Abstract
The balance between the pro-apoptotic lipids ceramide and sphingosine and the pro-survival lipid sphingosine 1-phosphate (S1P) is termed the “sphingosine rheostat”. Two isozymes, sphingosine kinase 1 and 2 (SK1 and SK2), are responsible for phosphorylation of pro-apoptotic sphingosine to form pro-survival S1P. We have previously reported the antitumor properties of an SK2 selective inhibitor, ABC294640, alone or in combination with the multikinase inhibitor sorafenib in mouse models of kidney carcinoma and pancreatic adenocarcinoma. Here we evaluated the combined antitumor effects of the aforementioned drug combination in two mouse models of hepatocellular carcinoma. Although combining the SK2 inhibitor, ABC294640, and sorafenib in vitro only afforded additive drug-drug effects, their combined antitumor properties in the mouse model bearing HepG2 cells mirrored effects previously observed in animals bearing kidney carcinoma and pancreatic adenocarcinoma cells. Combining ABC294640 and sorafenib led to a decrease in the levels of phosphorylated ERK in SK-HEP-1 cells, indicating that the antitumor effect of this drug combination is likely mediated through a suppression of the MAPK pathway in hepatocellular models. We also measured levels of S1P in the plasma of mice treated with two different doses of ABC294640 and sorafenib. We found decreases in the levels of S1P in plasma of mice treated daily with 100 mg/kg of ABC294640 for 5 weeks, and this decrease was not affected by co-administration of sorafenib. Taken together, these data support combining ABC294640 and sorafenib in clinical trials in HCC patients. Furthermore, monitoring levels of S1P may provide a pharmacodynamic marker of ABC294640 activity.
</br> See commentary: Sphingosine kinase: A promising cancer therapeutic target
Introduction
Hepatocellular carcinoma (HCC) is one of the most common solid tumors, and the third most common cause of cancer related deaths in humans.Citation1 It is also one of the deadliest, as the annual incidence almost equals the annual mortality, and its incidence is on the rise in developed countries. People with high risk for developing HCC are those who have hepatitis B or C, or hereditary hemochromatosis as well as those with chronic alcoholic cirrhosis.Citation2,Citation3 The median survival after diagnosis is approximately 6–20 months, depending on stage at diagnosis. Systemic chemotherapy only provides response rates of 0–25% and, until recently, there was no evidence that systemic chemotherapy improved overall survival in HCC patients.Citation4 Characterization of the pathology of HCC and the molecular pathways responsible for hepatocarcinogenesis showed that tumor cells overexpress several tyrosine kinase receptors, and have a highly vascular phenotype.Citation5 Molecular pathways associated with hepatocarcinogenesis include the Ras/Raf/MAP/ERK, the PI3K/Akt/mTOR, the Wnt/β-catenin and the JAK/STAT pathways.Citation6 Overall, HCC has presented a very difficult problem in the design of successful chemotherapy.
A clinical trial reported in 2007 showed efficacy of the multikinase inhibitor sorafenib in hepatocellular carcinoma, with a 44% improvement in overall survival in patients who received sorafenib compared to placebo (p = 0.0001).Citation4,Citation7,Citation8 Both median survival and time to progression showed 3 month improvements, and sorafenib obtained FDA approval for the treatment of advanced HCC at the end of 2007.Citation9 Ongoing clinical trials are seeking to increase the efficacy of sorafenib by combining it with other chemotherapy drugs. For example, a randomized, double-blind, phase II trial combining sorafenib with doxorubicin indicted increased overall survival and progression-free survival in patients receiving sorafenib plus doxorubicin compared with those receiving doxorubicin alone.Citation10 Because sorafenib is a targeted drug, it is likely that its combination with other agents that target molecular alterations in HCC will provide optimal therapies for these patients.
Sphingolipids, structural constituents of plasma membrane, have recently received considerable attention as targets for anticancer drug development due to their involvement in the regulation of cell survival and apoptosis.Citation11–Citation14 Three signaling sphingolipids, pro-apoptotic sphingosine and ceramide and pro-survival sphingosine 1-phosphate (S1P) can be manipulated pharmacologically to tip the balance (named “sphingolipid rheostat”) toward tumor cell apoptosis/senescence or survival.Citation15–Citation17 This manipulation can be achieved with small molecules that are designed to target either ceramidaseCitation18,Citation19 or sphingosine kinase (SK).Citation20,Citation21 Ceramidases hydrolyze ceramides and therefore tip the balance toward cell survival, and SKs phosphorylate sphingosine to yield S1P. So far, two sphingosine kinase isozymes have been discovered, sphingosine kinase 1 (SK1) and sphingosine kinase 2 (SK2).Citation22 Most scientific endeavor has been focused toward understanding of the biological function of SK1. Much less is known about the biological function of SK2 and its contribution to tumor development.
