Abstract
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase belonging to the HER family of receptor tyrosine kinases. Receptor activation upon ligand binding leads to down stream activation of the PI3K/AKT, RAS/RAF/MEK/ERK and PLCγ/PKC pathways that influence cell proliferation, survival and the metastatic potential of tumor cells. Increased activation by gene amplification, protein overexpression or mutations of the EGFR has been identified as an etiological factor in a number of human epithelial cancers (e.g., NSCLC, CRC, glioblastoma and breast cancer). Therefore, targeting the EGFR has been intensely pursued as a cancer treatment strategy over the last two decades. To date, five EGFR inhibitors, including three small molecule tyrosine kinase inhibitors (TKIs) and two monoclonal antibodies have gained FDA approval for use in oncology. Both approaches to targeting the EGFR have shown clinical promise and the anti-EGFR antibody cetuximab is used to treat HNSCC and CRC. Despite clinical gains arising from use of cetuximab, both intrinsic resistance and the development of acquired resistance are now well recognized. In this review we focus on the biology of the EGFR, the role of EGFR in human cancer, the development of antibody-based anti-EGFR therapies and a summary of their clinical successes. Further, we provide an in depth discussion of described molecular mechanisms of resistance to cetuximab and potential strategies to circumvent this resistance.
Introduction
Approximately 40 years ago, Graham Carpenter performed experiments identifying the presence of specific binding receptors for EGF on human fibroblast cells.Citation1 In 1975, Carpenter and co-workers identified the epidermal growth factor receptor (EGFR) as a 170 KDa membrane protein that increased 32P incorporation in response to EGF treatment of A431 epidermoid carcinoma cells.Citation2 In 1984, a group of collaborators isolated, cloned and sequenced the human EGFR from normal placental cells and A431 tumor cells.Citation3 During this same time frame, it was discovered that modification of proteins by phosphorylation on tyrosine residues might be a critical step in tumorigenesis.Citation4,Citation5 Shortly after these discoveries EGFR was recognized as a receptor protein tyrosine kinase. This two-decade effort led to the identification of the prototypical receptor tyrosine kinase (RTK) and its ligand. The identification of EGFR as a RTK contributed to pivotal studies advancing our understanding of RTK activationCitation6,Citation7 and phosphorylation. Elucidation of EGFR regulation of downstream signaling also contributed to understanding critical pathways involved in cell proliferation and survival.
During the 1980s, several reports described the overexpression of EGFR in a variety of epithelial tumors supporting the hypothesis that dysregulated EGFR expression and signaling play a critical role in the etiology of human cancers. These findings led to hallmark studies designed to target EGFR via two fundamental approaches. The first approach was the development of an antibody directed against the EGFR extracellular domain. The second approach focused on the rational design of anti-EGFR small-molecule tyrosine kinase inhibitors. Both targeting approaches have proved clinically useful, however, resistance (intrinsic and acquired) to both modalities is a serious treatment issue. Understanding the molecular mechanisms of resistance to EGFR inhibitors is vitally important and will lead to improvement of these promising molecular targeting agents and increased benefit to patients. In this review, we focus on the biology of EGFR, the role of EGFR in human cancer, the development of antibody-based anti-EGFR therapies, and a summary of their clinical successes. Further, we provide an in depth discussion of known molecular mechanisms of resistance to the EGFR antibody cetuximab and potential strategies to overcome resistance to antibody therapy.
EGFR Biology
Aberrant expression or activity of the EGFR has been identified as an important biological factor in many human epithelial cancers including head and neck squamous cell carcinoma (HNSCC), non-small cell lung cancer (NSCLC), colorectal cancer (CRC), breast, pancreatic and brain cancer. EGFR is a member of the EGF receptor tyrosine kinase family, which consists of the EGFR (ErbB1/HER1), HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4). These receptors contain an extracellular ligand-binding domain (domains I–IV), a single membrane-spanning region, a juxtamembrane nuclear localization signal (NLS), a cytoplasmic tyrosine kinase domain (TKD) and a C-terminal tail housing several tyrosine residues for propagating down stream signaling.
HER receptors are ubiquitously expressed in various cell types, but primarily include those of epithelial, mesenchymal and neuronal origin. Under homeostatic conditions, receptor activation is tightly regulated by the availability of ligands, which collectively form the EGF growth factor family. This family is divided into three distinct groups. The first includes EGF, transforming growth factor alpha (TGFα) and amphiregulin (AR), which all bind specifically to EGFR. The second group includes betacellulin (BTC), heparin-binding EGF (HB-EGF) and epiregulin (EPR), which bind to both EGFR and HER4. The third group is composed of the neuregulins (NRG1-4) and is further subdivided based on their ability to bind both HER3 and HER4 (NRG1 and NRG2) or only HER4 (NRG3 and NRG4) (reviewed in ref. Citation8 and Citation9). HER2 has no known ligand.Citation10
Ligand precursors are cleaved by ADAM proteases at the cell surface and are subsequently secreted. EGFR ligands can participate in autocrine, paracrine, juxtacrine and/or endocrine activation of EGFR.Citation11 Ligand binding to the leucine-rich repeats in domains I and III of the EGFR extracellular domain triggers a conformational change in the receptor that exposes the dimerization loop (domain II) to other receptors on the cell surface (reviewed in ref. Citation12). Exposure of domain II allows for homo- or heterodimerization with other HER family members, activating EGFR kinase function. This induces both autophosphorylation and transphosphorylation of the C-terminal cytoplasmic tails of the receptor pairs. HER3 is the only family member that lacks intrinsic kinase activity,Citation13 however, downstream signaling is readily achieved through heterodimerization.Citation14
Phosphorylated cytoplasmic tails serve as docking sites for numerous proteins that contain Src Homology 2 (SH2) and phosphotyrosine binding domains. EGFR activation stimulates many complex intracellular signaling pathways tightly regulated by the presence and identity of ligand, heterodimer composition and availability of phosphotyrosine-binding proteins. The three primary signaling pathways activated by EGFR include the RAS/RAF/MEK/ERK, PI3K/AKT and PLCγ/PKC axes; however, SRC tyrosine kinases and STAT activation have also been well documented. The intersections of those pathways are shown in .
(right) illustrates the RAS/RAF/MEK/ERK pathway, which leads to cell proliferation and is a central element in many human tumors (reviewed in ref. Citation15). After activation and subsequent autophosphorylation, C-terminal phospho-tyrosine residues on RTKs including PDGFR, VEGFR, HER and FGFR act as binding sites for the SH2-domain-containing protein Grb2. Grb2 recruits the guanine nucleotide exchange factor SOS via its SH3 domain, and promotes binding of GTP to Ras, a small G-protein responsible for activation of the MAPK cascade. Ras-GTP initiates this cascade by binding to and activating the RAF kinase (MAPKKK). Activated RAF in turn binds to and phosphorylates MEK (MAPKK), which then phosphorylates ERK1/2 (MAPK). Upon activation, ERK kinases can translocate to the nucleus and activate several other kinases including MNK1 and MNK2, MSK1 and MSK2, and RSK. MAPK can also phosphorylate several transcription factors including Elk-1, peroxisome-proliferator-activated receptor γ (PPARγ), signal transducer and activator of transcription 1 and 3 (STAT1 and STAT3), C-myc and AP-1. Activation of transcription factors leads to an increased transcription of genes involved in cellular proliferation, most notably cyclin D1.
Growth factor binding to RTKs also initiates the PI3K/AKT/mTOR pathway (, left) (reviewed in ref. Citation16). Activated RTKs can recruit PI3K to the cell membrane. PI3K phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) yields the second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 serves as a membrane-docking site for the serine/threonine protein kinase AKT, which binds to PIP3 with high affinity through its pleckstrin homology (PH) domain. Once positioned on the plasma membrane, AKT is phosphorylated by two kinases, phosphoinositide dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2), leading to its full activation. Phosphorylated AKT regulates a variety of different substrates, influencing cell survival, proliferation and cellular metabolism. One main effector of AKT activation is the mammalian target for rapamycin complex 1 (mTORC1). The activation of mTORC1 is regulated by AKT via the tuberous sclerosis protein complex 2 (TSC2). Phosphorylation of TSC2 by AKT cancels inhibition of the small G-protein, Ras homolog enriched in brain (RHEB), allowing binding to the mTOR catalytic domain, activating the intrinsic kinase function of mTOR. mTOR is a serine/threonine kinase having two well studied substrates, p70-S6 kinase 1 and 4E-BP1. Phosphorylation of p70-S6 kinase 1 by mTOR leads to activation and initiation of protein synthesis via the S6 ribosomal subunit. Additionally, phosphorylation of 4E-BP1 cancels inhibition of the translation initiation factor eIF4E. Overall, the activation of protein synthesis is pivotal to cancer cell growth and survival, consistent with the anticancer effects of the mTOR inhibitor rapamycin. PIP3 is dephosphorylated to yield PIP2 by the tumor suppressor protein PTEN (phosphatase and tensin homologue deleted in chromosome 10), which attenuates AKT signaling.
The protein kinase C (PKC) pathway (, left) also plays an important role in mediating the effects of activated growth factor receptors including EGFR (reviewed in ref. Citation17–Citation20). After EGFR activation, phospholipase C (PLC) interacts with phospho-tyrosine sites on EGFR via its SH2 domain. This leads to the specific phosphorylation of PLC by EGFR and dissociation from EGFR. Activated PLC in turn interacts with the plasma membrane, most likely mediated by PIP3 via a PH domain, where it cleaves PIP2 to inositol triphosphate (IP3) and diacyglycerol (DAG). IP3 can diffuse into the cytosol and bind IP3 receptors on the endoplasmic reticulum (ER) to induce calcium influx into the cell from the ER. DAG remains in the membrane where it can activate PKC via a conformational change that removes the pseudosubstrate region from the catalytic active site. Activated PKC is a potent serine/threonine kinase second messenger, capable of phosphorylating a plethora of substrates ultimately leading to complex cellular process including proliferation, apoptosis, cell survival and cell migration.
