Abstract
Many solid tumors and hematologic malignancies lack expression of the enzyme methylthioadenosine phosphorylase (MTAP), due either to deletion of the MTAP gene or to methylation of the MTAP promoter. In cells that have MTAP, its natural substrate, methylthioadenosine (MTA), generated during polyamine biosynthesis, is cleaved to adenine and 5-methylthioribose-1-phosphate. The latter compound is further metabolized to methionine. Adenine and methionine are further metabolized and hence salvaged. In MTAP-deficient cells, however, MTA is not cleaved and the salvage pathway for adenine and methionine is absent. As a result, MTAP-deficient cells are more sensitive than MTAP-positive cells to inhibitors of de novo purine synthesis and to methionine deprivation. The challenge has been to take advantage of MTAP deficiency, and the changes in metabolism that follow, to design a strategy for targeted treatment. In this review, the frequency of MTAP-deficiency is presented and past and recent strategies to target such deficient cells are discussed, including one in which MTA is administered, followed by very high doses of a toxic purine or pyrimidine analog. In normal host cells, adenine, generated from MTA, blocks conversion of the analog to its toxic nucleotide. In MTAP-deficient tumor cells, conversion proceeds and the tumor cells are selectively killed. Successful mouse studies using this novel strategy were recently reported.
Introduction
Since the publication in 1988 of a review on tumors lacking MTAP,Citation1 both the results of a clinical trial,Citation2 and new information from many sources on the incidence of MTAP-deficiency, have been reported, prompting the present review. The focus here is on the potential for selectively targeting these tumors with inhibitors of de novo purine synthesis and on a new strategy using toxic purine and pyrimidine analogs.
The MTAP gene, located at chromosomal locus 9p21, is flanked by CDKN2A and miR-31 and the gene is frequently co-deleted, in many different tumors, with the CDKN2A and CDKN2B genes that encode the tumor suppressors p15, p16, p19 and with the genes for interferons alpha and beta that lie telomeric to miR-31.Citation3–Citation16 Selective MTAP deficiency, without co-deletion of the CDKN2 genes, has also been reported, due either to selective deletion of the MTAP locus or to methylation of the MTAP promoter.Citation17–Citation19 In normal cells, MTAP cleaves MTA, generated during the biosynthesis of polyamines, to adenine and 5-methylthioribose-1-phosphate (). The latter compound is further metabolized to methionine and adenine is converted to AMP. Cells lacking MTAP, however, are unable to salvage adenine or methionine from endogenous MTA. As a consequence, they are more sensitive to inhibitors of de novo purine synthesis than cells with intact MTAP, and are also more sensitive to methionine starvation.Citation20,Citation21 MTAP deficiency occurs frequently in both solid tumors and hematologic malignancies.Citation3–Citation16,Citation22 Solid tumors in which a high percentage lack MTAP include mesothelioma, non-small cell lung cancer (NSCLC), gliomas and pancreatic cancer (). MTAP gene deletions were also noted in 9 of 54 ampullary cancers and 4 of 33 biliary cancers.Citation5 In another series, MTAP deficiency was found in 10 of 28 biliary tract cancers.Citation16 In all of these solid tumors, loss of MTAP protein expression, detected by a monoclonal anti-MTAP antibody, was associated with loss of p16 protein expression.Citation5
Initial estimates of MTAP deficiency in NSCLC, by quantitative PCR-ELISA, were 44% in adenocarcinoma and 29% in squamous cell carcinoma,Citation18 while a larger series showed that 17% of patients with NSCLC were MTAP-negative.Citation6 A recent series of patients, screened by an immunohistochemical assay, showed that a lower percentage of patients with mesothelioma and pancreatic tumors lacked MTAP.Citation2 Other solid tumors reported to lack MTAP include soft tissue sarcoma, esophageal cancer, endometrial cancer, chondrosarcoma, osteosarcoma, gastrointestinal stromal tumors and chordoma (). Several reports have linked MTAP deficiency with increased aggressiveness of solid tumors. Compared to benign nevi, MTAP expression was reduced in primary malignant and metastatic melanoma.Citation17
Lack of MTAP expression is also common in hematologic malignanciesCitation22–Citation28 (). Using a probe that encompassed MTAP, CDKN2A and CDKN2B genes, 6 of the 16 diffuse large cell lymphomas and 1 of 9 of low-grade lymphomas had deletions of this region. Deletions were more common in the transformed lymphomas. In childhood B-lineage ALL, MTAP was inactive in 16% of patients. In mantle cell lymphoma (MCL), lack of MTAP expression was noted in 8 of 52 tumors. Patients with MCL and MTAP gene deletions had a shorter overall survival (mean, 16 months) than did patients with wild-type MTAP (mean, 64 months). Although ascribed to MTAP deficiency, the association with p16 and p19 deletions, known to affect prognosis, made it difficult to attribute the difference in survival and progression of tumors to MTAP deficiency alone. The recent findings that the MTAP gene may be inactivated by methylation would indicate that tests for genomic deletion may underestimate the prevalence of MTAP deficiency in tumors and that the test for MTAP deficiency should be lack of protein expression.