Recently, we reported the antitumor properties of a first-in class inhibitor of SK2, named ABC294640.Citation21,Citation23,Citation24 This compound was found to suppress both the pro-survival MAPK/ERK pathway and the anti-apoptotic Akt pathway in various tumor cell lines. Moreover, a delay in tumor growth was observed in several mouse tumor models, such as an allograft model of mammary adenocarcinoma and xenograft models of pancreatic adenocarcinoma and kidney carcinoma.Citation23,Citation24 This delay was further potentiated by co-administration of sorafenib, indicating that this drug-drug combination can be further exploited in clinics.Citation24 In the kidney carcinoma model, ABC294640 was shown to induce autophagy both in vivo and in vitro, and this mechanism appears to be a major contributor to tumor cell death.Citation23 This is important in treatments of HCC because intrinsic or acquired drug resistance is commonly seen in these patients.Citation4 Large HCCs commonly develop areas of hypoxic cells which may contribute to drug resistance (for example by decreasing drug delivery), but those cells may be more susceptible to activation of autophagy which can lead to terminal self-digestion in cells that are deprived of nutrients.Citation25 Clearly, to extend the life of patients with advanced HCC, alternatives to traditional cytotoxic chemotherapy agents must be explored.
Herein, we report that combining of sorafenib with ABC294640 in three hepatocellular carcinoma cell lines, Hep G2, SK-HEP1 and Hep 3b2.1-7, leads to additive toxicity in vitro. Downregulation of p-ERK and increases in the markers of autophagy were observed in cells exposed to the test compounds. In two mouse models of HCC, further delay in tumor growth was observed when sorafenib was combined with ABC294640, indicating that ABC294640 can be used in the treatment of HCC both alone and in combination with sorafenib.
Results
In vitro combined anticancer effects of ABC294640 and sorafenib.
We have previously reported that combining SK inhibitors and sorafenib leads to synergistic toxic effects in kidney carcinoma and pancreatic adenocarcinoma cells.Citation24 To assess if combining ABC294640 and sorafenib leads to synergistic cytotoxicity in HCC cells, SK-HEP-1, Hep G2 or Hep 3b2.1-7 cells were plated in 96-well plates (3,000 cells per well) and exposed to various concentrations of ABC294640, sorafenib or combinations. After 72 hr of exposure, cell survival was measured by the standard sulforhodamine B assay. As shown in , IC50 values for ABC294640 were approximately 70, 30 and 35 µM for SK-HEP-1, Hep G2 and Hep 3b2.1-7, respectively. Sorafenib was more potent than the SK inhibitor, with IC50 values of 5, 2 and 1.5 µM for SK-HEP-1, Hep G2 and Hep 3b2.1-7 cells, respectively. These data indicate that sorafenib is more efficient in killing HCC cells in vitro and ABC294640 is less toxic in vitro. To assess combined drug effects, we exposed HCC cells to the constant molar ratio of ABC294640 and sorafenib of 5:1. After 72 hr of drug exposure, we assessed cell viability and calculated the Combination Index (CI) to determine whether this drug combination resulted in additive (CI = 1.0), synergistic (CI < 1.0) or antagonistic (CI > 1.0) toxicity. Slight to moderate synergism was observed in SK-HEP-1 cells, and overall additive drug-drug effects were observed in Hep G2 and Hep 3b2.1-7 cells.
To assess how combining ABC294640 and sorafenib affects cell apoptosis, we examined activation of caspases 3 and 7 and measured genomic DNA fragmentation by flow cytometry (). We observed no changes in activities of caspases 3 and 7 in SK-HEP-1 cells that were exposed to the combination of ABC294640 and sorafenib, and only a moderate activation was observed in Hep3b2.1-7 cells exposed to 50 µM ABC294640 and 5 µM sorafenib alone or in combination. However, substantial increases in caspases 3 and 7 were observed in HepG2 cells, indicating that the apoptotic response measured by the activation of caspases 3 and 7 varies among HCC cells. Quantification of fragmented genomic DNA by flow cytometry confirmed the lack of apoptosis in SK-HEP-1 cells, but an induction of apoptosis measured by genomic DNA fragmentation in Hep3b2.1-7 cells was observed after treatment with both compounds (). Interestingly, apoptosis in these cells was also induced by low concentrations of ABC294640. Taken together, these data indicate significant contribution of apoptosis induced by ABC294640 alone or in combination with sorafenib in certain HCC tumor cells.
We have reported previously that ABC294640 induces autophagy in cancer cells, and that this is a major cell death mechanism in certain cancer cells.Citation23 In addition, sorafenib was also shown to induce autophagy in several tumor cell systems.Citation26 Therefore, to determine if ABC294640 and/or sorafenib induce autophagy in these cells, we examined two markers of autophagy, levels of LC3-II and beclin-1 in HCC cells (). Exposure of all three cell lines to either ABC294640 or sorafenib led to increases in the levels of LC3-II and beclin-1, both of which were used previously used as markers for activation of autophagy with SK inhibitors. Therefore, ABC294640 or sorafenib elevate autophagy in these HCC cells.
