Abstract
We have previously cloned and characterized a novel p53 and DNA damage-regulated gene named PDRG1. PDRG1 was found to be differentially regulated by ultraviolet (UV) radiation and p53. In this study, we further investigated stress regulation of PDRG1 and found it to be selectively regulated by agents that induce genotoxic stress (DNA damage). Using cancer profiling arrays, we also investigated PDRG1 expression in matching normal and tumor samples representing various malignancies and found its expression to be upregulated in multiple malignancies including cancers of the colon, rectum, ovary, lung, stomach, breast and uterus when compared to their respective matched normal tissues. Western blot and immunohistochemical analyses were also performed on select specimen sets of colon cancers and matching normal tissues and the results also indicated PDRG1 overexpression in tumors relative to normal tissues. To gain insight into the function of PDRG1, we performed PDRG1 knockdown in human colon cancer cells and found its depletion to result in marked slowdown of tumor cell growth. These results suggest that PDGR1 may be linked to cell growth regulation. Yeast two-hybrid screen also led to the identification of PDCD7, CIZ1 and MAP1S as PDRG1-interacting proteins that are involved in apoptosis and cell cycle regulation which further implicate PDRG1 in controlling cell growth regulation. Taken together, our results indicate that PDRG1 expression is increased in multiple human malignancies suggesting it to be a high-value novel tumor marker that could play a role in cancer development and/or progression.
Introduction
Tumorigenesis is a multistep process that involves abnormalities in various oncogenes, tumor suppressor genes, growth factors and their receptors.Citation1 Although significant progress has been made in recent years to better understand the molecular pathogenesis of human malignancies, the exact molecular mechanisms are still unclear. Identification of novel molecular markers, particularly those exhibiting altered expression during the progressive transition from normal to premalignant to malignant stages, is likely to reveal important insights into these mechanisms and facilitate the development of clinical assays capable of detecting the malignant potential of premalignant lesions.
We have previously reported the identification and initial characterization of a novel p53 and DNA damage-regulated gene 1 (PDRG1).Citation2 PDRG1 was found to be differentially regulated by ultraviolet (UV) radiation and p53. Interestingly, PDRG1 mRNA was upregulated by ultraviolet radiation (UV), but downregulated by tumor suppressor p53. We have also noted that p53-null cells constitutively express higher levels of PDRG1 and that p53 downregulated PDRG1 gene promoter region suggesting that p53 negatively regulated PDRG1 at the transcriptional level.Citation2
Signaling molecules involved in the DNA damage response are important candidates for tumor marker studies because DNA damage response and cancer development are closely linked and DNA, the carrier of genetic information, appears to be the most precious possession of a cell. However, DNA is vulnerable to damage caused by a variety of toxic insults including those mediated by intrinsic and extrinsic genotoxic agents. Cellular response to DNA damage (genotoxic stress) is complex and involves a variety of molecules.Citation3 Defects in DNA response pathway could result in accumulation of mutations leading to genomic instability which may be one of the underlying causes of cancer development.
p53 is believed to play a key role in guarding the genome. To prevent the potential deleterious consequences of DNA damage, p53 serves as a signal-converging node for signals emanating from damage sensors and directing cells to either undergo growth arrest to repair the damaged-DNA or commit suicide when the damage is beyond repair.Citation4 Loss of p53 function would leave the cells vulnerable to genetic damage and subsequently cancer development. Indeed, p53 inactivation is associated with a wide variety of human cancers.
The function of PDRG1 remains to be elucidated and because PDRG1 is regulated by UV and p53, it is possible that it could be linked to regulating cellular stress response and cancer development and progress. We undertook this study to gain further insight into the function of PDRG1 and our results indicate that it is selectively regulated by agents that induce genotoxic stress. Our results further indicate that PDRG1 is overexpressed in multiple human malignancies and that knockdown of PDRG1 in human colon cancer cells affects their growth. Yeast two-hybrid screening also led us to identify PDCD7, CIZ1 and MAP1S as PDRG1-interacting proteins that are involved in apoptosis and cell cycle regulation which further support the role of PDRG1 in controlling cell growth regulation. Collectively, our results indicate that PDRG1 expression is increased in multiple human malignancies suggesting it to be a high-value novel tumor marker that could play a role in cancer development and/or progression.