A recent study of MTAP knockout mice showed that mice homozygous for the MTAP null allele had an embryonic lethal phenotype, while mice heterozygous for MTAP appeared to be indistinguishable from wild-type mice, but died prematurely with lymphomas of T-cell origin. MTAP expression, measured by immunohistochemistry staining with an antibody to MTAP protein, and levels of MTAP RNA, were significantly reduced, compared to normal controls, in the tissues of these mice infiltrated with lymphoma. The investigators of this study proposed that the MTAP gene might be a tumor suppressor, independent of CDKN2A.Citation29
Information on MTAP-deficiency in primary breast cancer is lacking, although many cell lines, including the often-studied MCF-7 and MDA-MB-231, are MTAP-deficient. In a recent report on primary breast cancer, the frequency of loss of heterozygosity (LOH) of an MTAP intragenic marker was 19/119.Citation30 Among informative cases for intragenic markers in CDKN2A and MTAP, there was a 90% concordant LOH.
MTAP-deficient Cells are Sensitive to Inhibitors of Purine Biosynthesis
Several studies have reported that MTAP-deficient tumor cells are as much as 20 times more sensitive than MTAP-positive cells to inhibitors of purine biosynthesis, such as methotrexate (MTX), 6-mercaptopurine, azaserine (a potent inhibitor of the initial step in purine biosynthesis) and L-alanosine.Citation10,Citation20,Citation31 These inhibitors have differing metabolic actions and hence the increase in sensitivity of MTAP-deficient cells to inhibitors varies. Thus MTX is considered to be primarily an inhibitor of dihydrofolate reductase, and polyglutamate forms of MTX are potent inhibitors of purine biosynthesis.Citation32 Both 6-mercaptopurine and 6-thioguanine (6-TG) are rapidly converted to nucleotides, and inhibit de novo purine synthesis. 6-TG, in addition, is incorporated into DNA, and cells lacking mismatch repair are more resistant to 6-TG treatment.Citation33 L-alanosine, an amino acid analog, is converted to L-alanosinyl-5-amino-4-imidazole carboxylic acid ribonucleotide, which inhibits the penultimate step from IMP to AMP. Hypoxanthine can reverse the action of some inhibitors of purine synthesis, such as azaserine. Hypoxanthine lies upstream of IMP and hence, in the presence of L-alanosine, hypoxanthine would not be converted to AMP, nor would it reverse the action of L-alanosine.
Perhaps the most convincing study showing the relationship of MTAP deficiency to sensitivity to purine and methionine depletion was that by Hori et al.Citation31 who transfected MTAP cDNA into an A549 lung cancer cell line lacking MTAP, thus minimizing additional genetic alterations that might affect chemotherapeutic sensitivity. Cells lacking MTAP protein were more sensitive to MTX, 5,10-dideazafolate (a purine synthesis inhibitor), L-alanosine, and also to methionine depletion. MTA was able to completely rescue the cell lines that contained MTAP, but not the cell lines deficient in MTAP, from these inhibitors and from methionine restriction.