While studying the combined drug effects of SK inhibitors and sorafenib in kidney carcinoma and pancreatic adenocarcinoma cells, we found a profound affect on suppression of p-ERK signaling in cells exposed to low concentrations of drug combinations. This was not surprising as both inhibitors exert their antitumor activity by decreasing signaling in MAPK pathway.Citation4,Citation17,Citation23 Firstly, we confirmed that SK-HEP-1 cells exposed to single drugs downregulate p-ERK (). Then, we assessed levels of p-ERK in SK-HEP-1 cells treated with combinations of ABC294640 and sorafenib and found that the combination of ABC294640 and sorafenib cooperate to decrease the pro-survival signaling of p-ERK. Therefore, ABC294640 and sorafenib additively increase cytotoxicity in HCC cells and exert combined effects on p-ERK signaling. In addition, we accessed LC3-II levels in SK-HEP-1 cells that were exposed to single drugs or their combinations. Our data reveal prominent increases in LC3-II in cells that are exposed to 50 µM ABC294640, but the levels of LC3-II did not increase in cells that were treated with combinations of both drugs, compared to the LC3-II levels in cells treated with ABC294640 only. This indicates that autophagy induced by ABC294640 is not potentiated by addition of sorafenib.
We also assessed levels of LC3-II in splenocytes that were exposed to ABC294640 () and in peripheral lymphocytes from mice treated with 50 mg/kg ABC294640 for 4 weeks (). We did not find any increase in the levels of LC3-II compared to the vehicle-treated cells/mice, indicating that ABC294640 does not induce autophagy in either normal splenocytes or peripheral lymphocytes.
In vivo antitumor effects of combination of ABC294640 with sorafenib.
Because ABC294640 and sorafenib exert additive drug effects in HCC cells and because this drug combination potentiated tumor growth inhibition in A-498 and Bxpc-3 xenograft models, we tested this combination in SK-HEP-1 and HepG2 xenograft models. HCC cells were implanted subcutaneously into SCID mice, and upon the development of measurable tumors, mice were randomized into groups (n = 7–8) and treated with vehicle (PEG-200/DMSO = 50/50), ABC294640, sorafenib or ABC294640 and sorafenib. Both ABC294640 and sorafenib were administered orally, either every day Monday–Friday at 50 mg/kg body weight (ABC294640), or Monday, Wednesday and Friday at 20 mg/kg body weight (sorafenib). Tumors were measured with calipers, and body weight was measured twice a week to assess systemic toxicity.
Administration of ABC294640 or sorafenib to mice reduced tumor growth in both SK-HEP-1 and HepG2 xenograft models ( and B). Combination of sorafenib with ABC294640 resulted in further reductions of tumor growth compared with either single agent in both HCC models. To assess the systemic toxicity of the treatments, we measured mouse total body weight, and found no significant weight loss for any treatment group ( and D).
Establishment of pharmacodynamic parameters for ABC294640 treatments.
It was reported previously that the plasma S1P levels in SK2-knockout mice are reduced ∼25% compared to the S1P levels in the wild-type mice.Citation27 Therefore, we hypothesized that mice treated with the SK2 inhibitor ABC294640 will exibit reduced levels of S1P in their plasma compared to the vehicle-treated mice. To assess the levels of S1P, we utilized the previously established pancreatic adenocarcinoma mouse model and administered two different concentrations of ABC294640 (50 or 100 mg/kg mouse weight) alone or in combination with sorafenib (10 or 20 mg/kg mouse weight) according to the schedule described above. After 5 weeks of treatment, we isolated mouse plasma and measured the levels of S1P according to a previously published procedure, and then correlated those data with the tumor growth reduction (). The maximum reductions in S1P levels (∼40% reduction compared to control) were found in mice treated daily with 100 mg/kg ABC294640. Simultaneous administration of sorafenib at either 10 or 20 mg/kg did not affect the levels of S1P compared to treatment with ABC294640 alone. These data were well correlated with the reduction in tumor size that was observed in xenograft models (). Therefore, it is possible that levels of S1P in plasma can be used as an indicator of drug action in patients treated with the SK2 inhibitor ABC294640.
We also assessed the levels of S1P in HepG2 tumors that were grown in xenograft models in mice treated with ABC294640 and/or sorafenib. After 4 weeks of treatment, mice were euthanized and tumor tissues were homogenized and S1P was extracted, derivatized and quantified by HPLC (). The data indicate that the greatest decrease in S1P levels is observed in the tumors that were grown in mice treated with both ABC294640 and sorafenib.
Discussion
More than 626,000 cases of HCC-related deaths are reported yearly worldwide, which ranks it as the third most common cause of death from cancer.Citation1 Pharmacologic treatment of HCC has proven very difficult. Administration of multikinase inhibitor sorafenib, which is the only FDA approved medication for treatment of this disease, leads to a modest increase the median duration of survival (7.9 month on placebo; 10.7 months on sorafenib). Therefore, there is unmet need for more effective drugs and/or combinations for the treatment of this deadly disease.
Sorafenib (Nexavar) is a small molecule inhibitor of tumor-cell proliferation and angiogenesis and inducer of apoptosis which has a potential for clinical applications in numerous tumors.Citation28–Citation30 Sorafenib is the inhibitor of the serine-threonine kinase Raf-1 and the receptor tyrosine kinase activity of vascular endothelial growth factor (VEGF) receptors 1, 2 and 3 and platelet-derived growth factor receptor β. The molecular pathogenesis of HCC is thought to be mediated by the Raf-1 and VEGF pathways, providing a rationale for investigating sorafenib for treatment of HCC. In preclinical HCC models, sorafenib reduces tumor growth, angiogenesis and tumor-cell signaling and it also increases tumor cell apoptosis.Citation31 Its success in clinical trials led to FDA approval of sorafenib for the treatment of renal cell carcinoma and advanced hepatocellular carcinoma. Clinical trials are currently assessing its efficacy in the treatment of pancreatic adenocarcinoma, lung and thyroid cancers, either alone or in combination with other chemotherapeutics.Citation32
Sphingolipids represent an attractive area for cancer drug development because two sphingolipid second messengers, ceramide and S1P, regulate apoptosis and cell survival. A plethora of studies in various transformed and non-transformed cell lines consistently indicate that S1P induces proliferation by stimulating cell proliferation pathways such as MAPK, and that S1P protects cells from apoptotic stimuli. The stimulation of MAP/ERK pathway by S1P provides rationale for testing SK inhibitors in HCC models, as this pathway is partially responsible for HCC pathogenesis.