Results
We have previously reported that PDRG1 mRNA was upregulated by UV.Citation2 Because UV is a potent genotoxic stress inducing agent, next we sought to investigate PDRG1 regulation in cellular stress response to genotoxic and non-genotoxic agents. For this purpose, we used adriamycin (doxorubicin), etoposide, camptothecin and UV as genotoxic agents while paclitaxel (taxol), thapsigargin and sulindiac sulfide as non-genotoxic agents. As shown in , in multiple cell lines, genotoxic stress inducing drugs upregulated PDRG1 expression whereas non-genotoxic stress inducing agents did not. These results, therefore indicate that PDRG1 is selectively regulated in response to genotoxic stress.
We have found that PDRG1 resides at the long arm of chromosome 20 in the cytogenetic region 20q11.2. A large body of evidence indicates high-frequency chromosome gains of 20q in cancers of the breast, lung, liver, colon and rectum.Citation5–Citation9 Furthermore, colorectal cancer patients with 20q gains have a significantly short survival time.Citation10 More recent evidence indicates that the cytogenetic region 20q11.2–20q13.2 is frequently amplified in human colorectal cancers.Citation11,Citation12 Evidence also suggests that this region is specifically amplified during progression from adenoma to carcinoma of the colon.Citation13 Additionally, frequency of amplification in this region has been reported to further increase in the metastatic lesions of colorectal cancers.Citation14 Thus, PDRG1 resides in the cytogenetic region that is frequently amplified in human colorectal cancers and perhaps in other types of malignancies. Furthermore, PDRG1 is transcriptionally downregulated by p53 and p53-null cells constitutively express higher levels of PDRG1.Citation2
It is now well-established that p53 is frequently inactive in a variety of human malignancies including colorectal cancers. Therefore, we sought to investigate the expression of PDRG1 in primary human malignancies and to that end, analyzed the expression pattern of PDRG1 in primary tumors and matching normal tissues using commercially available cancer profiling arrays. As shown in , PDRG1 expression was indeed upregulated in a variety of human cancer tissues when compared with matching normal tissues. The overall results are summarized in and indicate that PDRG1 expression is upregulated in tumors of the colon (82%), rectum (78%), ovary (71%), lung (70%), stomach (68%), breast (63%) and uterus (62%) when compared with their matching normal tissues. Thus, PDRG1 expression was highly elevated in various human malignancies, a finding which implicated its role in cancer development and/or progression. The clinicopathologic features of the tumors exhibiting PDRG1 upregulation are presented in Supplemental Tables 1–7.
Our preceding results show that PDRG1 expression was increased in multiple human malignancies at the mRNA levels. Next, we sought to validate these results at the protein levels and to that end we selected human colon cancer as a representative malignancy.
First, we used seven matched pairs of normal and colon cancer tissues and performed Western blot analysis to investigate PDRG1 expression pattern at the protein level. Consistent with our results at the mRNA levels, PDRG1 protein levels were significantly higher in all seven cancer samples when compared to the matching normal tissues (). We also performed immunohistochemical analysis to investigate the level and distribution of PDRG1 in tumors and normal tissues using specimen sections of matched colorectal normal and tumor samples. As shown in , PDRG1 was predominantly expressed by epithelial cells and the tumor sample exhibited a significant increase in PDRG-specific staining compared to the matching normal tissue.
Our results thus far indicated PDRG1 to be overexpressed in human malignancies, a finding which could link PDRG1 to cancer development and/or progression. To gain some insight into this issue, we also investigated the effect of PDRG1 depletion on human cancer cell growth. To that end, shRNA approach was employed to knockdown the expression of PDRG1 in RKO human colon cancer cells and cell proliferation was analyzed by MTT assays. Results in indicate that two different PDRG1 shRNAs which targeted different regions of the PDRG mRNAs efficiently depleted the expression of PDRG. Next, we analyzed the growth pattern of PDRG1-proficient and -deficient cells and as shown in , PDRG1 depletion significantly affected cell growth, a finding that could link PDRG1 to modulating cell growth regulation.