A new and less well-characterized EGFR signaling network, the nuclear EGFR signaling network (), has emerged over the last decade and been implicated in both cancer progression and response to molecular targeting agents. EGFR is consistently detected in the nuclei of cancer cells, primary tumor specimens, and other highly proliferative tissues.Citation21–Citation25 Increased nuclear EGFR localization correlates with poor clinical outcome in patients with breast cancer,Citation26 oropharyngeal SCCCitation27 and ovarian cancer.Citation28 Recent reports have characterized a novel nuclear localization sequence in EGFR and its family members.Citation29–Citation31 Furthermore, transport mechanisms for EGFR to the nucleus have been described previously.Citation32 These mechanisms involve ligand binding, dimerization, activation and internalization. Endosomal sorting to the ER allows EGFR to associate with the Sec61 translocon for transport from the ER to the cytoplasm.Citation33 Here, EGFR binds importin β, which facilitates movement into the nucleus.Citation32 Nuclear EGFR is known to regulate the promoters of several target genes including, Cyclin D1,Citation23 iNOS,Citation29 B-myb,Citation34 Aurora Kinase ACitation35 and COX2.Citation31 Mechanisms of EGFR-mediated gene regulation involve direct interaction of EGFR with STAT3 to regulate iNOSCitation29 and COX2,Citation31 promoters, with STAT5 for regulation of the Aurora Kinase A promoter,Citation35 and with E2F1 transcription factors for regulation of the B-Myb promoter.Citation34 Nuclear EGFR has also recently been shown to function as a tyrosine kinase in the nucleus, phosphorylating and stabilizing PCNA and thus enhancing the proliferative potential of cancer cells.Citation36 In addition to ligand induced translocation of the EGFR to the nucleus, radiation induces EGFR transport to the nucleus mediated by the Src family kinases (SFKs).Citation37 Cetuximab, a monoclonal antibody targeting EGFR, also leads to EGFR translocation to the nucleus.Citation38 Collectively, these findings suggest that EGF ligand, radiation and cetuximab enhance the nuclear accumulation of EGFR. As data accrues suggesting a functional impact of nuclear EGFR, it becomes important to understand the extent to which nuclear EGFR may contribute to cancer growth and progression, as well as its effect on the therapeutic response to EGFR targeted therapies.
Attenuation of EGFR signaling is as crucial to the biology of EGFR as activation. The attenuation of the EGFR involves numerous processes including receptor ubiquitinylation, dephosphorylation, depletion of ligand access, receptor trafficking to the lysosome and subsequent destruction.Citation39 Clathrin-mediated endocytosis represents the major pathway by which activated surface associated EGFR is quickly internalized and degraded.Citation40–Citation44 This process is well characterized and involves numerous steps.
Ligand binding and dimerization of EGFR results in phosphorylation of tyrosine residues on EGFR's C-terminal domain. Two of these residues, Y1068 and Y1086, serve as the major binding site for the Grb2 adaptor protein (). The SH2 domain of Grb2 binds to phospho-tyrosines on EGFR, while SH3 domains of Grb2 bind other proteins coupling them to EGFR. One of the most recognized Grb2-interacting proteins is casitas B-lineage lymphoma (Cbl), a ring finger-containing E3 ubiquitin ligase. Cbl can directly bind to EGFR Y1045 and recruit E2 enzymes (UbcH4/5) to its ring domain to promote EGFR ubiquitination. Ubiquitinated EGFR is quickly recognized by the ubiquitin-binding domains of epsin, Eps15, Eps15R and Hrs/Hgs,Citation45,Citation46 proteins strongly associated with clathrin. Clathrin-coated vesicles containing ubiquitinated EGFR fuse with early endosomes and interact with the endosomal sorting complex required for transport (ESCRT), which directs the ubiquitinated EGF:EGFR complex to the lysosome for degradation.
Endocytosis of EGFR doesn't always lead to its destruction; EGFR can be recycled back to the cell surface, remain active in intracellular compartments, or completely circumvent endocytosis.Citation39 Modulation of EGFR trafficking arises both from mutations and from overexpression and/or dimerization with other HER family members.Citation47–Citation53 Recently, HER2 overexpression was shown to alter EGFR degradation by competitively binding endosomal retention components preventing their binding to EGFR.Citation54
The Role of EGFR in Human Cancers
Over the past three decades, numerous reports described overexpression, increased activity or mutations of EGFR in various human epithelial tumors, strengthening the view that deregulated EGFR activity may be a causative factor in the etiology of human epithelial cancers. Most notably, deregulated EGFR activity has been linked to the development, progression and metastatic spread of HNSCC, NSCLC, CRC, breast, ovarian, cervical, bladder, pancreatic, gastric, endometrial and brain cancer.Citation3,Citation55–Citation71 In many of these cancers, EGFR expression is associated with decreased overall survival rates.Citation72 Modes of deregulated EGFR activity have included increased stimulation of the EGFR through ligand binding,Citation73 point mutations within the receptor,Citation74 deletion mutants of exons 2–7 of the EGFR (EGFRvIII),Citation75 impaired downregulation of the EGFR,Citation76 and gene amplification of the EGFR loci.Citation75 Collectively, these findings demonstrate the importance of EGFR in human cancers and the need to develop inhibitors that effectively target the activity of this RTK.
EGFR Targeted Antibodies: Discovery and Function
Targeting EGFR has been intensely pursued over the last three decades as a treatment strategy for cancer. From these efforts two fundamental approaches have proven useful. One approach involves the use of small molecule tyrosine kinase inhibitors (TKIs) that bind to the ATP-binding site in the tyrosine kinase domain (TKD) of EGFR. To date, three anti-EGFR TKIs, erlotinib (OSI-774, Tarceva), gefitinib (ZD1839, Iressa) and lapatinib (GW572016, Tykerb) are FDA-approved for use in oncology.
A second approach uses monoclonal antibodies (mAbs) to target the extracellular domain of EGFR to block natural ligand binding. In 1983, Sato et al. isolated four mouse (M) hybridomas secreting immunoglobulin G (IgG) against the EGFR on A431 cells. Three of the antibodies, M225 IgG, M528 IgG and M579 IgG blocked 95% of EGF binding to human A431 cells as well as competing with each other in binding assays. These three antibodies could also bind HeLa cells and foreskin fibroblasts. Further, each antibody could immunoprecipitate EGFR from A431 cells, but not from three rodent lines tested demonstrating their specificity. Finally, each antibody effectively blocked EGF-induced phosphorylation of the receptor resulting in reduced proliferative potential of the cell lines examined.Citation77–Citation80
M225 has slightly more effective anti-EGFR activity than M528, which is in turn more effective than M579. The phase I trial with M225 was successful, but all patients produced human-anti-mouse antibodies (HAMA). Therefore, M225 was converted to a human:murine chimera, C225, with an IgG1 Fc isotype (). The IgG1 Fc isotype was chosen for its potential to enhance the immune contribution to C225 antitumor effects (Dr. John Mendelsohn, MD Anderson Cancer Center, personal communication).
Subsequently, the anti-EGFR antibody C225 IgG1 was developed for clinical use. Cetuximab (ICM-225, Erbitux™) is a human/murine chimeric monoclonal antibody that works by binding to the extracellular domain III of EGFR. This interaction partially blocks the ligand-binding domain and sterically hinders the correct extended conformation of the dimerization arm on domain II.Citation81 Thus, cetuximab prevents both ligand binding and the proper exposure of the EGFR dimerization domain, preventing dimerization with other HER family members. In 2006, another monoclonal antibody directed against EGFR and having a similar mode of action was made; panitumumab (Vectibix) differs from cetuximab in that it is a fully humanized antibody.Citation82
In addition to blocking ligand binding and dimerization, cetuximab effectively blocks EGFR phosphorylation, promotes EGFR internalizationCitation83 and reduces cellular proliferation in a variety of cancer models ().Citation84,Citation85 Cetuximab induces arrest in the G1 phase of the cell cycle by increasing levels of p27kip1 (). This in turn results in an increase in the dimerization of p27Kip1-Cdk2 complexes, which ultimately prevents exit from G1.Citation86–Citation88
In vivo experiments corroborated the p27Kip1 findings as well as a reduction in PCNA in human tumor xenografts.Citation89 Furthermore, cetuximab decreases cancer cell metastasis in various studies via downregulation of pro-angiogenic factors and matrix metalloproteinases responsible for cell adhesion ().Citation90–Citation93 Cetuximab can induce the upregulation of various proapoptotic factors such as Bax, in addition to the downregulation of Bcl2, leading to the activation of caspases ().Citation94–Citation97 Cetuximab can also induce antibody-dependent cellular cytotoxicity in vivo by recruiting immune cells to tumor cells decorated by cetuximab ().Citation98,Citation99 Collectively, cetuximab results in several biological effects that have impact on the growth and spread of multiple human tumors.
Cetuximab in the Clinic
Cetuximab has exhibited promising antitumor activity in clinical trials as either monotherapy or in combination with chemotherapy and/or radiation, particularly in the settings of metastatic CRC (mCRC)Citation100–Citation105 and HNSCC ().Citation106–Citation110 In 2004, the FDA approved cetuximab for use in patients with EGFR-expressing mCRC refractory to irinotecan-based chemotherapy.Citation111 Since this approval, several extensive clinical trials have supported the use of cetuximab in the setting of mCRC.