The increased sensitivity of MTAP-deficient tumor cells to inhibitors of purine biosynthesis has important implications for clinical applications, as was recognized in the design of a trial described below in reference Citation2.
Clinical Studies Targeting Tumors that Lack MTAP
To date, in the only clinical trial reported in patients whose tumors specifically lacked MTAP, the purine synthesis inhibitor L-alanosine was used.Citation2 Previous Phase II studies with this drug did not take into account MTAP status, and no appreciable therapeutic activity was noted in these trials.Citation34–Citation37 Patients with advanced refractory tumors—NSCLC, mesothelioma, soft tissue sarcoma, pancreatic cancer and osteosarcoma—that were shown by immunohistochemistry to lack MTAP expression were treated with a continuous infusion of L-alanosine, at a starting dose of 80 mg/m2 daily for 5 days, repeated every 21 days. Stable disease lasting a median of 4 months was recorded in 5 of 13 patients with mesothelioma, and 8 of 42 patients with other histologies. While this recent study in patients with MTAP-deficient tumors was therefore not very promising, as compared to treatment with cisplatin and pemetrexed that produces tumor regressions in 30% of patients with mesothelioma,Citation38 why—given the preclinical data—did it not work better? As the investigators of this study noted, no pharmacodynamic studies were performed on tumors to show that L-alanosine entered cells and inhibited purine biosynthesis. It is also possible that tumors may have salvaged enough adenine or adenosine from blood or the microenvironment to compete with L-alanosine for activating enzymes.
A modified trial might produce better results. Since tumors lacking MTAP have increased sensitivity to L-alanosine, perhaps lower, non-toxic doses, administered for longer periods, would be more effective. Other trials to consider could involve the choice of a different inhibitor of purine biosynthesis, combined with a cytocidal drug. Patients with mesothelioma and osteosarcoma have been shown to respond to antifolates, and the combination of pemetrexed and cisplatin is an FDA-approved treatment for mesothelioma. Another study, perhaps, would target these tumors with the combination of MTX and a thiopurine and compare the response of patients whose tumors lacked or did not lack MTAP.Citation39 Given the substantial number of hematologic tumors with MTAP deficiency, and the known response of these tumors to MTX and thiopurines, another important study would be to determine if MTAP deficiency correlates with increased response and survival to treatments containing MTX alone and in combination with other drugs, specifically with 6-mercaptopurine, as used in maintenance therapy in patients with acute lymphocytic leukemia, and in patients with T-cell lymphomas.
MTA as a Protective Agent
MTA is derived as a byproduct of the synthesis of spermidine and spermine. As mentioned earlier, MTA is rapidly cleaved by MTAP, an enzyme ubiquitously expressed in tissues, generating adenine and 5-methylthioribose-1-phosphate. The latter substance is converted to methionine through a complex set of reactions (). Thus the metabolism of MTA is an important salvage pathway that generates purines and methionine. In the absence of MTAP, MTA may accumulate and inhibit, by feedback, spermine and, to a lesser extent, spermidine biosynthesis and ornithine decarboxylase, in part by its metabolite, 4-methylthio-2-oxobutanate.Citation40 Tumor cells lacking MTAP may secrete MTA instead of metabolizing it.