Our preceding results indicated that PDRG1 was overexpressed in human malignancies, and that depletion of PDRG1 slowed down tumor cell growth. To gain further insight into the potential mechanisms by which PDRG1 could play such a role, we performed a yeast two-hybrid screening to identify the interaction partners of PDRG1. This screening led us to identify three proteins including PDCD7 (programmed cell death 7), CIZ1 (Cip1 interacting zinc finger) and MAP1S (microtubules-associated protein 1S). PDCD7 has been implicated in modulating apoptosis and also reported as a component of U12-type spliceosome.Citation15 CIZ1, via its interaction with p21, is believed to modulate cell cycle progression by shuttling p21 and itself between nucleus and cytoplasm.Citation16 It has also been reported that CIZI promotes mammalian DNA replication and thus, plays a role in S-phase progression of the cell cycle.Citation17 MAP1S, on other hand, is a microtubule-binding protein that has been shown to interact with RASSF1A tumor suppressor protein to modulate cell cycle progression.Citation18,Citation19
Using PDCD7 as a representative protein, we also performed co-immunoprecipitation assay to investigate its interactions with PDRG1 in mammalian cells. For that purpose, HEK-293T cells were transiently co-transfected with HA-tagged PDRG1 expression vector along with myc-tagged PDCD7 or tag-alone control vector and cell lysates were then analyzed by protein co-immunoprecipitation assays. As shown in , myc-tagged PDCD7, but not the myc-tag alone, co-immunoprecipitated with HA-tagged PDRG1 indicating that PDRG1 specifically interacted with PDCD7 in 293T cells.
Discussion
In the current study, we have found that PDRG1 is selectively regulated in response to genotoxic stress. It is interesting that genotoxic stress upregulates while p53 downregulates PDRG1 expression. Because genotoxic stress regulation of PDRG1 occurs in both p53-positive and -negative cells, it is likely that genotoxic stress-mediated upregulation of PDRG1 occurs in a p53-independent manner. Furthermore, PDRG1 gene promoter harbors Oct1-binding sites and Oct1 strongly upregulates PDRG1 expression.Citation2 Oct1 is positively regulated in response to genotoxic stress and therefore, it is possible that, following genotoxic stress, strong positive regulation by Oct1 could override the negative effect of p53 on PDRG1 in p53 wild-type background. We have also found that PDRG1 is overexpressed in multiple human malignancies and that knockdown of PDRG1 in human colon cancer cells affects their growth.
Yeast two-hybrid screening also led us to identify PDCD7, CIZ1 and MAP1S as PDRG1-interacting proteins that are involved in apoptosis and cell cycle regulation, which further support the role of PDRG1 in controlling cell growth regulation. Together these results suggest PDRG1 to be a high-value novel tumor marker that could play a role in cancer development and/or progression. Our finding that knockdown of PDRG1 in human colon cancer cells affects their growth highlights its potential role in modulating cancer cell growth. Previously, we had found that overexpression of PDRG1 in HCT116 human colon cancer cells decreases clonogenic survival in response to UV irradiation.Citation2 Such an outcome could occur due to overexpression, which may not be perceived as a very physiologically-relevant approach to evaluate PDRG's role in cellular sensitivity to DNA damage. Further indepth studies are, therefore, needed to explore how PDRG1 depletion would affect cellular sensitivity to genotoxic stress.
In a recent study that used mass spectrometry analysis, PDRG1 was identified as a subunit of a prefoldin complex.Citation20 Prefoldin is a heterohexameric molecular chaperone complex that binds to nascent unfolded polypeptides and then delivers them to a group II chaperonin for proper folding.Citation20 Tubulin and actin are its best known substrates.Citation21 MAP1S, the PDRG1 binding partner identified in our yeast two-hybrid screening, is also a microtubule-associated protein.Citation18,Citation22 Together, this information would suggest that PDRG1 might be involved in the regulation of microtubules. Microtubule dynamics are essential to a number of cellular functions including cell cycle progression. Studies on the potential involvement of PDRG1 in modulating microtubule dynamics would likely shed more light on PDRG's cellular functions and its role in cell growth control.