We have shown previously that combining the dual SK1/SK2 inhibitor ABC294375 or the SK2-selective inhibitor ABC294640 with sorafenib in kidney carcinoma and pancreatic adenocarcinoma results in synergistic cytotoxicity in vitro and further delay of tumor growth in xenograft models compared to the single agents.Citation24 These cooperative effects between SK inhibitors and sorafenib were associated with increases in genomic DNA fragmentation and caspase 3/7 activation. Downregulation of proliferative MAPK signaling in A-498 and Bxpc-3 tumor cells was also observed for the combination of these agents. These results led to a hypothesis that combining sorafenib with the SK2 selective inhibitor ABC294640 will lead to more efficient induction of apoptosis and decreased proliferative signaling in other cancer models, particularly HCC.
In the present work, we tested the aforementioned combination of the two kinase inhibitors in preclinical models of HCC. We used three HCC cell lines, and found that combining sorafenib with ABC294640 leads to an overall additive cell toxicity in vitro in HepG2 and Hep3b2.1-7 cells, and a slight to moderate synergism in SK-HEP-1 cells. Increases in both caspase 3/7 activities and genomic DNA fragmentation were observed in HepG2 and Hep3b2.1-7 cells exposed to ABC294640 plus sorafenib, but not in SK-HEP-1 cells. In Hep3b2.1-7 cells, levels of caspase 3/7 activity and genomic DNA fragmentation in combination-treated cells were comparable to the levels found in single drug-treated cells. When HCC cells were treated with sorafenib or ABC294640, increases in LC3 cleavage and upregulation of beclin-1 were observed for all three cell lines. These data indicate that ABC294640 and sorafenib induce autophagy in HCC cells. In contrast, a downregulation of MAP/ERK kinase pathway signaling was observed in SK-HEP-1 cell line, but no additional increase in cleavage of LC3-II or apoptosis was observed in ABC294640 plus sorafenib-treated cells, indicating that neither autophagy nor apoptosis are responsible for the combined drug cytotoxic effects in this cell line. Loss of viability without activation of apoptosis or markers of autophagy is consistent with necrotic cell death.Citation33 Therefore, it is likely that the combined drug effect on cytotoxicity in this cell line is due to the programmed necrosis, a cell death pathway associated with elevated ceramide.Citation34
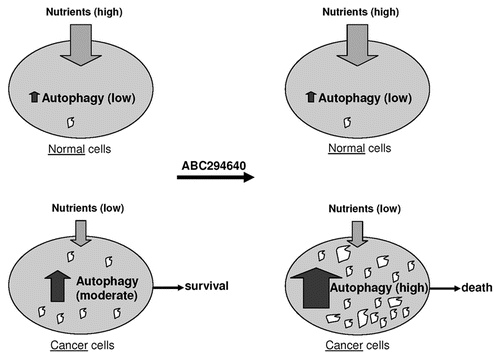
Interestingly, no increases in LC3-II were observed in splenocytes that were exposed in vitro to ABC294640 for as long as 48 hr, or in peripheral lymphocytes of mice treated with ABC294640. These results suggest that compounds that induce autophagy in tumor cells (such as ABC294640) are potential cancer chemotherapeutics with minimal effect on normal cells. It is likely that cells that are rapidly dividing and hypoxic cells are in a higher demand for nutrient supplies compared to quiescent cells, and that the former often resort to upregulation of autophagy as a means for survival and maintenance of homeostasis. Cells that constitute normal tissues do not divide as often, and probably rarely upregulate autophagy. When such cells encounter autophagy-inducing compounds, they are less prone to start autophagic self digestion compared to tumor cells. The possible consequences of this mechanism are depicted on the .
Administration of ABC294640 or sorafenib to mice bearing either SK-HEP-1 or HepG2 tumor xenografts resulted in tumor growth inhibition without overt toxicity to mice. When ABC294640 and sorafenib were combined, further delays of tumor growth were observed. These data indicate that ABC294640 has antitumor activity in HCC xenograft models, and that this antitumor activity can be further potentiated by administration of sorafenib.
To establish pharmacodynamic biomarkers for ABC294640 action, we measured plasma levels of the enzymatic product of SK enzymes, S1P. Mice bearing pancreatic adenocarcinoma cells (Bxpc-3) were treated for five weeks with ABC294640 (50 or 100 mg/kg) or sorafenib (10 or 20 mg/kg) or with combinations. At the end of the treatment, tumors sizes were measured and S1P was extracted from plasma and quantified. The data show good correlation between the tumor growth delay and S1P levels in plasma. Therefore, we conclude that this measurement may be useful as a pharmacodynamic biomarker for ABC294640.
