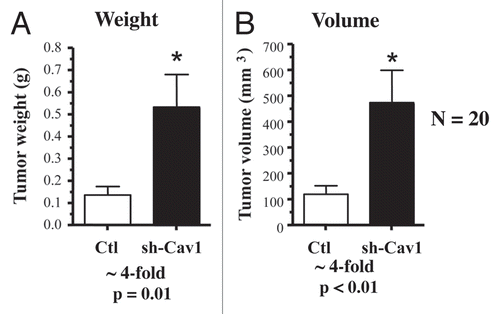
Figure 3 Targeted knock-down of Cav-1 in stromal fibroblasts does not affect tumor angiogenesis. Frozen sections from the tumors were cut and immuno-stained with anti-CD31 antibodies, and vessel density was quantitated (A). Note that no significant increases in vessel density were observed, suggesting that the tumor promoting effects of the Cav-1 knock-down fibroblasts we observe are independent of angiogenesis (n.s., not significant). Representative images are shown in (B). Ctl, control sh-RNA; KD, harboring sh-RNA targeting Cav-1 (knock-down).
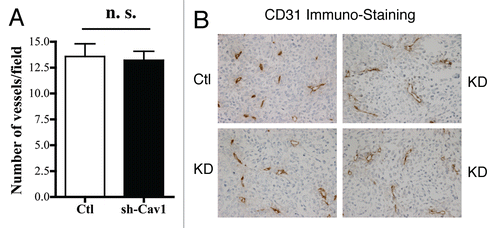
Figure 4 Recombinant overexpression of eNOS in fibroblasts does not promote tumor growth. Control or eNOS-overexpressing fibroblasts (300,000 cells) were co-injected with MDA-MB-321 cells (1 million cells) in the flanks of nude mice. After 4 weeks post-injection, the tumors were harvested. Note that no significant differences were noted between control fibroblasts and eNOS-overexpressing fibroblasts. N = 10 flank injections for each experimental group. n.s., not significant. EV, empty vector; eNOS, stably overexpressing eNOS. (A) tumor weight; (B) tumor volume.
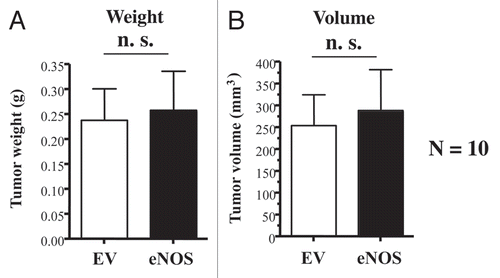
Figure 5 Mitochondrial SOD2 significantly reverts the tumor promoting phenotype of Cav-1 deficient fibroblasts. We have previously shown that loss of Cav-1 increases ROS production in stromal fibroblasts. To combat the resulting oxidative stress, we stably overexpressed SOD2 in Cav-1 knock-down fibroblasts, using a lenti-viral vector with puromycin resistance. Cav-1 knock-down cells were transfected with the empty vector alone, in parallel. Then, these two fibroblast lines were co-injected with MDA-MB-231 cells into the flanks of nude mice. Note that overexpression of SOD2, a mitochondrial enzyme that deactivates super-oxide, is sufficient to reduce the tumor promoting effects of Cav-1 knock-down fibroblasts by nearly 2-fold. An asterisk indicates that p = 0.01 (B). The overexpression of SOD2 was validated by western blot analysis (A). The expression of b-actin is shown as a control for equal protein loading. N ≥ 9 flank injections for each experimental group.
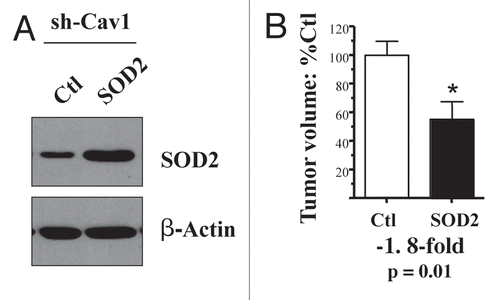
Figure 6 Cytoplasmic soluble SOD1 does not revert the tumor promoting phenotype of Cav-1 deficient fibroblasts. To combat the oxidative stress, we stably overexpressed SOD1 in Cav-1 knock-down fibroblasts, using a lenti-viral vector with puromycin resistance. Cav-1 knock-down cells were transfected with the empty vector alone, in parallel. Then, these two fibroblast lines were co-injected with MDA-MB-231 cells into the flanks of nude mice. Note that overexpression of SOD1, a cytoplasmic soluble enzyme that deactivates super-oxide, is not sufficient to reduce the tumor promoting effects of Cav-1 knock-down fibroblasts. The overexpression of SOD1 was validated by western blot analysis (A). The expression of β-actin is shown as a control for equal protein loading. N ≥ 8 flank injections for each experimental group (n.s., not significant).