The molecular basis for PDRG1 overexpression in human malignancies remains to be elucidated. We have found that PDRG1 resides at the long arm of chromosome 20 in the cytogenetic region 20q11.2. This region is frequently amplified in human colorectal cancers and perhaps in other types of malignancies.Citation5–Citation9 It is possible that increased expression could result due to PDRG1 gene amplification. We have previously reported that p53-null cells constitutively express higher levels of PDRG1.Citation2 Because p53 negatively regulates PDRG1 expression and the fact that p53 is inactive in a large number of various human tumors, it is also possible that PDRG1 expression is increased as a consequence of p53 inactivation due to either p53 mutation or p53 gene deletion.
As shown in current study, PDRG1 is highly expressed in multiple human malignancies highlighting its value as a novel tumor marker. Our current findings have laid the groundwork for more in-depth studies to further investigate the exact function of PDRG1 and firmly establish its potential as a high-value tumor marker.
Material and Method
Cell lines and culture conditions.
Human cancer cell lines HCT15 and RKO (colon), DU145 (prostate), HS578T (breast) as well as HEK-293T (human embryonic kidney cells), were regularly maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Gemeni Bioproducts Inc., Calabasas, CA). Mouse fibroblast NIH3T3 cells were maintained in DMEM (Cellgro, Mediatech, Herndon, VA) supplemented with 10% calf serum (Invitrogen, Grand Island, NY).
Plasmids.
For yeast two-hybrid screening, we generated PDRG1 and Gal4 DNA binding domain (DNA-BD) fusion protein, the open reading frame (ORF) of PDRG1 was first amplified using primers: 5′-GGA ATT CCA TAT GCT ATC ACC CGA GGC AGA-3′ and 5′-CGC GGA TCC TCA TCC TTT CAA GAT GAC CTT GAG A-3′, then digested with Nde I and BamH I and subcloned in frame at carboxal terminal of Gal4 DNA binding domain (DNA-BD) in pGBKT7 vector (a kind gift from Dr. Friedman, SUNY Upstate Medical University). The resulting plasmid was named pBD-PDRG1 and was sequenced to confirm the in-frame fusion of PDRG1 and DNA-BD. HA-tagged PDRG1 expression vector (pCEP-PDRG) was generated by subcloning of PDRG1 whole ORF into the mammalian expression vector pCEP4 as described previously in reference Citation2. The full-length PDCD7 ORF was generated using two ESTs (BU855083 and BC016992, ATCC, Manassas, VA) that contain overlapped partial PDCD7 ORF. 5′ part of PDCD7 ORF was amplified by primers: 5′-CCG CTC GAG CCA CCA TGG CCC TGC CAC CAT TCT T-3′ and 5′-AGG GCT CTC TCT TCA GCT TC-3′ using EST BU855083 as template. 3′ part of PDCD7 ORF was amplified by primers: 5′-TGT CCT CCA GCC TCA GCA GA-3′ and 5′-CCG GAA TTC AAT GCA GCT TAA CAG CAG TTG-3′ using EST BC016992 as template. The amplified 5′ part of PDCD7 ORF was digested with Xho I and BsmA I, and the amplified 3′ part of PDCD7 ORF was digested with EcoR I and BsmA I. Both digested fragments were mixed and ligated with Xho I and EcoR I digested pDsRed N1 vector. The resulting plasmid was sequenced to confirm that it contains correct the full-length PDCD7 ORF sequence. The mammalian myctagged PDCD7 expression vector was constructed by subcloning the full-length PDCD7 ORF in pDsRed N1 vector into pCEP4 vector using primers: 5′-CGG GGT ACC AAG CTT ACC ATG GAA CAA AAA CTC ATC TCA GAA GAG GAT CTG ATG GCC CTG CCA CCA TTC TT-3′ and 5′-CCG CTC GAG CTC GAG TTA CTA ATG CAG CTT AAC AG-3′. The resulting plasmid was named pCEP-PDCD7 and was sequenced to confirm that it contains the correct myctagged PDCD7 sequence.