In summary, we demonstrate that simultaneous targeting of the sphingolipid and MAPK pathways using SK inhibitors and sorafenib has a potential for further exploration in clinics. Growth suppression is associated with increases in tumor cell apoptosis and downregulation of MAP/ERK signaling pathway. In addition, measuring of plasma S1P levels can be used to monitor antitumor effectiveness of ABC294640.
Materials and Methods
Cell lines and animals.
Human hepatocellular adenocarcinoma cells (SK-HEP-1), human hepatocellular carcinoma cells (Hep G2 and Hep 3b2.1-7) and pancreatic adenocarcinoma Bxpc-3 cells were purchased from American Type Culture Collection, and grown in RPMI 1640 medium containing 10% Fetal Bovine Serum (Invitrogen; Carlsbad, CA) and 50 µg/ml gentamycin sulfate. SCID mice were purchased from the National Institutes of Health (Bethesda, MD).
Compounds.
Sorafenib and ABC294640 were synthesized as described previously in reference Citation24.
Cell proliferation and apoptosis assays.
Cell proliferation was measured using the standard sulforhodamine B assay and combined-effects analyses, based on the method of Chou and Talalay,Citation35 were conducted to establish whether combinations of ABC294640 and sorafenib result in synergism, additivity or antagonism using CalcuSyn software (Biosoft, Ferguson, MO). For quantification of genomic DNA fragmentation and cell cycle analyses, cells were exposed to various concentrations of ABC294640, and/or sorafenib for 48 hr, washed twice with PBS and incubated in 0.5 ml of PI staining solution (50 µg/ml of propidium iodide, 40 µg/ml RNase A in PBS) for 30 min at 37°C. Cell cycle distributions were analyzed with a Becton Dickinson FACSCalibur Analytical Flow Cytometer at the MUSC flow cytometry facility. The activities of caspases 3 and 7 were measured using the Caspase-Glo 3/7 Assay (Promega, Madison, WI) according to manufacturer's instructions. Briefly, SK-HEP-1, Hep G2 or Hep 3b2.1-7 cells were seeded in white 96-well plates at a density of 10,000 cells per well and 4–6 hr later exposed to the test compounds. After 48 hr, 100 µl of the caspase reagent was added and plates were incubated at room temperature for 30 min. After incubation, luminescence was measured using a Molecular Devices SpectraMax M5 plate reader. Cells exposed to cisplatin were used as a positive control for caspases 3 and 7 activation, and cells exposed to the vehicle were considered to be negative controls.
Isolation of peripheral blood lymphocytes and splenocytes.
Blood from animals (0.5 ml) was collected by retro-orbital bleeding of anesthetized animals and transferred into 0.5 ml of EDTA (3 mg/ml, pH 7.2) and 0.5 ml of Dextran T500 solution was added (2% Dextran T500). The mixture was incubated for 20 min at 37°C. After the red blood cells (RBC) clumped and settled, the top layer was transferred to a new tube and cells were pelleted at 1,500 rpm. 250 µl of ACK buffer (8.3 g NH4Cl and 1 g KHCO3 per 1 dm3) was added to cell pellet and incubated for 5 min at room temperature. This suspension was diluted with 3 ml of PBS, spun down (5 min 3,000 rpm), and the cell pellet was lysed and subjected to western blot analyses as described below. In order to isolate splenocytes, mice were sacrificed, their spleens removed, passed through 40 mm nylon inserts (BD Biosciences, San Jose, CA), and cells were collected in 50 ml tubes. After two washes with 5 ml PBS, cells were centrifuged (1,800 rpm for 5 min) and residual red cells were lysed by 2.5 ml of ACK buffer for 3 min at room temperature. ACK buffer was neutralized with 12.5 ml PBS and cells were precipitated by centrifugation, washed twice with PBS, resuspended in RPMI 1640 medium containing 10% Fetal Bovine Serum and 50 µg/ml gentamycin sulfate, and exposed to ABC294640 for 4, 24 or 48 hr.
Western blot analyses.
Whole cell lysates were prepared by lysing cells after drug treatments, proteins were separated by polyacryamide gel electrophoresis and western blotting was carried out as previously reported. ERK and p-ERK antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA); LC3 antibody was from Novus Biologicals (Littleton, CO); and beclin antibody was from Abcam (Cambridge, MA). Proteins were visualized by enhanced chemiluminescence using anti-rabbit or anti-mouse horseradish peroxidase-conjugated IgG (Pierce; Appleton, WI). Equal loading was confirmed by probing the blots with mouse anti-actin antibody (Abcam, Cambridge, MA).
Antitumor studies.