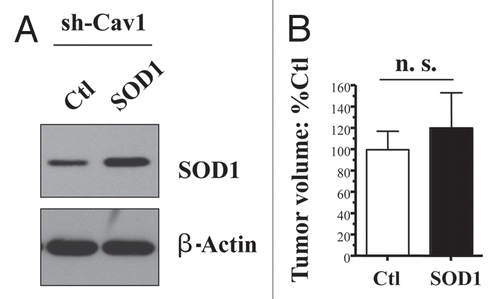
Table 1 Proteomic analysis of hTERT Cav-1 knock-down (KD) fibroblasts
Table 2 Intersection of Cav-1 deficient fibroblast proteomics with the transcriptome of human breast cancer tumor stroma
Table 3 Proteomic analysis of hTERT eNOS fibroblasts
Table 4 Intersection of eNOS fibroblast proteomics with the transcriptome of human breast cancer tumor stroma
Acknowledgements
M.P.L. and his laboratory were supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. F.S. was supported by grants from the Breast Cancer Alliance (BCA), and a Research Scholar Grant from the American Cancer Society (ACS). R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
References
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001; 1:46 - 54
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 2002; 70:537 - 546
- Ronnov-Jessen L, Bissell MJ. Breast cancer by proxy: can the microenvironment be both the cause and consequence?. Trends Mol Med 2009; 15:5 - 13
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121:335 - 348
- Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 2006; 5:1597 - 1601
- Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med 2010; 2:211 - 230
- Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Allen KG, et al. Human Breast Cancer-Associated Fibroblasts (CAFs) Show Caveolin-1 Downregulation and RB Tumor Suppressor Functional Inactivation: Implications for the Response to Hormonal Therapy. Cancer Biol Ther 2008; 7:1212 - 1225
- Witkiewicz AK, Casimiro MC, Dasgupta A, Mercier I, Wang C, Bonuccelli G, et al. Towards a new “stromal-based” classification system for human breast cancer prognosis and therapy. Cell Cycle 2009; 8:1654 - 1658
- Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8:1167 - 1175
- Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of Stromal Caveolin-1 Expression Predicts Poor Clinical Outcome in Triple Negative and Basal-like Breast Cancers. Cancer Biol Ther 2010; 10:135 - 143
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An Absence of Stromal Caveolin-1 Expression Predicts Early Tumor Recurrence and Poor Clinical Outcome in Human Breast Cancers. Am J Pathol 2009; 174:2023 - 2034
- Sloan EK, Ciocca D, Pouliot N, Natoli A, Restall C, Henderson M, et al. Stromal Cell Expression of Caveolin-1 Predicts Outcome in Breast Cancer. Am J Pathol 2009; 174:2035 - 2043
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: Emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther 2010; 10:537 - 542
- Di*Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An Absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial akt activation. Cell Cycle 2009; 8:2420 - 2424
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: a new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and dcis therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, hif1 induction and nf.b activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle 2010; 9:2201 - 2219
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1alpha functions as a tumor promoter in cancer associated fibroblasts and as a tumor suppressor in breast cancer cells: autophagy drives compartment-specific oncogenesis. Cell Cycle 2010; 9:3534 - 3551
- Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: a simple solution to the autophagy paradox. Cell Cycle 2010; 9:4297 - 4306
- Witkiewicz AK, Freydin B, Chervoneva I, Potoczek MB, Rizzo W, Rui R, et al. Stromal CD10 and SPARC expression in ductal carcinoma in situ (DCIS) patients predicts disease recurrence. Cancer Biol Ther 2010; 10:391 - 396
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009; 457:910 - 914
- Persoon-Rothert M, Egas-Kenniphaas JM, van de, Valk-Kokshoorn EJ, Buys JP, van der Laarse A. Oxidative stress-induced perturbations of calcium homeostasis and cell death in cultured myocytes: role of extracellular calcium. Mol Cell Biochem 1994; 136:1 - 9
- Mattson MP. Oxidative stress, perturbed calcium homeostasis and immune dysfunction in Alzheimer's disease. J Neurovirol 2002; 8:539 - 550
- Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev 2010; 131:517 - 526
- Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: The calcium connection. Biochim Biophys Acta 2010; 1797:607 - 618
- Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, et al. The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta 2010; 1797:1018 - 1027
- Masamune A, Watanabe T, Kikuta K, Satoh K, Shimosegawa T. NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2008; 294:99 - 108
- Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol 2002; 36:200 - 209
- Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factorbeta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005; 97:900 - 907
- Gailit J, Marchese MJ, Kew RR, Gruber BL. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol 2001; 117:1113 - 1119
- Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15:1077 - 1081
- Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, et al. NAD(P)H oxidase mediates TGFbeta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol 2010; 21:93 - 102
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001
- Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010; 9:1960 - 1971
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 2001; 276:38121 - 38138
- Garcia-Cardena G, Martasek P, Siler-Masters BS, Skidd PM, Couet JC, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin: Functional Significance of the NOS caveolin binding domain in vivo. J Biol Chem (Communication) 1997; 272:25437 - 25440
- Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010; 9:2412 - 2422
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010; 2:185 - 199
- Bravard A, Sabatier L, Hoffschir F, Ricoul M, Luccioni C, Dutrillaux B. SOD2: a new type of tumor-suppressor gene?. Int J Cancer 1992; 51:476 - 480
- Behrend L, Mohr A, Dick T, Zwacka RM. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer cells. Mol Cell Biol 2005; 25:7758 - 7769
- St Clair D, Zhao Y, Chaiswing L, Oberley T. Modulation of skin tumorigenesis by SOD. Biomed Pharmacother 2005; 59:209 - 214
- Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother 2005; 59:143 - 148
- Venkataraman S, Jiang X, Weydert C, Zhang Y, Zhang HJ, Goswami PC, et al. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005; 24:77 - 89
- Tsanou E, Ioachim E, Briasoulis E, Damala K, Charchanti A, Karavasilis V, et al. Immunohistochemical expression of superoxide dismutase (MnSOD) antioxidant enzyme in invasive breast carcinoma. Histol Histopathol 2004; 19:807 - 813
- Pani G, Colavitti R, Bedogni B, Fusco S, Ferraro D, Borrello S, et al. Mitochondrial superoxide dismutase: a promising target for new anticancer therapies. Curr Med Chem 2004; 11:1299 - 1308
- Duan H, Zhang HJ, Yang JQ, Oberley LW, Futscher BW, Domann FE. MnSOD upregulates maspin tumor suppressor gene expression in human breast and prostate cancer cells. Antioxid Redox Signal 2003; 5:677 - 688
- Oberley LW. Anticancer therapy by overexpression of superoxide dismutase. Antioxid Redox Signal 2001; 3:461 - 472
- Zhong W, Oberley LW, Oberley TD, St. Clair DK. Suppression of the malignant phenotype of human glioma cells by overexpression of manganese superoxide dismutase. Oncogene 1997; 14:481 - 490
- Yan T, Oberley LW, Zhong W, St. Clair DK. Manganese-containing superoxide dismutase overexpression causes phenotypic reversion in SV40-transformed human lung fibroblasts. Cancer Res 1996; 56:2864 - 2871
- Li JJ, Oberley LW, St. Clair DK, Ridnour LA, Oberley TD. Phenotypic changes induced in human breast cancer cells by overexpression of manganese-containing superoxide dismutase. Oncogene 1995; 10:1989 - 2000
- Borrello S, De Leo ME, Galeotti T. Defective gene expression of MnSOD in cancer cells. Mol Aspects Med 1993; 14:253 - 258
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 2009; 29:1575 - 1591
- Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, et al. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab 2009; 94:5155 - 5162
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 2009; 29:4467 - 4483
- Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest 2005; 115:1163 - 1176
- Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, et al. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 2003; 22:6408 - 6423
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 3514
- Marouga R, David S, Hawkins E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal Bioanal Chem 2005; 382:669 - 678
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008; 14:518 - 527