MTA at high doses, in experimental models, has been reported to have protective effects against liver damage and, even when given intraperitoneally to rodents over extended periods, MTA showed no toxicity.Citation41,Citation42 MTA has also been administered orally to 50 volunteers at 600 mg daily for one month and to 10 volunteers at 1,600 mg daily for one month, without toxicity.Citation43,Citation44 Clinical trials of MTA alone, as an anti-inflammatory agent and as an inhibitor of melanoma and colon cancer growth, have been proposed.Citation45–Citation47
Selective Killing of Tumors Deficient in MTAP: A New Strategy
Strategies to take advantage of MTAP-deficiency in many tumors were proposed as far back as 1981. These include inhibition of de novo purine synthesis, and methionine deprivation by a methioninase.Citation21,Citation48 In a new proposal,Citation49 MTAP-deficient tumors are treated with both MTA and a drug which requires phosphoribosylation for conversion to its toxic nucleotide (). These drugs include adenine analogs, such as 2,6-diaminopurine, 6-methylpurine and 2-fluoroadenine, and two analogs that are in clinical use: 6-TG and 5-fluorouracil (5-FU). In normal host cells, when MTA is administered, substantial adenine is produced by the action of MTAP. Adenine then competes effectively with a co-administered drug for phosphoribosylation by 5-phosphoribosyl-1-pyrophosphate (PRPP). Hence the drug is not converted to its nucleotide, as it must be for toxic activity. Tumor cells lacking MTAP, however, cannot derive adenine from MTA. Hence PRPP remains at adequate levels, and the co-administered drug can be readily converted to its toxic nucleotide. A high degree of selectivity of the treatment is assured by the clear difference between tumor and host cells in MTAP activity. The method may be potentiated by providing MTA in advance of the drug, so as to decrease PRPP levels in host cells before exposure to the drug. In a pilot study of the strategy, intraperitoneal pre-treatment of Swiss-Webster mice with MTA protected them from subsequent lethal doses of 6-TG.Citation50,Citation51 In a second study,Citation52 NOD-SCID mice bearing the MTAP-negative CCRF-CEM human T-cell leukemia were given MTA (100 mg/kg) intraperitoneally, followed one hour later by 6-TG (75 mg/kg), on days 1, 4 and 7. By day 13, the tumors had regressed completely, with minimal loss of weight, although the tumors did return in the weeks following ( and B). Animals treated with 6-TG (75 mg/kg) alone on days 1, 4 and 7 had marked tumor regression but died of toxicity by day 13. The results of this latter study demonstrate proof-of-concept: MTA protected the host from the cytocidal drug 6-TG, while the tumor was not protected. Moreover, the protection of the host mice by MTA from high—in fact supralethal—doses of 6-TG suggests that 6-TG might be safely used at levels that far exceed the current maximum tolerated dose for humans, presently limited by toxicity to bone marrow. 6-TG is primarily used in treatment of leukemia and lymphoma but, if the host were protected by MTA, much higher doses of 6-TG would be tolerated, and higher doses may show antitumor effects even in solid tumors lacking MTAP.
This treatment strategy applies as well to MTA and 5-FU. 5-FU is converted to its toxic nucleotide by orotate phosphoribosyltranferase, with PRPP serving as the donor of the phosphoribosyl group (as is the case with 6-TG). Two experiments with similar results have been performed that clearly demonstrate that MTA can protect mice from lethal doses of 5-FU. The results of the second experiment are shown in . In groups 3 and 4, MTA protected all mice from the lethal effect of 5-FU. There was very little weight loss in groups 3 and 4, although group 4 had more weight loss than group 3 (data not shown). Historically the maximum tolerated dose of 5-FU using this schedule is 75 mg/kg (q4dx4). In this experiment, therefore, the tolerated dose of 5-FU was increased from 75–200 mg/kg.
One caveat to this treatment strategy might be that when MTA is administered, sufficient adenine would be generated by normal tissues expressing MTAP, and utilized by an MTAP-deficient tumor, consuming PRPP and thus decreasing the activation of purine analogs or 5-FU. There may be an optimal dose of MTA, therefore, for protection of normal tissues without compromising antitumor effects.