Xenograft HCC tumor models of either SK-HEP-1 or HepG2 cells were established by subcutaneous injection of 5 × 106 cells as previously described in reference Citation36. Animal care and procedures were in accordance with guidelines and regulations of the Medical University of South Carolina. Animals were housed under 12 hr light/dark cycles, with food and water provided ad libitum. Upon detection of tumors (2 and 4 weeks for SK-HEP-1 and Hep G2 models, respectively), mice were randomized into treatment groups (n = 7–8). Mice were then treated daily Monday–Friday with 50 mg/kg of ABC294640 dissolved in vehicle consisting of 50% water and 50% PEG-200, and/or 20 mg/kg sorafenib every other day. Whole body weight and tumor size measurements were performed twice a week, and tumor volume was calculated using the equation: (length × width2)/2. After 4–5 weeks of treatment, three animals from each cohort were sacrificed and tumors were excised, fixed in paraformaldehyde or snap frozen and stored at −80°C.
Measurment of S1P leves in plasma, tissues and cell lysates.
The levels of S1P were quantified by HPLC using the modified one-step extraction procedure and derivatization of S1P by napthalene-2,3-dicarboxaldehyde (NDA) (Sigma-Aldrich, St. Louis, MO) as described previously in reference Citation37. C-17 S1P (Avanti Polar Lipids, Alabaster, AL) was used as the internal control. Briefly, 50 µL of plasma or 100 µL of the tumor tissue lysate were extracted with 300 or 450 µL of chloroform:methanol (1:2, v/v) and sonicated for 10 min. After the addition of 200 or 300 µL of both 1 M NaCl and chloroform and 20 or 30 µL of concentrated HCl, samples were vortexed for 60 sec and centrifuged at 13,000x g for 5 min. The organic phase was transferred to a new tube and the aqueous phase was re-extracted, and the combined organic phases were dried in a SpeedVac concentrator. The dried lipid extracts were dissolved in 20 µL of methanol to which 40 µL of the NDA derivatization reaction mixture (containing 20 µL of 50 mM borate buffer, pH 9.0, 10 µL 5 mM NDA and 10 µL of 5 mM NaCN) was added. This mixture was then incubated for 10 min at 50°C, and diluted 1:3 with ethanol and centrifuged at 13,000x g (5 min). The samples were separated and quantified by HPLC (Waters, Milford, MA), using a Synergi Fusion-RP (30 mm × 4.6 mm) column (Phenomenex, Torrance, CA USA). Separation was performed using 5 mM KH2PO4 (mobile phase A), MeOH (mobile phase B) and CH3CN (mobile phase C), with the following program: 0–8 min 45-30% A, 40–55% C; 8–10 min 30-5% A, 55–80% C; 10–12 min 5–45% A, 80-40% C at a flow rate of 1 ml/min. Mobile phase B was maintained at 15%. The fluorescent derivatives were monitored at the excitation and emission wavelengths of 252 and 483 nm, respectively. The quantities of S1P and C-17 S1P were calculated using the Waters Millennium software (Waters, Milford, MA).
Conflicts of Interest
Charles D. Smith is the Chief Executive Officer and President of Apogee Biotechnology Corporation and owns stock in this entity.
Abbreviations
| Ras/Raf/MAP/ERK | = | rat sarcoma/rat sarcoma-activated factor/mitogen activated protein kinase/extracellular regulated kinase |
| PI3K/Akt/mTOR | = | the phosphatidylinositide-3-kinase/protein kinase B/mammalian target of rapamycin |
| JAK/STAT | = | janus kinase/signal transducers and activators of transcription |
Figures and Tables
Figure 1 Cytotoxicities of ABC294640 and sorafenib (A) and combination indexes for combined drugs (B). The indicated HCC cells were exposed to ABC294640 (solid squares), sorafenib (solid circles) or combinations (empty triangles) for 72 hr. Standard SRB assays were performed to assess cytotoxicity. Combination indices were calculated as described in the Materials and Methods section. A combination index of: 1.0 indicates additive cytotoxicity; <1.0 indicates synergy; and >1.0 indicates antagonism. Data represent the mean ± standard error for three independent experiments.
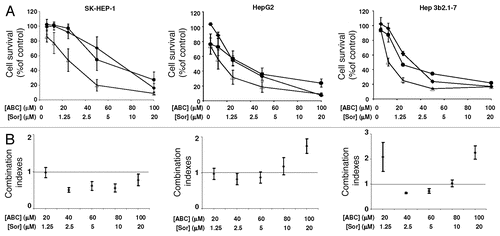
Figure 2 Effects of ABC294640 alone or in combination with sorafenib on apoptosis. SK-HEP-1 (A), HepG2 (B) or Hep 3b2.1-7 (C) cells were treated with the indicated concentrations of ABC294640 and/or sorafenib for 48 hr. Caspases 3/7 activity was measured by luminescence as described in the Materials and Methods section (left parts). Data represent mean ± standard error for three independent experiments. Cisplatin (Cis-DDP) was used as a positive control. For DNA fragmentation analyses (right parts), cells were harvested and nuclei were stained with propidium iodide and the DNA content was analyzed by flow cytometry as described in the Materials and Methods section.