In 1997–98, a phase I trial of HNSCC enrolled 16 patients with locoregionally advanced tumors and provided the first clinical demonstration that adding cetuximab to radiation may improve tumor response and disease control.Citation112 Despite the absence of true phase II data, a phase III trial that enrolled 424 patients was carried out between 1999–2002 that confirmed a 10% overall survival advantage for patients receiving cetuximab in combination with curative radiation for advanced HNSCC compared with radiotherapy alone.Citation107
Although not yet approved by the FDA for use in advanced NSCLC, cetuximab has undergone clinical evaluation in this setting. A series of phase II trials suggested the benefit of cetuximab in combination with platinum doubles in the first-line treatment setting.Citation113–Citation117 Two phase III trials have been reported, including the FLEX and BMS099 trials.Citation118,Citation119 The FLEX trial demonstrated an improvement in overall survival with the addition of cetuximab to first-line cisplatin and vinorelbine. The BMS099 trial evaluated the addition of cetuximab to carboplatin/taxane in the first-line setting and identified an improvement of overall response rate, but not a statistically significant improvement in progression-free survival (PFS).
Predictive Biomarkers of Cetuximab Response
Since the FDA approval of cetuximab and its associated clinical successes, intense investigations have been made to find markers in patient tumors that could predict individual responses to cetuximab therapy and positive clinical benefit. It was believed that expression levels of EGFR would serve as a simple predictive biomarker for the likelihood of response to cetuximab therapy. This would be akin to women with breast cancer who have high HER2 expression and are more likely to respond to trastuzumab anti-HER2 therapy. However, early clinical studies did not confirm a correlation between EGFR expression level by immunohistochemistry (IHC) and clinical response to EGFR inhibitor therapy.Citation100 Chung et al. confirmed that several CRC patients who received cetuximab exhibited a major objective response despite the absence of measureable EGFR. These studies suggested that IHC-based assays measuring EGFR expression do not serve as robust predictors for response to cetuximab therapy and that expression levels of the EGFR alone are not a reliable predictor of response to cetuximab therapy.
EGFR mutations as a predictor of response.
In 2004, a series of landmark papers identified EGFR mutations in the tyrosine kinase domain in NSCLC patients, which predicted response to the TKIs erlotinib and gefitinib.Citation120–Citation122 These gain-of-function mutations conferred dependence of tumor cells on the mutated EGFR kinase and rendered them more sensitive to erlotinib and gefitinib than tumors without these mutations. Soon after this finding it was reported that following initial sensitive response, patients harboring these mutations in the catalytic domain acquired resistance within 6–12 months of TKI therapy. Pao et al. reported that NSCLC patients with acquired resistance to gefitinib or erlotinib contain a secondary mutation in exon 20 of EGFR, which leads to substitution of methionine for threonine at position 790 (T790M) in the kinase domain.Citation123,Citation124 Despite these findings for EGFR TKIs and NSCLC, no EGFR mutations have yet been identified that reliably predict response to antibody-based EGFR therapies.Citation125,Citation126
EGFR gene copy number as a predictor of response.
Although mutations within the EGFR do not predict response to cetuximab therapy, increased copy number of the EGFR gene is associated with response in CRC.Citation127 Large CRC clinical cohorts confirmed the relationship between EGFR gene amplification and clinical response to cetuximab.Citation128–Citation131 However, increased copy number of the EGFR gene does not lead to increased expression of EGFR in these patients,Citation127,Citation130 and how EGFR gene copy number correlates with improved response is unknown. In lung cancer, 30–60% of patients with advanced NSCLC have increased EGFR gene copy number according to FISH analysis and respond to cetuximab plus chemotherapy.Citation132 However, the BMS-099 trial concluded that patient response rate to cetuximab was not dependent on EGFR gene copy number.Citation133 Thus, predictive response to cetuximab based on EGFR gene copy number remains controversial at this time.
EGFR ligand status as a predictor of response.
Increased expression levels of the EGFR ligands AR and EPR in CRC tumors are positively associated with response to cetuximab.Citation134 These findings centered on the mRNA levels within the tumor itself, versus circulating serum levels. Increased mRNA expression correlates with improved disease control and PFS. These findings suggest that EGFR ligands may drive tumor proliferation in an autocrine/paracrine fashion and that cetuximab may be able to block ligand binding to the receptor.
KRAS mutations as a predictor of response.
One of the most notable predictive biomarkers of response to cetuximab is the mutational status of the KRAS gene. KRAS is a small GTPase responsible for coupling EGFR to the RAF/MEK/ERK pathway. KRAS binding to GTP leads to conformational changes in RAF and activation of the down stream-signaling pathway. Although KRAS is a GTPase, its catalytic activity is slow and dramatically enhanced by accessory proteins called GTPase activating proteins (GAPs) that convert KRAS-GTP to KRAS-GDP and thus turn off RAF-mediated signaling. Mutations in codon 12 or 13 of KRAS impair the intrinsic GTPase activity and confer resistance to GAPs, thereby causing cancer-associated mutant Ras proteins to accumulate in the active, GTP-bound conformation.Citation135,Citation136 Lievre et al. reported that KRAS with mutations at codon 12 or 13 might be predictive of resistance to cetuximab therapy. In this report, they analyzed 30 patients with metastatic CRC treated with cetuximab for the presence of KRAS, BRAF and PIK3CA mutations. KRAS mutations were found in 43% of tumors (13 tumors), and were significantly associated with resistance to cetuximab therapy (p = 0.002).Citation137 Further, Di Fiore et al.Citation97 studied 59 patients with chemorefractory mCRC treated with cetuximab plus chemotherapy and found that KRAS mutations were highly predictive of resistance to cetuximab plus chemotherapy.Citation138 A larger study was performed to measure the KRAS mutation status in 113 patients with irinotecan-refractory mCRC treated with cetuximab. The authors reported that wild-type KRAS is a strong predictor of significant increase in overall survival (p < 0.001) in this cohort of patients.Citation139 Following this work, Van Cutsem et al. investigated the efficacy of cetuximab plus irinotecan, fluorouracil and leucovorin (FOLFIRI) as first-line treatment for mCRC and looked for associations between the mutation status of KRAS and clinical response to cetuximab. In this study, 599 patients received cetuximab plus FOLFIRI, and 599 received FOLFIRI alone. First-line treatment with cetuximab plus FOLFIRI reduced the risk of disease progression compared with FOLFIRI alone, and the benefit of cetuximab was limited to patients with KRAS wild-type tumors.Citation105 Since the publication of these studies, several additional clinical trials have further strengthened these findings.Citation140–Citation144 This collective body of work has led to a Provisional Clinical Opinion from ASCO in 2009 stating that all patients with mCRC who are candidates for anti-EGFR antibody therapy should have their tumor tested for KRAS mutations in a clinical laboratory improvement amendments (CLIA)-accredited laboratory. If codons 12 or 13 of KRAS are mutated, patients with mCRC should not receive anti-EGFR antibody therapy as part of their treatment.Citation145 However, some patients carrying KRAS mutant tumors have been reported to respond to EGFR antibodies.Citation128,Citation143,Citation146,Citation147 How to distinguish these individuals from other KRAS mutant patients is worthy of clinical investigation.
BRAF mutations as a predictor of response.
BRAF is a serine-threonine kinase belonging to the RAF family of protein kinases. GTP bound KRAS activates BRAF,Citation148 which in turn activates the MEK pathway.Citation149,Citation150 Initial work indicated that BRAF mutations impair response to EGFR antibodies in CRC.Citation151 This investigation indicated that clinically responsive tumors had wild-type BRAF, whereas approximately 14% of non-responders had a mutation at valine 600 (V600E). Several reports revealed that BRAF gene mutations are relatively rare events in lung cancer and HNSCC.Citation152–Citation157
IGF1R gene expression status in response to cetuximab.
Fei et al. investigated the correlation between gene expression levels of IGF pathway components in 70 pre-treatment mCRC patients and the subsequent clinical benefit from cetuximab. In this cohort, patients with tumors expressing high level of IGF1R had longer PFS with cetuximab than patients with low expression levels of these genes. Additionally, patients with KRAS wild-type tumors with high expression of IGF1R had a significantly higher rate of disease control (62.5%) and longer PFS than patients with low expression (26.3% disease control).Citation158 These data suggested that IGF pathway activation might play an important role in predicting positive responders to cetuximab therapy.
Mechanisms of Resistance to EGFR Targeted Antibodies
Angiogenesis.
Several lines of investigation have identified inhibition of angiogenesis as part of the antitumor effect of cetuximab ().Citation159–Citation161 Thus, cetuximab resistance is partly due to the ability of cells to re-activate pro-angiogenic factors via alternate pathways. In 2001, Viloria-Petit et al. demonstrated that tumors resistant to EGFR blocking antibodies have increased VEGF production,Citation162 a potent stimulator of neovascularization.Citation163,Citation164 Researchers established A431 squamous cell carcinoma xenografts in mice and subsequently treated them with three EGFR blocking antibodies: cetuximab, hR3 or mR3. Complete regression for each of these three antibodies was obtained after eight treatments, at which point treatment was terminated. Tumor recurrence was observed as soon as 18 days, and up to 4.5 months post treatment. Twenty percent of cetuximab-treated mice suffered tumor recurrence, while 40% of hR2 and 80% of mR3 treated mice suffered recurrence. All mice with recurrent tumors were subsequently treated with a second round of therapy. Six anti-EGFR antibody resistant variant cell lines were established from resistant recurrent tumors to hR2 and mR3 antibodies (cetuximab resistant cell lines were not established). When these cell lines were tested for response to hR2 and mR3 in vivo, they demonstrated increased growth, and decreased response rates compared to A431 parental tumors. Immunoblot analysis of the variant cells demonstrated that EGFR levels remained similar to those of parental cell lines, arguing against the possibility that changes in EGFR expression influence the resistant phenotype. Notably, resistant variant cells expressed two-fold more VEGF mRNA and protein than parental cells. When the A431 parental cell line was engineered to overexpress VEGF and injected into mice, the resulting tumors were resistant to all three anti-EGFR antibodies and demonstrated increased vascularization within the tumor itself. In support of these findings, another study used the VEGF inhibitor (DC101) combined with cetuximab and demonstrated that it had greater efficacy in treatment of gastric cancer xenografts than either agent alone, both in the inhibition of tumor growth and the induction of apoptosis.Citation165 Overall, these studies provide evidence for the role of VEGF and angiogenesis in resistance to EGFR targeted antibodies.