Conclusions
Since the subject of MTAP-deficiency in tumors was last reviewed over twenty years ago, the variety of tumors discovered to have this deficiency has increased steadily. The list now includes many solid tumors and hematologic malignancies with a dire prognosis. The challenge has been to take advantage of MTAP-deficiency to design a strategy for targeted treatment. This review discusses early proposals, and one clinical trial that involved administering an inhibitor of de novo purine synthesis but was not successful. Several other possible strategies are offered. In one, MTA is administered, followed by very high doses of a toxic purine or pyrimidine analog. In normal host cells, adenine, generated from MTA, blocks conversion of the analog to its toxic nucleotide. In MTAP-deficient tumor cells, conversion proceeds and the tumor cells are selectively killed. To optimize doses and timing of MTA for protection of normal cells, without compromising antitumor effects in MTAP deficient tumors, further studies are planned. If warranted, clinical studies would follow.
Figures and Tables
Figure 1 MTAP metabolic pathway. In normal cells, MTAP cleaves MTA, a by-product of polyamine biosynthesis, into adenine and 5-methylthioribose-1-phosphate (MTR-1-P). Adenine is converted to AMP by the ubiquitous enzyme adenine phosphoribosyltransferase (APRT), with phosphoribosyl-1-pyrophosphate (PRPP) serving as donor of the phosphoribosyl group. MTR-1-P is converted by a series of steps to methionine. AMP is also produced in cells by de novo purine biosynthesis. In addition to APRT, other cellular phosphoribosyltransferases, such as hypoxanthine-guanine phosphoribosyltransferase and orotate phosphoribosyltranferase, convert purines and pyrimidines to nucleotides.Citation49
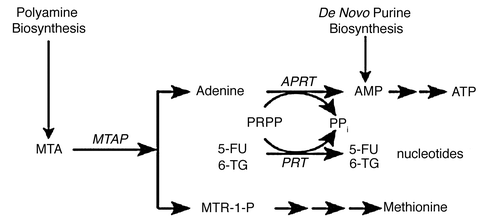
Figure 2 MTA protects host tissues but not MTAP-deficient tumor cells from 6-TG toxicity. (A) NOD-SCID mice (N = 6, per group) were inoculated with one million CCRF-CEM human lymphoblastic leukemia cells and when tumor size was 500 mm3 the mice were treated with either 6-TG, MTA or the combination of the two drugs (MTA followed one hour later by 6-TG) or saline, ♦ control; □, MTA; 100 mg/kg; ▴, 6-TG, 75 mg/kg; X, MTA, 100 mg/kg plus 6-TG, 75 mg/kg. (B) Effect of treatments on body weights. Control and MTA treated mice were sacrificed by day 13 due to large tumors. 6-TG treated mice also died by day 13, due to 6-TG toxicity, while marked tumor regression with minimal toxicity was noted (<10% weight loss) in mice treated with MTA followed one hour later by 6-TG. Vertical bars: SD. (♦, control; □, MTA; 100 mg/kg; ▴, 6-TG, 75 mg/kg; X, MTA, 100 mg/kg plus 6-TG, 75 mg/kg).