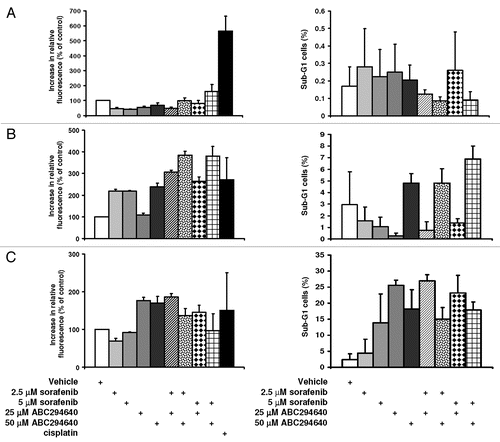
Figure 3 Effects of ABC294640 or sorafenib on autophagy markers in HCC cells. SK-Hep-1, HepG2 or Hep 3b2.1-7 cells were exposed to ABC294640 or sorafenib for 48 hr, except where indicated. Cell lysates were then fractionated by SDS-PAGE, and probed with antibodies to detect LC3-II, beclin-1, actin, p-ERK or ERK as described in the Materials and Methods section.
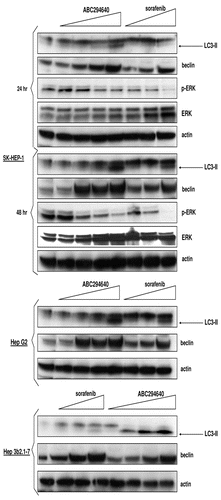
Figure 4 Effects of ABC294640, sorafenib or combinations on autophagy and MAP /ERK signaling in tumor cells and splenocytes. SK-HEP-1 cells (A) were exposed to ABC294640, sorafenib or combinations for 48 hr. Splenocytes (B) or peripheral lymphocytes (C) were isolated from either drug naïve mice (splenocytes) or drug-treated mice (peripheral lymphocytes) as described in Materials and Methods section. In (B), splenocytes were exposed to increasing concentrations of ABC294640 and harvested at indicated time points. Cell lysates were then fractionated by SDS-PAGE, and probed with antibodies to detect LC3-II, actin, p-ERK and ERK as described in the Materials and Methods section.
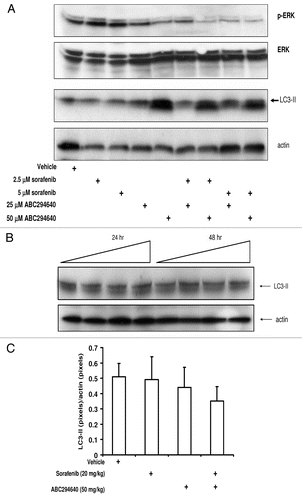
Figure 5 Effects of SK inhibitors and sorafenib on the growth of HCC cells in xenograft models. Female SCID mice (n = 7–8 per group) were injected subcutaneously with HepG2 (A) or SK-HEP-1 cells (B) suspended in PBS. After palpable tumors were formed, the animals were treated 5 days per week with vehicle (solid squares), 50 mg/kg of ABC294640 (solid triangles), three times per week with 20 mg/kg sorafenib (open squares) or combinations (open circles). Values represent the mean ± standard error tumor volume normalized to treatment day 1 for each mouse (A and B). The corresponding average mouse weights (mean ± standard error) for the treatment groups are shown in (C and D).
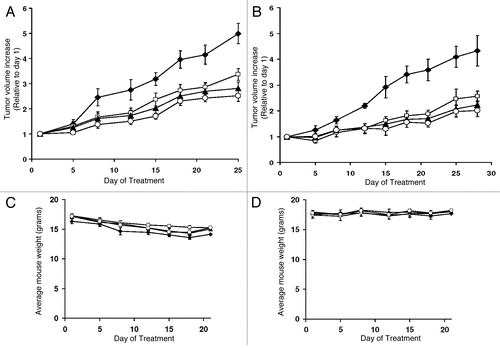
Figure 6 Comparison between plasma levels of S1P and tumor growth delay. (A) Mice bearing Bxpc-3 adenocarcinoma xenografts were treated for 5 weeks with the drugs indicated. At the end of the treatments, S1P was extracted from plasma and quantified as described in Materials and Methods (top). Tumors were measured and their size was normalized to the vehicle-treated mice (bottom). (B) After four weeks of treatment, HepG2 xenografts were excised from mice and S1P was extracted as described in Materials and Methods. After derivatization with a fluorescent tag, products were separated and quantified by HPLC.
Figure 7 Rationale for stimulating autophagy in cancer chemotherapy. The supply of nutrients is different in normal tissue cells (top) and cancer cells (bottom). Cancer cells can utilize various means to maintain stable metabolism and maintain homeostasis, including upregulation of autophagy. Upon exposure to autophagy-stimulating molecules such as ABC2946490, cancer cells likely further upregulate this pathway, leading to a high level of self-digestion in tumor cells. Empty spaces in these cells represent autophagic vacuoles where programed and non-programed lysosomal digestion of cytoplasmic constituents takes place. The consequence of “over-digestion” will be non-apoptotic cell death (bottom right).
Acknowledgements
Funding was provided by National Institutes of Health grant R01 CA122226 to C.D.S.