Further studies focused on angiogenesis have been reported. A study by Ciardiello et al. demonstrated the importance of VEGF signaling in resistance to EGFR targeted antibodies.Citation166 Researchers treated human GEO colon tumor xenografts with either cetuximab or ZD1839 (gefitinib) for four weeks, at which time complete regression of all tumors was noted. Mice taken off therapy after complete regression grew recurrent tumors within 3–5 weeks, while mice kept on therapy had prolonged tumor inhibition with recurrent tumor growth after 11–12 weeks; all recurrent tumors reached the growth rate of untreated controls. EGFR inhibitor resistant recurrent tumors were excised, and two cell lines were established in vitro. Immunoblot analysis of these resistant variants showed a 5–10 fold increase in the expression of COX-2, phosphorylated MAPK and VEGF, while EGFR expression levels remained constant. In addition, resistant variants secreted more VEGF into surrounding media compared to sensitive controls. Researchers showed that the dual VEGFR-EGFR tyrosine kinase inhibitor ZD6474 (vandetanib) was able to overcome resistance to EGFR targeted agents. Not only did mice initially treated with ZD6474 alone remain devoid of recurrent tumors through the end of the experimental period (23 weeks), but xenografts of resistant variants were also sensitive to ZD6474. Overall, these data support a role of VEGF signaling in the development of cetuximab resistance, and provide a rationale for targeting VEGF signaling pathways in cetuximab-resistant tumors.
In 2008, Bianco et al. used a variety of different cancer cell lines (breast, colon and prostate) with differing levels of EGFR expression. In vivo resistance to EGFR inhibitors was established for each cell line using a similar approach as Ciardiello et al.Citation166 This work confirmed the finding that EGFR resistance can be overcome with ZD6474 treatment, which reduced phosphorylation of AKT and p70-S6 kinase in resistant cells. Resistant cells also overexpressed VEGFR-1 at the mRNA and protein level. In addition, ZD6474 reduced the phosphorylation of both VEGFR-1 and VEGFR-2 significantly in resistant cell lines. To further support the role of VEGR-1 in resistance to EGFR inhibitors, siRNA directed towards VEGFR-1 re-sensitized resistant cell lines to EGFR inhibitors; siRNA towards VEGFR-2 restored sensitivity to EGFR inhibitors to a lesser degree. Overexpression of VEGFR-1 in ZD1839 sensitive lines conferred resistance to subsequent ZD1839 treatment. In wound-healing assays, resistant cells had up to 50% more migratory capability compared to parental cells, and treatment with ZD6474 reduced wound closure efficiency, and thus inhibited the migratory potential of resistant cells. Overall, this study provides further evidence for the role of VEGF and VEGFRs in a tumor's ability to increase its migratory potential, stimulate downstream signaling molecules, and induce EGFR inhibitor resistance in various types of cancers.Citation167
Dysregulation of EGFR internalization and degradation.
Dysregulated EGFR internalization and degradation has been shown to play a role in cetuximab resistance. In a study by Lu et al. a DiFi colorectal cancer cell line was subjected to increasing concentrations of cetuximab over time, producing a cetuximab-resistant cell line DiFi5.Citation168 DiFi5 cetuximab-resistant cells exhibited polyubiquitinated EGFR, decreased total and cell surface levels of EGFR and increased expression of SFKs compared to parental, cetuximab-sensitive controls. In addition, DiFi5 cetuximab-resistant cells showed high association with the ubiquitin E3 ligase Cbl, consistent with the high level of ubiquitinated EGFR. Contrary to previous work demonstrating that SFKs can downregulate Cbl to promote EGFR persistence,Citation169 Cbl was more associated with EGFR in cetuximab-resistant cells despite high SFK expression levels. The decrease in EGFR stability in this resistant model still needs to be further investigated, however authors postulate that alternative receptors/pathways are responsible for the observed continuation of cell growth and survival.Citation168
In another model of acquired resistance to cetuximab, various cetuximab-resistant clones were established from the NSCLC cell line NCI-H226 by exposing these cells to increasing concentrations of cetuximab over a six-month time course. Using this model, Wheeler et al. showed that resistant cell lines have increased surface levels of EGFR compared to the parental cetuximab-sensitive line.Citation170 These findings were confirmed using immunofluorescence (IF) microscopy and flow cytometry. In contrast to the study by Lu et al.Citation168 this study demonstrated that EGFR does not seem to be associated with c-Cbl in cetuximab-resistant lines, while the parental cetuximab-sensitive line maintains this association. These data support a previously published mechanism for EGFR degradation via c-Cbl recruitment to tyrosine 1045.Citation171 Overall, these data suggest that impaired EGFR internalization and degradation may lead to increased EGFR expression and activity manifesting in cetuximab resistance.Citation170
Oncogenic shift.
The increased activation of various RTKs plays a role in cetuximab resistance in an NSCLC in vitro model.Citation170 The cetuximab-sensitive NSCLC cell line, NCI-H226, and the HNSCC cell line, UMSCC-1, were treated continuously with increasing concentrations of cetuximab to establish several cetuximab-resistant clones. Using an antibody-based RTK array, the levels of phosphorylated RTKs were compared between the cetuximab-resistant and the parental cetuximabsensitive cell lines in both NSCLC and HNSCC models. In both cetuximab-resistant models, protein levels of the HER family members EGFR, HER2 and HER3 were upregulated, as well as the MET receptor compared to parental controls. Western blot analysis confirmed increased protein levels of these RTKs in the NSCLC model. Immunoprecipitaton of EGFR in resistant clones showed a steady-state increase in binding to HER2, HER3 and cMET in resistant lines. In addition, the monoclonal antibody 2C4 (Pertuzumab), which inhibits HER2 heterodimerization, was able to produce potent anti-proliferative effects in NSCLC cetuximab-resistant lines when treated simultaneously with cetuximab. This also corresponded with more potent knockdown of HER3 and AKT expression. Overall, these data suggest that resistant NSCLC and HNSCC cells may rely on the activation of various RTKs and their subsequent heterodimerization with EGFR to bypass cetuximab blockade.Citation170
Subcellular localization of EGFR.
The EGFR has been shown to be localized to subcellular compartments including the endosome, the mitochondriaCitation172 and the nucleus. Nuclear EGFR has been detected in a variety of cancer types including brain,Citation173 breast,Citation174–Citation177 bladder,Citation178 ovary,Citation179 NSCLCCitation180 and HNSCCCitation181 and serves as a prognostic factor in HNSCC, ovarian and breast cancers.Citation26–Citation28 In 2009, Li et al. demonstrated in an in vitro model of cetuximab resistance that resistant clones had increased levels of nuclear EGFR, determinded via biochemical fractionization and IF.Citation182 Increased nuclear EGFR coincided with increased expression levels of genes known to be regulated by nuclear EGFR: Cyclin D1,Citation181 B-mybCitation177 and PCNA,Citation183 demonstrating a correlation of nuclear EGFR and increased expression of known nuclear EGFR gene targets. In addition, cetuximab-resistant cells had increased SFK expression.Citation184 Based on this knowledge, researchers postulated that SFKs may influence nuclear EGFR and modulate cetuximab resistance. Using the pan-SFK inhibitor, Dasatinib, a 2.7–5.5-fold decrease in nuclear EGFR was detected with a significant increase in surface associated EGFR levels measured via flow cytometry. Dasatinib was able to re-sensitize cetuximab resistant clones to cetuximab; growth proliferation was only mildly affected by dasatinib treatment alone. Overall, these data suggest that SFKs are necessary for translocation of EGFR to the nucleus and protection from cetuximab. The authors concluded that SFK inhibition in combination with cetuximab may have clinical benefit in the future.
In another study, mammary-derived growth inhibitor (MDGI) was shown to confer cetuximab resistance in both breast and lung cancer cell models by stimulating the intracellular localization of EGFR.Citation185 MDGI is an intracellular fatty acid binding protein known to modulate fatty acid metabolism, in addition to having tumor suppressor activity in various cancer models.Citation186–Citation189 It is expressed in 40% of human breast cancers and 85% of human lung cancers; however, expression is lost in cell culture models possibly by epigenetic silencing mechanisms.Citation185 Experiments investigating the role of MDGI and cetuximab resistance are based on the use of cell lines stably transfected with MDGI grown in three-dimensional cell culture models, or inoculated into mouse mammary fat pads. Cells transfected with MDGI exhibit a reduced level of cell surface EGFR and labeled EGF on the cell surface as measured by flow cytometric analysis. In addition, there was an increase in intracellular biotin-labeled EGFR as detected by western blot analysis. Increased intracellular trafficking of EGFR appeared to be independent of other HER family members, or increased caveolin activity. Interestingly, EGFR remained phosphorylated within intracellular compartments in MDGI-transfected cells. Lung cancer cells transfected with MDGI are resistant to cetuximab in the 3D matrigel model, while GFP-transfected control cells remain sensitive. Both GFP and MDGI transformants remain sensitive to EGFR TKIs in 3D matrigel, demonstrating the ability of TKIs to penetrate intracellular compartments containing the majority of EGFR. Researchers further demonstrate this finding in vivo by inoculating mouse mammary fat pads with a breast cancer cell line stably expressing MDGI or GFP. MDGI-expressing cells were resistant to cetuximab treatment, and grew much faster then the GFP control tumors. Overall, this study identifies MDGI as a determinant of intracellular localization of EGFR, and provides a rationale for considering MDGI expression levels as a biomarker for cetuximab sensitivity.