Lentivirus-mediated shRNA silencing.
Scramble shRNA construct were purchased from Addgene. All other shRNA constructs were from Open Biosystems (Huntsville, AL). The 21-bp nucleotide sequences targeting human PDRG1 used in this study were #1, 5′-GAT GGT TTG CTT CGG GAA CAT-3′.
#2, 5′-GCT TCG GGA ACA TGT TTA TCA-3′.
Viruses were produced in HEK-293T cells using lipofectamine 2000 (Invitrogen) according to Addgene's protocol. Lentivirus particle was added to the cells in DMEM containing ∼8 µg/ml polybrene. Cells were cultured with 2 µg/ml puromycin (Sigma) for 7 days prior to analyses.
MTT assay.
MTT proliferation assays were done as we have previously described in reference 23. Briefly, cells were incubated with 1 mg/ml MTT (Sigma) for 3 hours, then the MTT precipitate was dissolved in isopropanol with 0.04 M HCl. Absorbance was read with a Bio-Rad SmartSpec 3100 at 570 nm with background subtraction read at 690 nm.
Northern blot analyses.
RNA extraction and Northern blot analyses were done as we have previously described in reference Citation2. Briefly, total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) and about 20 µg of total RNA for each sample were fractionated on 1.2% agarose gel and transferred to Nytran SuperCharge membranes (Schleicher & Schuell, Keene, NH). Human PDRG1 probes were labeled with 32P and prehybridizations and hybridizations were done in QuikHyb Solution (Stratagene, La Jolla, CA) at 65°C. To detect PDRG1 expression in paired human normal and cancer samples, Cancer Profiling Array I and Cancer Profiling Array II were purchased from Clontech, BD Biosciences, and probed with PDRG1 cDNA probe as described above.
Yeast two-hybrid screening.
Yeast two-hybrid screening was done using MATCHMAKER GAL4 Two-Hybrid System 3 (Clontech Laboratories Inc., Palo Alto, CA) per the company's manual. In brief, pBD-PDRG1 vector was co-transformed with pACT2 vector based human normal testis MATCHMAKER cDNA library (Clontech) into AH109 yeast cells. The transformed yeast cells were spread on minimal synthetic dropout medium (SD medium) agar plates without Adenine (Ade), Histidine (His), Leucine (Leu) and Tryptophan (Trp), but containing X-α-Gal. After 3 or 7 days incubation at 30°C, blue clones grown on SD medium agar plate were selected, then pACT2 plasmids in the yeast were rescued and sequenced to identify the inserts using Blast online comparison.
Immunoprecipitation and western blot analyses.
Immunoprecipitation were done as we have previously described in reference Citation24. Briefly, HEK-293T cells were transiently co-transfected with pCEP-PDRG1 and pCEP-PDCD7 or pCEP4 vectors using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Cells were harvested and lysed in lysis buffer (50 mmol/L Tris-HCl (pH 7.5), 100 mmol/L NaCl, 1% chaps, 10% glycerol, 0.5 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 µg/mL leupeptin, 1 mmol/L NaF and 1.5 µg/mL aprotinin). About 0.5 mg of 1 mg/ml total cell lysate was mixed with 2 µg of anti-myc monoclonal antibody 9E10 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and rotated for about 2 hours at 25 rpm in 4°C cold room. Then 100 µl of protein G-bead slur was washed with lysis buffer and added to total cell lysate. After 3–4 hours rotation, protein G-bead was pelleted by brief centrifuge and washed with lysis buffer three times. Bound protein was washed off by adding 100 µl 2x SDS-PAGE gel loading buffer and resolved by SDS-PAGE. Western blot analyses was performed as previously described in reference Citation25. HA-tagged PDRG1 was detected using a rat monoclonal anti-HA antibody 3F10 (Roche, Indianapolis, IN). Myctagged protein was detected using mouse monoclonal anti-myc antibody 9E10.
Immunohistochemical staining.