References
- Thomas MB, Jaffe D, Choti MM, Belghiti J, Curley S, Fong Y, et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 2010; 28:3994 - 4005
- McClune AC, Tong MJ. Chronic hepatitis B and hepatocellular carcinoma. Clin Liver Dis 2010; 14:461 - 476
- Manizate F, Hiotis SP, Labow D, Roayaie S, Schwartz M. Liver functional reserve estimation: state of the art and relevance to local treatments. Oncology 2010; 78:131 - 134
- Thomas MB, O'Beirne JP, Furuse J, Chan AT, Abou-Alfa G, Johnson P. Systemic therapy for hepatocellular carcinoma: cytotoxic chemotherapy, targeted therapy and immunotherapy. Ann Surg Oncol 2008; 15:1008 - 1014
- Hopfner M, Schuppan D, Scherubl H. Growth factor receptors and related signalling pathways as targets for novel treatment strategies of hepatocellular cancer. World J Gastroenterol 2008; 14:1 - 14
- Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010; 29:4989 - 5005
- Yau T, Chan P, Epstein R, Poon RT. Management of advanced hepatocellular carcinoma in the era of targeted therapy. Liver Int 2009; 29:10 - 17
- Zhu AX, Clark JW. Commentary: Sorafenib use in patients with advanced hepatocellular carcinoma and underlying Child-Pugh B cirrhosis: evidence and controversy. Oncologist 2009; 14:67 - 69
- Carr BI, Carroll S, Muszbek N, Gondek K. Economic evaluation of sorafenib in unresectable hepatocellular carcinoma. J Gastroenterol Hepatol 2010; 25:1739 - 1746
- Abou-Alfa GK, Johnson P, Knox JJ, Capanu M, Davidenko I, Lacava J, et al. Doxorubicin plus sorafenib vs. doxorubicin alone in patients with advanced hepatocellular carcinoma: a randomized trial. Jama 2010; 304:2154 - 2160
- Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res 2009; 50:91 - 96
- Meacham WD, Antoon JW, Burow ME, Struckhoff AP, Beckman BS. Sphingolipids as determinants of apoptosis and chemoresistance in the MCF-7 cell model system. Exp Biol Med (Maywood) 2009; 234:1253 - 1263
- Ozbayraktar FB, Ulgen KO. Molecular facets of sphingolipids: mediators of diseases. Biotechnol J 2009; 4:1028 - 1041
- Dumitru CA, Weller M, Gulbins E. Ceramide metabolism determines glioma cell resistance to chemotherapy. J Cell Physiol 2009; 221:688 - 695
- Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta 2006; 1758:2016 - 2026
- Lavieu G, Scarlatti F, Sala G, Levade T, Ghidoni R, Botti J, et al. Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision?. Autophagy 2007; 3:45 - 47
- Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, apoptosis. Biochim Biophys Acta 2002; 1585:193 - 201
- Holman DH, Turner LS, El-Zawahry A, Elojeimy S, Liu X, Bielawski J, et al. Lysosomotropic acid ceramidase inhibitor induces apoptosis in prostate cancer cells. Cancer Chemother Pharmacol 2008; 61:231 - 242
- Chan SY, Hilchie AL, Brown MG, Anderson R, Hoskin DW. Apoptosis induced by intracellular ceramide accumulation in MDA-MB-435 breast carcinoma cells is dependent on the generation of reactive oxygen species. Exp Mol Pathol 2007; 82:1 - 11
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 2003; 63:5962 - 5969
- French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, et al. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther 2010; 333:129 - 139
- Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol 2006; 39:113 - 131
- Beljanski V, Knaak C, Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther 2010; 333:454 - 464
- Beljanski V, Knaak C, Zhuang Y, Smith CD. Combined anticancer effects of sphingosine kinase inhibitors and sorafenib. Invest New Drugs 2010; In press
- Tafani M, Schito L, Anwar T, Indelicato M, Sale P, Di Vito M, et al. Induction of autophagic cell death by a novel molecule is increased by hypoxia. Autophagy 2008; 4:1042 - 1053
- Park MA, Zhang G, Martin AP, Hamed H, Mitchell C, Hylemon PB, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther 2008; 7:1648 - 1662
- Kharel Y, Lee S, Snyder AH, Sheasley-O'neill SL, Morris MA, Setiady Y, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem 2005; 280:36865 - 36872
- Josephs DH, Ross PJ. Sorafenib in hepatocellular carcinoma. Br J Hosp Med (Lond) 71:451 - 456
- Lekili M, Muezzinoglu T, Nese N, Temeltas G. Sorafenib in metastatic renal cell carcinoma with sarcomatoid differentiation. J Chin Med Assoc 73:262 - 264
- Smit EF, Dingemans AM, Thunnissen FB, Hochstenbach MM, van Suylen RJ, Postmus PE. Sorafenib in patients with advanced non-small cell lung cancer that harbor K-ras mutations: a brief report. J Thorac Oncol 5:719 - 720
- Strumberg D. Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 2005; 41:773 - 784
- Hasskarl J. Sorafenib. Recent Results Cancer Res 2010; 184:61 - 70
- Siskind LJ, Mullen TD, Romero Rosales K, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, et al. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Chem 2010; 285:11818 - 11826
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 2004; 16:663 - 669
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440 - 446
- Ma Y, Wang J, Liu L, Zhu H, Chen X, Pan S, et al. Genistein potentiates the effect of arsenic trioxide against human hepatocellular carcinoma: Role of Akt and nuclear factorkappaB. Cancer Lett 2010;
- He X, Huang CL, Schuchman EH. Quantitative analysis of sphingosine-1-phosphate by HPLC after napthalene-2,3-dicarboxaldehyde (NDA) derivatization. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:983 - 990