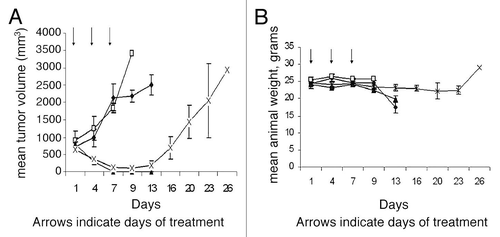
Table 1 MTAP deficiency in solid tumors
Table 2 MTAP deficiency in hematologic malignancies
Table 3 Effect of MTA on the toxicity of 5-FU in NCr-nu/nu mice
References
- Carson DA, Nobori T, Kajander EO, Carrera CJ, Kubota M, Yamanaka H. Methylthioadenosine (MeSAdo) phosphorylase deficiency in malignancy. Adv Exp Med Biol 1988; 250:179 - 185
- Kindler HL, Burris HA III, Sandler AB, Oliff IA. A phase II multicenter study of L-alanosine, a potent inhibitor of adenine biosynthesis, in patients with MTAP-deficient cancer. Invest New Drugs 2009; 27:75 - 81
- Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase and the alpha and beta-interferons in human pancreatic cell carcinoma cell lines and its implications for chemotherapy. Cancer Res 1996; 56:1083 - 1090
- Illei PB, Rusch VW, Zakowski MF, Ladanyi M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res 2003; 9:2108 - 2113
- Hustinx SR, Hruban RH, Leoni LM, Iacobuzio-Donahue C, Cameron JL, Yeo CJ, et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in peri-ampullary cancer: a potential new target for chemotherapy. Cancer Biol Ther 2005; 4:83 - 86
- Watanabe F, Takao M, Inoue K, Nishioka J, Nobori T, Shiraishi T, et al. Immunohistochemical diagnosis of methylthioadenosine phosphorylase (MTAP) deficiency in non-small cell lung cancer. Lung Cancer 2009; 63:39 - 44
- García-Castellano JM, Villanueva A, Healey JH, Sowers R, Cordon-Cardo C, Huvos A, et al. Methylthioadenosine phosphorylase gene deletions are common in osteosarcoma. Clin Cancer Res 2002; 8:782 - 787
- Miyazaki S, Nihioka J, Shiraishi T, Matsumine A, Uchida A, Nobori T. Methylthioadenosine phosphorylase deficiency in Japanese osteosarcoma patients. Int J Oncol 2007; 5:1069 - 1076
- Chow WA, Bedell V, Gaytan P, Borden E, Goldblum J, Hicks D, et al. Methylthioadenosine phosphorylase gene deletions are frequently detected by fluorescence in situ hybridization in conventional sarcomas. Cancer Genet Cytogenet 2006; 166:95 - 100
- Li W, Su D, Mizobuchi H, Martin DS, Gu B, Gorlick R, et al. Status of methylthioadenosine phosphorylase and its impact on cellular response to L-alanosine and methylmercaptopurine riboside in human soft tissue sarcoma cells. Oncol Res 2004; 14:373 - 379
- Nobori T, Karras JG, Della Ragione F, Waltz TA, Chen PP, et al. Absence of methylthioadenosine phosphorylase in human gliomas. Cancer Res 1991; 51:3193 - 3197
- Huang HY, Li SH, Yu SC, Chou FF, Tzeng CC, Hu TH, et al. Homozygous deletion of MTAP gene as a poor prognosticator in gastrointestinal stromal tumors. Clin Cancer Res 2009; 15:6963 - 6972
- Wong YF, Chung TK, Cheung TH, Nobori T, Chang AM. MTAP gene deletion in endometrial cancer. Gynecol Obstet Invest 1998; 45:272 - 276
- Powell EL, Leoni LM, Canto MI, Forastiere AA, Iocobuzio-Donahue CA, Wang JS, et al. Concordant loss of MTAP and p16/CDKN2A expression in gastroesophageal carcinogenesis: evidence of homozygous deletion in esophageal noninvasive precursor lesions and therapeutic implications. Am J Surg Pathol 2005; 29:1497 - 1504
- Sommer J, Itani DM, Homlar KC, Forastiere AA, Iocobuzio-Donahue CA, Wang JS, et al. Methylthioadenosine phosphorylase and activated insulin-like growth factor-1 receptor/insulin receptor: potential targets in chordoma. J Pathol 2010; 220:608 - 617
- Karikari CA, Mullendore M, Eshleman JR, Argani P, Leoni LM, Chattopadhyay S, et al. Homozygous deletions of methylthioadenosine phosphorylase in human biliary tract cancers. Mol Cancer Ther 2005; 4:1860 - 1866
- Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R, et al. Characterization of methylthioadenosine phosphorylase expression in malignant melanoma. Am J Pathol 2003; 163:683 - 690
- Schmid M, Malicki D, Nobori T, Rosenbach MD, Campbell K, Carson DA, et al. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC). Oncogene 1998; 17:2669 - 2675
- Ishii M, Nakazawa K, Wada H, Nishioka J, Nakatani K, Yamada Y, et al. Methylthioadenosine phosphorylase gene is silenced by promoter hypermethylation in human lymphoma cell line DHL-9: another mechanism of enzyme deficiency. Int J Oncol 2005; 26:985 - 991
- Chen ZH, Olopade OI, Savarese TM. Expression of methylthioadenosine phosphorylase cDNA in p16-, MTAP-malignant cells: restoration of methylthioadenosine phosphorylase-dependent salvage pathways and alterations of sensitivity to inhibitors of purine de novo synthesis. Mol Pharmacol 1997; 52:903 - 911
- Tisdale MJ. Methionine synthesis from 5′-methylthioadenosine by tumour cells. Biochem Pharmacol 1983; 32:2915 - 2920
- Harasawa H, Yamada Y, Kudoh M, Sugahara K, Soda H, Hirakata Y, et al. Chemotherapy targeting methylthioadenosine phosphorylase (MTAP) deficiency in adult T-cell leukemia (ATL). Leukemia 2002; 16:1799 - 1807
- M'Soka TJ, Nishioka J, Taga A, Kato K, Kawasaki H, Yamada Y, et al. Detection of methylthioadenosine phosphorylase (MTAP) and p16 gene deletion in T-cell acute lymphoblastic leukemia by real time quantitative PCR assay. Leukemia 2000; 14:935 - 940
- Traweek ST, Riscoe MK, Ferno AJ, Braziel RM, Magenis RE, Fitchen JH. Methylthioadenosine phosphorylase deficiency in acute leukemia: pathologic, cytogenetic and clinical features. Blood 1988; 71:1568 - 1573
- Hori Y, Hori H, Yamada Y, Carrera CJ, Tomonaga M, Kamihira S, et al. The methylthioadenosine phosphorylase gene is frequently co-deleted with the p16INK4A gene in acute type adult T-cell leukemia. Int J Cancer 1998; 75:51 - 56
- Dreyling MH, Roulston D, Bohlander SK, Vardiman J, Olopade OI. Codeletion of CDKN2 and MTAP genes in a subset of non-Hodgkin's lymphoma may be associated with histologic transformation from low-grade to diffuse large cell lymphoma. Genes Chromosomes Cancer 1998; 22:72 - 78
- Mirebeau D, Acquaviva C, Suciu S, Bertin R, Dastugue N, Robert A, et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica 2006; 91:881 - 885
- Marcé S, Balague O, Colomo L, Martinez A, Holler S, Villamor N, et al. Lack of methylthioadenosine phosphorylase expression in mantle cell lymphoma is associated with shorter survival: implications for a potential targeted therapy. Clin Cancer Res 2006; 12:3754 - 3761
- Kadariya Y, Yin B, Tang B, Shinton SA, Quinlivan EP, Hua X, et al. Mice heterozygous for germ-line mutations in methylthioadenosine phosphorylase (MTAP) die prematurely of T-cell lymphoma. Cancer Res 2009; 69:5961 - 5969
- Vieira de Oliveira SF, Oliveira MM, Urban CA, de Lima RS, Cavalli IJ, Ribeiro EM. Lack of association between LOH in the 9p region and clinicopathologic parameters in primary breast cancer. Cancer Genet Cytogenet 2010; 200:23 - 27
- Hori H, Tran P, Carrera CJ, Hori Y, Rosenbach MD, Carson DA, et al. Methylthioadenosine phosphorylase cDNA transfection alters sensitivity to depletion of purine and methionine in A549 lung cancer cells. Cancer Res 1996; 56:5653 - 5658
- Allegra CJ, Drake JC, Jolivet J, Chabner BA. Inhibition of phosphoribosylaminoimidazolecarboximide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc Natl Acad Sci USA 1985; 82:4881 - 4885