Frozen primary colon normal and tumor tissues from same patient were sectioned (6 µm) then fixed with 4% paraformaldehyde for 30 min. Endogenous peroxidase was quenched with hydrogen peroxide and the sections were then blocked in PBS with 10% goat sera for 20 min and incubated sequentially with anti-PDRG1 antibody for ∼1 hour, biotinylated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA) for 45 min and ABC reagent (Vector laboratories) for 30 min. After several washes, the sections were then incubated with peroxidase substrate DAB solution (Vector laboratories) and counterstained with hematoxylin.
Abbreviations
| PDRG1 | = | p53 and DNA damage-regulated gene 1 |
| UV | = | ultraviolet |
| PDCD7 | = | programmed cell death 7 |
| MAP1S | = | microtubules-associated protein 1S |
| HEK293T | = | human embryonic kidney 293T cells |
Figures and Tables
Figure 1 PDRG expression is selectively upregulated by genotoxic stress. (A) HCT15 and NIH3T3 cells were untreated as control (C) or treated with 2 µM adramycin (Adr), 30 µM etoposide (Etop) or 20 J/m2 ultraviolet radiation (UV) for 24 hours. Total RNA preparation and northern blot analyses were done as described in Material and Methods. A human PDRG cDNA probe was used for the blot contains RNA from HCT15 cells, and a mouse PDRG cDNA probe was used for the blot containing RNA from NIH3T3 cells. The integrity of total RNA and comparable loading are shown by ethidium bromide staining of the gel prior to transfer. (B) DU145 and HS578T cells were untreated as control (C) or treated with adramycin (Adr), etoposide (Etop), camtothecin (Cam), Taxol, thapsigargin (TG) or sulindac sulfide (SD) and then harvested after 24 hours or at indicated time points. Total RNA was extracted and northern blot analyses of PDRG mRNA levels were done using human PDRG cDNA probe as described in Material and Methods. Ethidium bromide staining of the gel shows integrity of RNA and comparable loading.
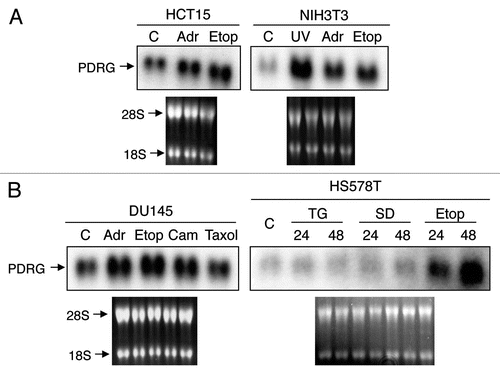
Figure 2 PDRG mRNA is overexpressed in human malignancies. Representative results showing PDRG expression in primary gastrointestinal tract tissue specimens. RNA samples representing matching normal (N) and tumor (T) specimens from colon, stomach and rectum were probed with PDRG cDNA probe as described in Material and Methods. Each pair indicates normal and tumor samples from a single patient. *Indicates higher PDRG mRNA level in tumor sample than that in the matching normal tissue sample.

Figure 3 PDRG1 protein is overexpressed in human colon cancer. Representative western blots showing PDRG expression in matching normal and tumor tissues. Protein samples prepared from primary matching normal (N) and tumor (T) tissues were subjected to western blotting using anti-PDRG antibody. Same blots were subsequently probed with anti-β-actin antibody to show loading in each lane.
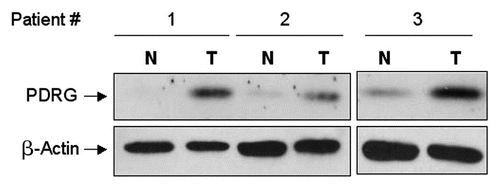
Figure 4 Immunohistochemical staining of PDRG in matched normal and cancer tissues from colon cancer patients. Frozen primary colon normal and tumor tissues from same patient were sectioned (6 µm) then fixed with 4% paraformaldehyde for 30 min. Endogenous peroxidase was quenched with hydrogen peroxide and the sections were then blocked in PBS with 10% goat sera for 20 min and incubated sequentially with anti-PDRG antibody for 1 hour, biotinylated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA) for 45 min and ABC reagent (Vector laboratories) for 30 min. After several washes, the sections were then incubated with peroxidase substrate DAB solution (Vector laboratories) and counterstained with hematoxylin. Brown color indicates the PDRG-specific signal. Photomicrographs from different regions captured at x10 and x20 magnifications are shown. Please note that PDRG1 is predominantly expressed by the epithelial cells as indicated by arrows and the tumor sample exhibits very strong PDRG-specific staining than the matching normal tissue. Insets show sections of enlarged images from x20 photomicrographs.