Epithelial to mesenchymal transition.
Epithelial to mesenchymal transition (EMT) has been described as a hallmark of cancer.Citation190 EMT is characterized by the loss of adherens junction proteins, such as E-cadherin; gain of cytoskeletal filaments, such as vimentin; and the subsequent increase in cell motility and invasiveness.Citation191,Citation192 These markers have been used by various investigators to characterize cells as either epithelial or mesenchymal; however, some researchers suggest that carcinomas do not fall into strict categories, and represent a mixed phenotype.Citation193,Citation194 In a study by Fuchs et al., 12 human hepatocellular carcinoma cell lines were characterized as either epithelial or mesenchymal by western blot analysis of E-cadherin and vimentin protein levels. Based on this classification, mesenchymal cell lines were shown to be resistant to cetuximab, erlotinib and gefitinib, while epithelial cell lines remained sensitive to these EGFR inhibitors. Analysis of downstream signaling pathways in all 12 cell lines demonstrated that mesenchymal cell lines had increased activation of AKT, STAT3 and integrin linked kinase (ILK); independent of total EGFR expression. ILK, a known activator of AKT,Citation195 seemed to play a role in resistance to EGFR inhibitors because an ILK inactive mutant increased the expression of E-cadherin, indicating a transition of mesenchymal cells back to epithelial cells. This transition was associated with decreased AKT signaling, and restoration of cellular sensitivity to EGFR inhibitors. This was further verified in a xenograft model, where cells became resensitized to gefitinib. Overall, EMT and ILK may play a role in resistance to cetuximab.Citation196
In 2010, mesenchymal-like squamous cell carcinomas were shown to be resistant to cetuximab treatment.Citation193 Using immunohistochemistry, flow cytometry and gene expression profiling, researchers identified SCC cell lines that were epithelial-like and mesenchymal-like based on E-cadherin and vimentin expression levels. In this system, researchers identified cell lines that had sub-populations of both high E-cadherin/low vimentin and low E-cadherin/high vimentin levles. Gene expression profiles of E-cadherin/high vs. E-cadherin/low sub-populations were compared, demonstrating that E-cadherin/low cells expressed various genes involved in EMT. Next, they compared this in vitro system to in vivo xenografts and clinical specimen tumor samples, which also demonstrated distinct E-cadherin/high and E-cadherin/low sub-populations. Using FACS, researchers separated out each sub-population and performed in vitro growth assays. E-cadherin/low cells exhibited attenuated growth compared to E-cadherin/high cells, and were also arrested in the G0 phase of the cell cycle. Thus, E-cadherin/low cells have a lower turnover rate compared to E-cadherin/high cells. Flow cytometry, IF microscopy and western blot analysis demonstrated that E-cadherin/low cells from both in vitro and in vivo models had decreased EGFR expression levels. EGF ligand was unable to stimulate the activation of EGFR downstream signaling pathways in E-cadherin/low cells, demonstrating that EGF ligand regulation of AKT and ERK activation was uncoupled from the EGFR in these cells. E-cadherin/low sub populations also remained resistant to cetuximab treatment in vitro and in vivo xenograft models. When xenografts of E-cadherin/high and low subpopulations were treated with cetuximab for 12 days, a two-fold enrichment of vimentin expressing cells was noted. Thus, it seems that tumors may escape from cetuximab by either (1) depleting epithelial-like cell types (as demonstrated by the loss of E-cadherin expressing subpopulations) or by (2) actively differentiating into mesenchymal-like cell types (as demonstrated by the increase in vimentin expressing subpopulations).
Constitutive activation of EGFR downstream effector molecules.
The constitutive activation of AKT due to the proteosomal degradation of PTEN induces resistance to cetuximab in the NSCLC cell line, NCI-HCC827.Citation197 Cetuximab-resistant HCC827 clones, produced via increased exposure to cetuximab over a six-month time period, demonstrated increased AKT activation, and markedly decreased protein levels of PTEN. In subsequent studies, researchers treated resistant cells with the proteosomal inhibitor MG-132, which prevented degradation of PTEN and reduced AKT activation. Western blot analysis of cells treated with MG-132 demonstrated that PTEN was polyubiquitinated, suggesting that PTEN was targeted for destruction. The AKT inhibitor, LY294002, combined with cetuximab was also able to block AKT activation even more potently that LY294002 alone. Overall, it appears that functional PTEN is necessary for the complete knockdown of AKT activity by cetuximab and PTEN destruction yields resistance to cetuximab.Citation197
The PI3K/AKT pathway is upregulated via increased activity of the intracellular kinase Src in cetuximab-resistant cells. In this study, Wheeler et al. used cetuximab-resistant clones derived from the NSCLC NCI-H226 cell line.Citation184 Cetuximab-resistant cells demonstrated a robust increase in SFK activity compared to sensitive parental control cells, possibly due to the steady state increase of EGFR in resistant lines. In addition, cetuximab-resistant cell lines had high AKT activity. The pan SFK inhibitor, dasatinib, decreased AKT activation along with the proliferative potential of cetuximab-resistant clones, demonstrating the importance of SFK signaling in resistance to cetuximab. When cetuximab-resistant cells were treated with both cetuximab and dasatinib simultaneously there were even more profound anti-proliferative effects, including enhanced decreases in downstream AKT signaling. Overall, dasatinib was able to re-sensitize resistant cells to cetuximab, providing a rationale for possible combinational therapies in the future.
Blockade of SFKs is also beneficial in the mutant KRAS setting of CRC. Clinical trials using cetuximab in patients with mCRC harboring a KRAS mutation demonstrated resistance to cetuximab therapy.Citation138,Citation145,Citation198 These trials have lead to the widely accepted view that KRAS mutation status is a predicative factor for response to cetuximab.Citation145,Citation199,Citation200 Since 30–40% of patients with CRC have a KRAS mutation, it is crucial that alternative treatment regimes be devised for these patients.Citation198 In 2010, Dunn et al. investigated whether SFK blockade could sensitize KRAS mutant CRC tumors to cetuximab. In this study, 16 CRCs lines were screened for expression of EGFR, SFKs, and the mutational status of KRAS. Nine of the 16 cell lines had mutations in KRAS at either codons 12 or 13, or BRAF mutations at codon 600. From these lines, three lines were chosen for continued studies based on their expression of EGFR and SFKs, KRAS mutational status, and ability to grow in vivo. KRAS mutated cell lines had a decreased response to cetuximab when plated on poly-d-lysine/laminin coated culture plates compared to wild-type KRAS cell lines, which demonstrated sensitivity. Importantly, the combination of cetuximab and dasatinib led to significant growth inhibition in KRAS mutant lines. These results encouraged researchers to perform a human phosphokinase array to identify potential mechanisms for growth inhibition. Unique kinase expression patterns were found for each KRAS mutant line treated with both cetuximab and dasatinib, however, all three lines demonstrated a common decreases in activity of the MAPK pathway, the β-catenin pathway, and of various STAT family transcription factors. Finally, researchers tested these findings in xenograft studies. Mice were injected with mutant KRAS cell lines, and treated with vehicle, cetuximab, dasatinib or the combination. These experiments demonstrated that the combinatorial treatment from initial tumor onset had statistically significant growth inhibitory effects in KRAS mutant xenografts. Overall the blockade of SFKs in mutant KRAS cell lines led to a decrease in key signaling pathways important for cell proliferation and survival, providing a rationale for treating KRAS mutant CRC patients with dasatinib and cetuximab in the clinic.Citation201
Increased expression of HER family growth factors.
In 2010, Hatakeyama et al. reported a role for microRNAs (miRs) in EGFR ligand suppression and cetuximab resistance. This study utilized cetuximab-resistant clone (1Cc8) derived from the HNSCC UMSCC-1 line after chronic, long-term exposure to cetuximab treatment. Using DNA microarray technology, differential gene expression patterns were compared between the UMSCC-1 parental line and 1Cc8. Examining differences in the EGFR signaling pathway revealed an increase in heparin-binding EGF (HB-EGF) in the cetuximab resistant line, 1Cc8. To confirm this result, HB-EGF and other growth factor ligands were measured via ELISA assay from conditioned media with or without cetuximab treatment in both UMSCC-1 and 1Cc8 cell lines. Indeed, 1Cc8 cells had increased HB-EGF in both untreated and cetuximab treated culture media. Additionally, 1Cc8 cells also had increased AR in both untreated and cetuximab treated culture media, while TGFα was only increased in cultured media treated with cetuximab. Stimulation of various cetuximab-sensitive cell lines with HB-EGF induced cetuximab resistance, as measured via in vitro MTS growth assay and colony formation assay. In addition, knocking down the expression of HB-EGF in 1Cc8 cells was able to resensitize these cells to cetuximab. These results suggested that deregulated ligand expression may play a role in cetuximab resistance, and led researchers to look at the expression levels of various miRs. Interestingly miR-212 was decreased 27-fold in 1Cc8 cells relative to the cetuximab sensitive UMSCC-1 cell line as measured by RT-PCR based array. To demonstrate direct interaction of miR-212 and HB-EGF, a mimic of miR-212 was used to downregulate expression of HB-EGF in various cell lines, assayed by luciferase reporter gene expression. Researchers also reported that the miR-212 mimic could resensitize 1Cc8 cells to cetuximab. Finally, experiments examined expression levels of HB-EGF, along with other EGFR ligands, in HNSCC tumor specimens taken at the time of diagnosis or recurrence. HB-EGF, TGFα and AR were expressed in all tumors, however HB-EGF was even more highly expressed in recurrent tumor specimens. HB-EGF levels in plasma samples from recurrent patients were five times higher than in patients with newly diagnosed tumors. Overall, the increased expression of HB-EGF along with the downregulation of specific miRs appear to play a role in cetuximab resistance.Citation202
Concluding Remarks
The discovery of the EGFR, the understanding of its signaling pathways and role in the etiology of human cancers, and ultimately the design of rational drugs to inhibit EGFR activity represents an incredible story of collaboration of scientists and clinicians over the last 30 years. Although cetuximab, an antibody directed against the EGFR, has had clinical success there is substantial room for progress in improving the effectiveness of this molecular therapeutic. Several key areas exist including (1) the identification of biomarkers that will accurately predict optimum responses to cetuximab, (2) the identification of other key signaling pathways that can be targeted to enhance cetuximab activity and (3) understanding the pathways involved in cetuximab resistance and how to circumvent them to increase survival of cancer patients. The lessons learned from the story of the EGFR and the development of cetuximab will pave the road for future scientific and clinical based investigations of other promising antibody-based therapies directed against RTKs in oncology.