- Swann PF, Waters TR, Moulton DC, Xu YZ, Zheng Q, Edwards M, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996; 273:1109 - 1111
- Rubin J, Hineman V, Moertel CG, Schutt AJ, Hahn RG. A phase II clinical trial of L-alanosine in advanced large bowel carcinoma. Am J Clin Oncol 1983; 6:191 - 193
- Creagan ET, Long HJ, Ahmann DL, Green SJ. Phase II evaluation of L-alanosine (NSC-153353) for patients with disseminated malignant melanoma. Am J Clin Oncol 1984; 7:543 - 544
- Creagan ET, Schutt AJ, Ingle JN, O'Fallon JR. Phase II clinical trial of L-alanosine in advanced upper aerodigestive cancer. Cancer Treat Rep 1983; 67:1047
- Von Hoff DD, Green SJ, Neidhart JA, Fabian C, Budd T, Boyd JF, et al. Phase II study of L-alanosine (NSC 153353) in patients with advanced breast cancer. A Southwest Oncology Group Study. Invest New Drugs 1991; 9:87 - 88
- Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: biochemical and cellular pharmacology, mechanisms and clinical applications. Mol Cancer Therapy 2007; 6:404 - 417
- Tan CT, Wollner N, Trippett T, Goker E, Tong WP, Kheradpour A, et al. Pharmacologic-guided trial of sequential methotrexate and thioguanine in children with advanced malignancies. J Clin Oncol 1994; 12:1955 - 1962
- Subhi AL, Tang B, Balsara BR, Altomare DA, Testa JR, Cooper HS, et al. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin Cancer Res 2004; 10:7290 - 7296
- Simile MM, Banni S, Angioni E, Carta G, De Miglio MR, Muroni MR, et al. 5′-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J Hepatol 2001; 34:386 - 394
- Wolford RW, Riscoe MK, Johnson L, Ferro AJ, Fitchen JH. Effect of 5′-methylthioadenosine (a naturally occurring nucleoside) on murine hematopoiesis. Exp Hematol 1984; 12:867 - 871
- Stramentinoli G, Gennari F. Adenosine derivatives of anti-inflammatory and analgesic activity, and therapeutic compositions which contain them as their active principle US patent 4454122 1984;
- Moratti E. Pharmaceutical compositions containing 5′-deoxy-5′-methylthioadenosine S-adenosylmethionine and their salts for reducing seborrhea US patent 5753213 1998;
- Moreno B, Hevia H, Santamaria M, Sepulcre J, Munoz J, Garcia-Trevijano ER, et al. Methylthioadenosine reverses brain autoimmune disease. Ann Neurol 2006; 60:323 - 334
- Andreu-Pérez P, Hernandez-Losa J, Moliné T, Gil R, Grueso J, Pujol A, et al. Methylthioadenosine (MTA) inhibits melanoma cell proliferation and in vivo tumor growth. BMC Cancer 2010; 10:265
- Li TW, Zhang Q, Oh P, Xia M, Chen H, Bemanian S, et al. S-Adenosylmethionine and methylthioadenosine inhibit cellular FLICE inhibitory protein expression and induce apoptosis in colon cancer cells. Mol Pharmacol 2009; 76:192 - 200
- Kamatani N, Nelson-Rees WA, Carson DA. Selective killing of human malignant cell lines deficient in methylthioadenosine phosphorylase, a purine metabolic enzyme. Proc Natl Acad Sci USA 1981; 78:1219 - 1223
- Lubin M, Lubin A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: a novel strategy. PLoS ONE 2009; 4:5735
- Lubin M, Lubin A. Strategy for selective killing of tumors deficient in methylthioadenosine phosphorylase (MTAP): a progress report. Proc Am Assoc Cancer Res 2009; 4:e5735
- Clarke DA, Hamilton LD, Philips FS, Sternberg SS. Effects of thioguanine in mammals. Cancer 1956; 9:1092 - 1101
- Bertino JR, Johnson-Farley N, Perez RP, Lubin A, Lubin M. Regression of a human T-cell leukemia lacking the methylthioadenosine phosphorylase (MTAP) gene without toxicity of 6-thioguanine (6TG) by pre-treatment with methylthioadenosine. Proc Am Assoc Cancer Res 2010; 5393