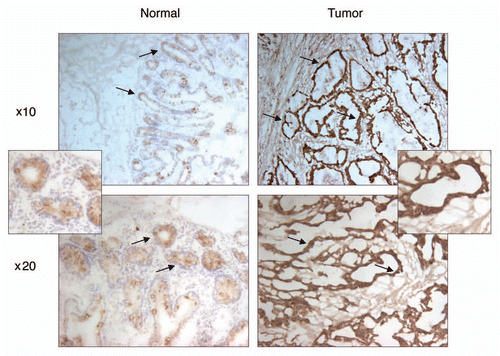
Figure 5 Depletion of PDRG suppresses tumor cell growth. Top, western blot analyses using anti-PDRG antibody show depletion of endogenous PDRG expression by lentiviral-based knockdown in the RKO cells; bottom, graphic representation of MTT assay depicting decreased cell proliferation due to PDRG knockdown in RKO cells. Cells were seeded at equal density and infected with lentiviral scrambled shRNA or PDRG-specific shRNAs. Nine days after infections, equal numbers of cells were reseeded onto 24-well plates (1 × 104 per well). Cell viability was determined by methyl thiazole tetrazolium (MTT) assay each day over a 7 day period. Points, mean of three independent experiments; bars, SE .
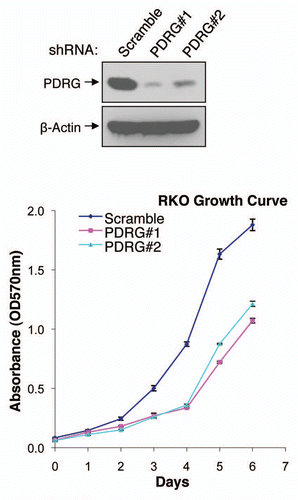
Figure 6 PDRG interacts with PDCD7 in mammalian cells. HE K 293T cells were transiently transfected with expression vector pCEP-PDRG1 and pCEP-PDCD7 or pCEP-PDRG1 and pCEP 4 vector without PDCD7 insert. After 24 hours, cells were harvested and lysed. About 500 total lysates were precipitated with anti-myc antibody (9E10) and protein G-bead. The precipitated proteins were resolved by SDS-PAGE along with 150 µg total cell lysates. HA-tagged PDRG was detected by western blotting with rat monoclonal anti-HA antibody and myc-tagged PDCD7 was detected using mouse monoclonal anti-myc antibody (9E10).
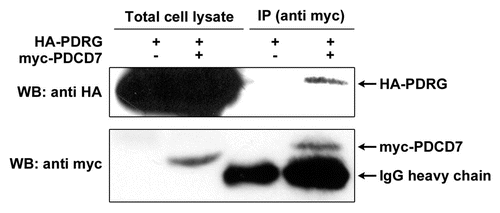
Table 1 PDRG expression in primary tumors
Additional material
Download Zip (97.4 KB)Acknowledgements
This work was supported in part by NIH grants ES005633-02, CA121850 and CA128096 to M.S.S. and Y.H. L.J. is supported by a pre-doctoral fellowship from the US Department of Defense (Grant BC073479).