Abbreviations
| AR | = | amphiregulin |
| BTC | = | betacellulin |
| Cbl | = | casitas B-lineage lymphoma |
| CRC | = | colorectal cancer |
| DAG | = | diacyglycerol |
| EGF | = | epidermal growth factor |
| EGFR | = | epidermal growth factor receptor |
| EMT | = | epithelial to mesenchymal transition |
| ER | = | endoplasmic reticulum |
| EPR | = | epiregulin |
| ERK | = | extracellular signal-regulated kinase |
| FACS | = | fluorescence-activated cell sorting |
| FDA | = | food and drug administration |
| FGFR | = | fibroblast growth factor receptor |
| GAPs | = | GTPase activating proteins |
| GSKα/β | = | glycogen synthase kinase alpha and beta |
| HAMA | = | human anti-mouse antibody |
| HB-EGF | = | heparin-binding epidermal growth factor |
| HER2 | = | human epidermal growth factor receptor 2 |
| HER3 | = | human epidermal growth factor receptor 3 |
| HNSCC | = | head and neck squamous cell carcinoma |
| IF | = | immunoflourescence |
| IGF1R | = | insulin-like growth factor receptor 1 |
| IgG | = | immunoglobulin G |
| IHC | = | immunohistochemistry |
| ILK | = | integrin linked kinase |
| IP3 | = | inositol triphosphate |
| mAb | = | monoclonal antibody |
| mCRC | = | metastatic colorectal cancer |
| MAPK | = | mitogen-activated protein kinase |
| MDGI | = | mammary-derived growth inhibitor |
| MEK | = | MAPK kinase |
| miRs | = | microRNAs |
| mTORC | = | mammalian target for rapamycin |
| MSK | = | mitogen and stress-activated protein kinase |
| nRTK | = | non-receptor tyrosine kinase |
| NLS | = | nuclear localization signal |
| NRG | = | neuregulins |
| NSCLC | = | non-small cell lung cancer |
| PI3K | = | phosphatidylinositol 3-kinase |
| PDGFR | = | platelet derived growth factor receptor |
| PDK1 | = | phosphoinositide-dependent kinase 1 |
| PFS | = | progression-free survival |
| PH | = | pleckstrin homology |
| PIP2 | = | phosphatidylinositol-4,5-bisphosphate |
| PIP3 | = | phosphatidylinositol (3,4,5)-trisphosphate |
| PLCγ | = | phospholipase C-gamma |
| PKC | = | protein kinase C |
| PPARγ | = | peroxisome-proliferator-activated receptor γ |
| PTEN | = | phosphatase and tensin homologue deleted in chromosome 10 |
| RSK | = | ribosomal s6 kinase |
| RTK | = | receptor tyrosine kinase |
| SFKs | = | Src-family kinases |
| SH2 | = | src homology |
| STAT | = | signal transducer and activator of transcription |
| TGFα | = | transforming growth factor receptor alpha |
| TUNEL | = | terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling |
| TKD | = | tyrosine kinase domain |
| TKIs | = | tyrosine kinase inhibitors |
| TSC2 | = | tuberous sclerosis protein complex 2 |
| VEGF | = | vascular endothelial growth factor |
| VEGFR | = | vascular endothelial growth factor receptor 2 |
Figures and Tables
Figure 1 EGFR biology. Ligand binding to domains I and III of the EGFR expose the dimerization loop in domain II and induce receptor homo- or hetero-dimerization. This process activates the intrinsic tyrosine kinase activity of the EGFR and leads to the phosphorylation of specific tyrosines on the cytoplasmic tails of the receptor pair. These phospho-tyrosines serve to recruit specific effector molecules containing SH2 or PTB domains. These effector molecules recruit and induce various signaling pathways including the PI3K/AKT (A), RAS/MAPK (A), PLCγ/PKC (B) and STAT (B) pathways. In addition, a less appreciated pathway, the nuclear EGFR signaling pathway (C), is initiated upon ligand binding and induces EGFR translocation to the nucleus where it behaves as a co-transcriptional activator regulating key genes such as Cox2, iNOS, B-myb, Aurora kinase A and Cyclin D1.
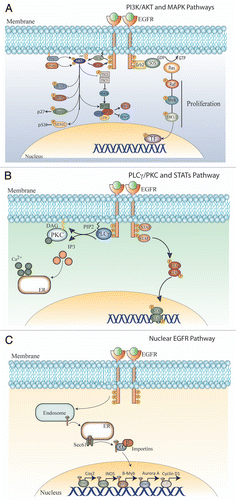
Figure 2 Structure and development of cetuximab. Three murine antibodies designated M225 IgG, M528 IgG and M579 IgG with activity against the EGFR were developed. Further testing identified M225 as being the most efficacious for anti-EGFR activity and was moved into Phase I clinical trials. Although successful, patients developed human-anti-mouse antibodies (HAMA) and therefore M225 was converted to a human:murine chimera, C225, with an IgG1 FC isotype.
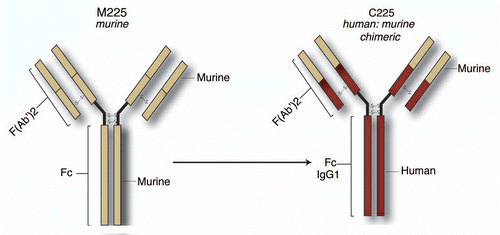
Figure 3 Mechanisms of action of cetuximab. (A) Cetuximab has a higher affinity for the EGFR than either TGFα or EGF and effectively blocks ligand binding and ligand induced EGFR phosphorylation.Citation77–Citation80 (B) Cetuximab has been noted to sterically hinder the binding of EGFR to other HER family members.Citation81 (C) Cetuximab promotes the internalization and degradation of the EGFR, abrogating its downstream signaling cascades.Citation83 (D) Cetuximab treatment of cancer cell lines and human tumor xenografts have shown a dramatic cell cycle arrest in the G1 phase of the cell cycle. Further investigations indicated that this was due to an increased expression of the cell cycle inhibitor p27Kip1. This increased expression led to the formation of p27Kip1-Cdk2 complexes and the prevention of cells from exiting the G1 phase of the cell cycle.Citation86–Citation88 (E) It has been noted that EGFR expressing tumor lines display a significant increase in pro-angiogenic factors leading to increased angiogenesis to the tumor. Treatment with cetuximab has been shown to dramatically decrease the expression of pro-angiogenic factors. In addition to decreased angiogenesis, there is evidence that cetuximab therapy may lead to decreased invasion and metastatic spread of tumor cell.Citation86,Citation94–Citation97 (F) Cetuximab treatment has also been noted to influence the balance of apoptosis and cell survival through modulation of the expression of Bax, which promotes apoptosis and Bcl2, which promotes survival. Treatment with cetuximab increased expression of Bax and decreased Bcl2.Citation95–Citation97 (G) Antibody-dependent cellular cytotoxicity mediated by cetuximab has also been noted in several studies.Citation98,Citation99
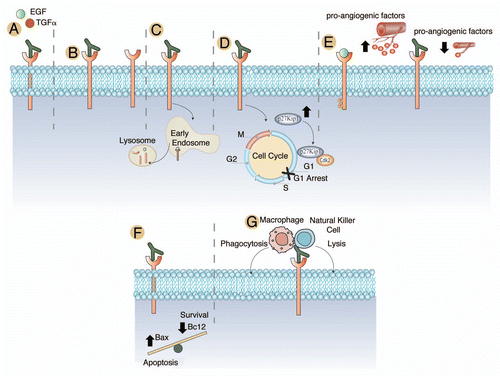
Table 1 Selected clinical trials of cetuximab
Table 2 Mechanisms of resistance to EGFR-targeted antibodies
Acknowledgements
We would like to thank Laura Hogan for her critical reading of the manuscript. This project was supported, in part, by grant P30CA014520 from the National Cancer Institute, grant 1UL1RR025011 from the Clinical and Translational Science Award program of the National Center for Research Resources, National Institutes of Health and by grant RSG-10-193-01-TBG from the American Cancer Society.