References
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer 2001; 1:157 - 162
- Luo X, Huang Y, Sheikh MS. Cloning and characterization of a novel gene PDRG that is differentially regulated by p53 and ultraviolet radiation. Oncogene 2003; 22:7247 - 7257
- Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol 2001; 3:277 - 286
- Jiang L, Sheikh MS, Huang Y. Decision Making by p53: Life versus death. Mol Cell Pharmacol 2010; 2:69 - 77
- Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Treat 2003; 78:337 - 345
- Wong MP, Fung LF, Wang E, Chow WS, Chiu SW, Lam WK, et al. Chromosomal aberrations of primary lung adenocarcinomas in nonsmokers. Cancer 2003; 97:1263 - 1270
- Raidl M, Pirker C, Schulte-Hermann R, Aubele M, Kandioler-Eckersberger D, Wrba F, et al. Multiple chromosomal abnormalities in human liver (pre)neoplasia. J Hepatol 2004; 40:660 - 668
- Schlegel J, Stumm G, Scherthan H, Bocker T, Zirngibl H, Ruschoff J, et al. Comparative genomic in situ hybridization of colon carcinomas with replication error. Cancer Res 1995; 55:6002 - 6005
- Verbeek W, Schulten HJ, Sperling M, Tiesmeier J, Stoop H, Dinjens W, et al. Rectal adenocarcinoma with choriocarcinomatous differentiation: clinical and genetic aspects. Hum Pathol 2004; 35:1427 - 1430
- De Angelis PM, Stokke T, Beigi M, Mjaland O, Clausen OP. Prognostic significance of recurrent chromosomal aberrations detected by comparative genomic hybridization in sporadic colorectal cancer. Int J Colorectal Dis 2001; 16:38 - 45
- Lukasova E, Kozubek S, Falk M, Kozubek M, Zaloudik J, Vagunda V, et al. Topography of genetic loci in the nuclei of cells of colorectal carcinoma and adjacent tissue of colonic epithelium. Chromosoma 2004; 112:221 - 230
- Nakao K, Mehta KR, Fridlyand J, Moore DH, Jain AN, Lafuente A, et al. High-resolution analysis of DNA copy number alterations in colorectal cancer by array-based comparative genomic hybridization. Carcinogenesis 2004; 25:1345 - 1357
- Boonsong A, Marsh S, Rooney PH, Stevenson DA, Cassidy J, McLeod HL. Characterization of the topoisomerase I locus in human colorectal cancer. Cancer Genet Cytogenet 2000; 121:56 - 60
- Knosel T, Schluns K, Stein U, Schwabe H, Schlag PM, Dietel M, et al. Chromosomal alterations during lymphatic and liver metastasis formation of colorectal cancer. Neoplasia 2004; 6:23 - 28
- Will CL, Schneider C, Hossbach M, Urlaub H, Rauhut R, Elbashir S, et al. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA 2004; 10:929 - 941
- Mitsui K, Matsumoto A, Ohtsuka S, Ohtsubo M, Yoshimura A. Cloning and characterization of a novel p21(Cip1/Waf1)-interacting zinc finger protein, ciz1. Biochem Biophys Res Commun 1999; 264:457 - 464
- Coverley D, Marr J, Ainscough J. Ciz1 promotes mammalian DNA replication. J Cell Sci 2005; 118:101 - 112
- Dallol A, Cooper WN, Al-Mulla F, Agathanggelou A, Maher ER, Latif F. Depletion of the Ras association domain family 1, isoform A-associated novel microtubule-associated protein, C19ORF5/MAP1S, causes mitotic abnormalities. Cancer Res 2007; 67:492 - 500
- Liu L, Vo A, Liu G, McKeehan WL. Putative tumor suppressor RASSF1 interactive protein and cell death inducer C19ORF5 is a DNA binding protein. Biochem Biophys Res Commun 2005; 332:670 - 676
- Sardiu ME, Cai Y, Jin J, Swanson SK, Conaway RC, Conaway JW, et al. Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci USA 2008; 105:1454 - 1459
- Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, et al. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 1998; 93:863 - 873
- Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, et al. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol 2005; 18:1632 - 1640
- Montalbano J, Lui K, Sheikh MS, Huang Y. Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J Biol Chem 2009; 284:18129 - 18142
- Rong R, Jiang LY, Sheikh MS, Huang Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007; 26:7700 - 7708
- He Q, Shi J, Jones S, An J, Liu Y, Huang Y, et al. Smac deficiency affects endoplasmic reticulum stress-induced apoptosis in human colon cancer cells. Mol Cell Pharmacol 2009; 1:23 - 28