References
- Carpenter G, Lembach KJ, Morrison MM, Cohen S. Characterization of the binding of 125I-labeled epidermal growth factor to human fibroblasts. J Biol Chem 1975; 250:4297 - 4304
- Carpenter G, King L Jr, Cohen S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 1978; 276:409 - 410
- Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984; 309:418 - 425
- Eckhart W, Hutchinson MA, Hunter T. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 1979; 18:925 - 933
- Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA 1980; 77:1311 - 1315
- Riedel H, Dull TJ, Schlessinger J, Ullrich A. A chimaeric receptor allows insulin to stimulate tyrosine kinase activity of epidermal growth factor receptor. Nature 1986; 324:68 - 70
- Schlessinger J. Signal transduction by allosteric receptor oligomerization. Trends Biochem Sci 1988; 13:443 - 447
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127 - 137
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5:341 - 354
- Klapper LN, Glathe S, Vaisman N, Hynes NE, Andrews GC, Sela M, et al. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci USA 1999; 96:4995 - 5000
- Singh AB, Harris RC. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 2005; 17:1183 - 1193
- Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009; 9:463 - 475
- Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL 3rd. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci USA 1994; 91:8132 - 8136
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J 1995; 14:4267 - 4275
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 2004; 4:937 - 947
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550 - 562
- Carpenter G, Ji Q. Phospholipase C-gamma as a signal-transducing element. Exp Cell Res 1999; 253:15 - 24
- Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol 2004; 5:785 - 790
- Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 2007; 7:281 - 294
- Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet?. Nat Rev Cancer 2007; 7:554 - 562
- Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology 1991; 13:15 - 20
- Cao H, Lei ZM, Bian L, Rao CV. Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta. Endocrinology 1995; 136:3163 - 3172
- Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3:802 - 808
- Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat 2006; 95:211 - 218
- Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer 2006; 94:184 - 188
- Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res 2005; 65:338 - 348
- Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, et al. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res 2005; 11:5856 - 5862
- Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog 2009; 48:610 - 617
- Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005; 7:575 - 589
- Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem 2007; 282:10432 - 10440
- Lo HW, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFRSTAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res 2010; 8:232 - 245
- Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem 2006; 98:1570 - 1583
- Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell 2007; 18:1064 - 1072
- Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog 2006; 45:10 - 17
- Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res 2008; 36:4337 - 4351
- Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol 2006; 8:1359 - 1368
- Dittmann K, Mayer C, Kehlbach R, Rodemann HP. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer 2008; 7:69
- Liao HJ, Carpenter G. Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor. Cancer Res 2009; 69:6179 - 6183
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Bio 2006; 7:505 - 516
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 2003; 5:461 - 466
- Mineo C, Gill GN, Anderson GW. Regulated migration of epidermal growth factor receptor from caveolae. J Biol Chem 1999; 274:30636 - 30643
- Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996; 274:2086 - 2089
- Yamabhai M, Anderson RGW. Second cysteine-rich region of epidermal growth factor receptor contains targeting information for caveolae/rafts. J Biol Chem 2002; 277:24843 - 24846
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 2002; 416:183 - 187
- de Melker AA, van der Horst G, Borst J. c-Cbl directs EGF receptors into an endocytic pathway that involves the ubiquitin-interacting motif of Eps15. J Cell Sci 2004; 117:5001 - 5012
- Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 2004; 23:2057 - 2070
- Bache KG, Slagsvold T, Stenmark H. Defective down-regulation of receptor tyrosine kinases in cancer. EMBO J 2004; 23:2707 - 2712
- Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: Quantitative analysis of HER2 overexpression effects. Cancer Res 2003; 63:1130 - 1137
- Worthylake R, Opresko LK, Wiley HS. ErbB-2 amplification inhibits downregulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem 1999; 274:8865 - 8874
- Baulida J, Kraus MH, Alimandi M, DiFiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem 1996; 271:5251 - 5257
- Sorkin A, Waters CM. Endocytosis of growth-factor receptors. Bioessays 1993; 15:375 - 382
- Wang ZX, Zhang LF, Yeung TK, Chen XM. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol Biol Cell 1999; 10:1621 - 1636
- Chung BM, Raja SM, Clubb RJ, Tu C, George M, Band V, et al. Aberrant trafficking of NSCLCassociated EGFR mutants through the endocytic recycling pathway promotes interaction with Src@. Bmc Cell Biol 2009; In press
- Hendriks BS, Wiley HS, Lauffenburger D. HER2-mediated effects on EGFR endosomal sorting: Analysis of biophysical mechanisms. Biophys J 2003; 85:2732 - 45
- Weichselbaum RR, Dunphy EJ, Beckett MA, Tybor AG, Moran WJ, Goldman ME, et al. Epidermal growth factor receptor gene amplification and expression in head and neck cancer cell lines. Head Neck 1989; 11:437 - 442
- Veale D, Ashcroft T, Marsh C, Gibson GJ, Harris AL. Epidermal growth factor receptors in non-small cell lung cancer. Br J Cancer 1987; 55:513 - 516
- Libermann TA, Razon N, Bartal AD, Yarden Y, Schlessinger J, Soreq H. Expression of epidermal growth factor receptors in human brain tumors. Cancer Res 1984; 44:753 - 760
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985; 313:144 - 147
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl 1985; 3:161 - 172
- Maurizi M, Almadori G, Ferrandina G, Distefano M, Romanini ME, Cadoni G, et al. Prognostic significance of epidermal growth factor receptor in laryngeal squamous cell carcinoma. Br J Cancer 1996; 74:1253 - 1257
- FischerColbrie J, Witt A, Heinzl H, Speiser P, Czerwenka K, Sevelda P, et al. EGFR and steroid receptors in ovarian carcinoma: Comparison with prognostic parameters and outcome of patients. Anticancer Res 1997; 17:613 - 619
- Kersemaekers AMF, Fleuren GJ, Kenter EG, Van den Broek LJCM, Uljee SM, Hermans J, et al. Oncogene alterations in carcinomas of the uterine cervix: Overexpression of the epidermal growth factor receptor is associated with poor prognosis. Clin Cancer Res 1999; 5:577 - 586
- Mellon K, Wright C, Kelly P, Horne CHW, Neal DE. Long-term outcome related to epidermal growth-factor receptor status in bladder-cancer. Journal of Urology 1995; 153:919 - 925
- Inada S, Koto T, Futami K, Arima S, Iwashita A. Evaluation of malignancy and the prognosis of esophageal cancer based on an immunohistochemical study (p53, E-cadherin, epidermal growth factor receptor). Surg Today 1999; 29:493 - 503
- Ohgaki H, Watanabe K, Tachibana O, Sato K, Kleihues P. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. J Neuropathol Exp Neurol 1996; 55:3
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37:9 - 15
- Nielsen JS, Jakobsen E, Holund B, Bertelsen K, Jakobsen A. Prognostic significance of p53, Her-2 and EGFR overexpression in borderline and epithelial ovarian cancer. Int J Gynecol Cancer 2004; 14:1086 - 1096
- Santini J, Formento JL, Francoual M, Milano G, Schneider M, Dassonville O, et al. Characterization, quantification and potential clinical-value of the epidermal growth-factor receptor in head and neck squamous-cell carcinomas. Head Neck 1991; 13:132 - 139
- Yasui W, Sumiyoshi H, Hata J, Kameda T, Ochiai A, Ito H, et al. Expression of epidermal growth-factor receptor in human gastric and colonic carcinomas. Cancer Res 1988; 48:137 - 141
- Piyathilake CJ, Frost AR, Manne U, Weiss H, Bell WC, Heimburger DC, et al. Differential expression of growth factors in squamous cell carcinoma and pre-cancerous lesions of the lung. Clin Cancer Res 2002; 8:734 - 744
- Scambia G, Panici PB, Ferrandina G, Battaglia F, Distefano M, Dandrea G, et al. Significance of epidermal growth-factor receptor expression in primary human endometrial cancer. Int J Cancer 1994; 56:26 - 30
- Nicholson RI, Gee JMW, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37:9 - 15
- Sizeland AM, Burgess AW. Anti-sense transforming growth factor alpha oligonucleotides inhibit autocrine stimulated proliferation of a colon carcinoma cell line. Mol Biol Cell 1992; 3:1235 - 1243
- Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA 1990; 87:4207 - 4211
- Malden LT, Novak U, Kaye AH, Burgess AW. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res 1988; 48:2711 - 2714
- Peschard P, Park M. Escape from Cbl-mediated down-regulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 2003; 3:519 - 523
- Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, et al. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem 1984; 259:7755 - 7760
- Kawamoto T, Sato JD, Le A, Polikoff J, Sato GH, Mendelsohn J. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci USA 1983; 80:1337 - 1341
- Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med 1983; 1:511 - 529
- Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res 1984; 44:1002 - 1007
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301 - 311
- Yang XD, Jia XC, Corvalan JRF, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hemat 2001; 38:17 - 23
- Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proc Natl Acad Sci USA 1986; 83:3825 - 3829
- Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res 1995; 1:1311 - 1318
- Sunada H, Magun BE, Mendelsohn J, Macleod CL. Monoclonal-antibody against epidermal growth-factor receptor is internalized without stimulating receptor phosphorylation. Proc Natl Acad Sci USA 1986; 83:3825 - 3829
- Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res 1999; 59:1935 - 1940
- Peng D, Fan Z, Lu Y, DeBlasio T, Scher H, Mendelsohn J. Anti-epidermal growth factor receptor monoclonal antibody 225 upregulates p27KIP1 and induces G1 arrest in prostatic cancer cell line DU145. Cancer Res 1996; 56:3666 - 3669
- Wu X, Rubin M, Fan Z, DeBlasio T, Soos T, Koff A, et al. Involvement of p27KIP1 in G1 arrest mediated by an anti-epidermal growth factor receptor monoclonal antibody. Oncogene 1996; 12:1397 - 403
- Baselga J. The EGFR as a target for anticancer therapy—focus on cetuximab. Eur J Cancer 2001; 37:16 - 22
- P OC, Rhys-Evans P, Modjtahedi H, Court W, Box G, Eccles S. Overexpression of epidermal growth factor receptor in human head and neck squamous carcinoma cell lines correlates with matrix metalloproteinase-9 expression and in vitro invasion. Int J Cancer 2000; 86:307 - 317
- P Oc, Rhys-Evans P, Court WJ, Box GM, Eccles SA. Differential modulation of proliferation, matrix metalloproteinase expression and invasion of human head and neck squamous carcinoma cells by c-erbB ligands. Clin Exp Metastasis 1999; 17:631 - 639
- Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ, et al. Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clin Cancer Res 2000; 6:1936 - 1948
- Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, et al. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 1999; 5:257 - 265
- Tortora G, Caputo R, Pomatico G, Pepe S, Bianco AR, Agrawal S, et al. Cooperative inhibitory effect of novel mixed backbone oligonucleotide targeting protein kinase A in combination with docetaxel and anti-epidermal growth factor-receptor antibody on human breast cancer cell growth. Clin Cancer Res 1999; 5:875 - 881
- Liu B, Fang M, Schmidt M, Lu Y, Mendelsohn J, Fan Z. Induction of apoptosis and activation of the caspase cascade by anti-EGF receptor monoclonal antibodies in DiFi human colon cancer cells do not involve the c-jun N-terminal kinase activity. Br J Cancer 2000; 82:1991 - 1999
- Mandal M, Adam L, Mendelsohn J, Kumar R. Nuclear targeting of Bax during apoptosis in human colorectal cancer cells. Oncogene 1998; 17:999 - 1007
- Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J. Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest 1995; 95:1897 - 1905
- Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 2007; 13:1552 - 1561
- Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci 2007; 98:1275 - 1280
- Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005; 23:1803 - 1810
- Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040 - 2048
- Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:2311 - 2319
- Borner M, Koeberle D, Von Moos R, Saletti P, Rauch D, Hess V, et al. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first-line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Ann Oncol 2008; 19:1288 - 1292
- Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 2009; 27:663 - 671
- Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360:1408 - 1417
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 1127
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006; 354:567 - 578
- Herbst RS, Arquette M, Shin DM, Dicke K, Vokes EE, Azarnia N, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol 2005; 23:5578 - 5587
- Baselga J, Trigo JM, Bourhis J, Tortochaux J, Cortes-Funes H, Hitt R, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol 2005; 23:5568 - 5577
- Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an eastern Cooperative Oncology Group study. J Clin Oncol 2005; 23:8646 - 8654
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351:337 - 345
- Robert F, Ezekiel MP, Spencer SA, Meredith RF, Bonner JA, Khazaeli MB, et al. Phase I study of antiepidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol 2001; 19:3234 - 3243
- Thienelt CD, Bunn PA Jr, Hanna N, Rosenberg A, Needle MN, Long ME, et al. Multicenter phase I/II study of cetuximab with paclitaxel and carboplatin in untreated patients with stage IV non-small-cell lung cancer. J Clin Oncol 2005; 23:8786 - 8793
- Rosell R, Robinet G, Szczesna A, Ramlau R, Constenla M, Mennecier BC, et al. Randomized phase II study of cetuximab plus cisplatin/vinorelbine compared with cisplatin/vinorelbine alone as first-line therapy in EGFR-expressing advanced non-small-cell lung cancer. Ann Oncol 2008; 19:362 - 369
- Robert F, Blumenschein G, Herbst RS, Fossella FV, Tseng J, Saleh MN, et al. Phase I/IIa study of cetuximab with gemcitabine plus carboplatin in patients with chemotherapy-naive advanced non-small-cell lung cancer. J Clin Oncol 2005; 23:9089 - 9096
- Herbst RS, Chansky K, Kelly K, Atkins JN, Davies AM, Dakhil SR, et al. phase II randomized selection trial evaluating concurrent chemotherapy plus cetuximab or chemotherapy followed by cetuximab in patients with advanced non-small cell lung cancer (NSCLC): Final report of SWOG 0342 2007; ASCO
- Butts CA, Bodkin D, Middleman EL, Englund CW, Ellison D, Alam Y, et al. Randomized phase II study of gemcitabine plus cisplatin or carboplatin [corrected], with or without cetuximab, as first-line therapy for patients with advanced or metastatic non small-cell lung cancer. J Clin Oncol 2007; 25:5777 - 5784
- Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009; 373:1525 - 1531
- Lynch TJ, Patel T, Dreisbach L, McCleod M, Heim WJ, Hermann RC, et al. Cetuximab and first-line taxane/carboplatin chemotherapy in advanced non-small-cell lung cancer: results of the randomized multicenter phase III trial BMS099. J Clin Oncol 2010; 28:911 - 917
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101:13306 - 13311
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 2139
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 1500
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:73
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352:786 - 792
- Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA. Responsiveness to cetuximab without mutations in EGFR. N Engl J Med 2005; 353:208 - 209
- Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst 2005; 97:1185 - 1194
- Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 2005; 6:279 - 286
- Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 2007; 97:1139 - 1145
- Personeni N, Fieuws S, Piessevaux H, De Hertogh G, De Schutter J, Biesmans B, et al. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res 2008; 14:5869 - 5876
- Cappuzzo F, Finocchiaro G, Rossi E, Janne PA, Carnaghi C, Calandri C, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 2008; 19:717 - 723
- Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol 2007; 25:3238 - 3245
- Hirsch FR, Herbst RS, Olsen C, Chansky K, Crowley J, Kelly K, et al. Increased EGFR gene copy number detected by fluorescent in situ hybridization predicts outcome in non-small-cell lung cancer patients treated with cetuximab and chemotherapy. J Clin Oncol 2008; 26:3351 - 3357
- Khambata-Ford S, Harbison CT, Hart LL, Awad M, Xu LA, Horak CE, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol 2010; 28:918 - 927
- Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007; 25:3230 - 3237
- Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987; 238:542 - 545
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295 - 308
- Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006; 66:3992 - 3995
- Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 2007; 96:1166 - 1169
- De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 2008; 19:508 - 515
- Punt CJ, Tol J, Rodenburg CJ, Cats A, Creemers GM, Schrama JG, et al. Randomized phase III study of capecitabine, oxaliplatin and bevacizumab with or without cetuximab in advanced colorectal cancer (ACC), the CAIRO2 study of the Dutch Colorectal Cancer Group (DCCG) 2008; ASCO Meeting Abstracts May 20 2008
- Van Cutsem E, Lang I, D'haens G, Moiseyenko V, Zaluski J, Folprecht G, et al. KRAS status and efficacy in the first-line treatment of patients with metastatic colorectal cancer (mCRC) treated with FOLFIRI with or without cetuximab: The CRYSTAL experience. ASCO Meeting Abstracts 2008; 2:2008
- Bokemeyer C, Bondarenko I, Hartmann J, De Braud F, Volovat C, Nippgen L, et al. KRAS status and efficacy of first-line treatment of patients with metastatic colorectal cancer (mCRC) with FOLFOX with or without cetuximab: The OPUS experience. ASCO Meeting Abstracts 2008; 4000
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757 - 1765
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:1626 - 1634
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009; 27:2091 - 2096
- Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 2009; 27:2622 - 2629
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to antiepidermal growth factor receptor antibody therapies. Cancer Res 2007; 67:2643 - 2648
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 1995; 14:3136 - 3145
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291 - 3310
- Niault TS, Baccarini M. Targets of Raf in tumorigenesis. Carcinogenesis 2010; 31:1165 - 1174
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008; 26:5705 - 5712
- Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R, et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat 2009; 30:1199 - 1206
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA 2009; 106:4519 - 4524
- Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002; 62:6997 - 7000
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 954
- Weber A, Langhanki L, Sommerer F, Markwarth A, Wittekind C, Tannapfel A. Mutations of the BRAF gene in squamous cell carcinoma of the head and neck. Oncogene 2003; 22:4757 - 4759
- Perrone F, Suardi S, Pastore E, Casieri P, Orsenigo M, Caramuta S, et al. Molecular and cytogenetic subgroups of oropharyngeal squamous cell carcinoma. Clin Cancer Res 2006; 12:6643 - 6651
- Fei Huang LAX, Khambata-Ford Shirin. Analysis of the correlation between gene expression of IGF-pathway components and benefit from cetuximab (Erbitux™) in metastatic colorectal cancer patients. AACR meeting Abstracts 2010;
- Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, et al. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 1999; 5:257 - 265
- Petit AMV, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, et al. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases downregulate vascular endothelial growth factor production by tumor cells in vitro and in vivo—Angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol 1997; 151:1523 - 1530
- Ciardiello F, Bianco R, Damiano V, Fontanini G, Caputo R, Pomatico G, et al. Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human GEO colon cancer cells. Clin Cancer Res 2000; 6:3739 - 3747
- Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: A role for altered tumor angiogenesis. Cancer Res 2001; 61:5090 - 5101
- Ferrara N. The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res Treat 1995; 36:127 - 137
- Fontanini G, Vignati S, Boldrini L, Chine S, Silvestri V, Lucchi M, et al. Vascular endothelial growth factor is associated with neovascularization and influences progression of non-small cell lung carcinoma. Clin Cancer Res 1997; 3:861 - 865
- Jung YD, Mansfield PF, Akagi M, Takeda A, Liu W, Bucana CD, et al. Effects of combination anti-vascular endothelial growth factor receptor and anti-epidermal growth factor receptor therapies on the growth of gastric cancer in a nude mouse model. Eur J Cancer 2002; 38:1133 - 1140
- Ciardiello F, Bianco R, Caputo R, Caputo R, Damiano V, Troiani T, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res 2004; 10:784 - 793
- Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res 2008; 14:5069 - 5080
- Lu Y, Li XQ, Liang K, Luwor R, Siddik ZH, Nuiis GB, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the Anti-EGFR monoclonal antibody cetuximab. Cancer Res 2007; 67:8240 - 8247
- Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: Implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci USA 2003; 100:2438 - 2443
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008; 27:3944 - 3956
- Grovdal LM, Stang E, Sorkin A, Madshus IH. Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res 2004; 300:388 - 395
- Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem 2009; 284:36592 - 36604
- Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively Activated STAT3 Frequently Coexpresses with Epidermal Growth Factor Receptor in High-Grade Gliomas and Targeting STAT3 Sensitizes Them to Iressa and Alkylators. Clin Cancer Res 2008; 14:6042 - 6054
- Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Tr 2006; 95:211 - 218
- Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia WY, Wei YK, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005; 7:575 - 589
- Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem 2006; 98:1570 - 1583
- Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinogen 2006; 45:10 - 17
- Kim J, Jahng WJ, Di Vizio D, Lee JS, Jhaveri R, Rubin MA, et al. The phosphoinositide kinase PIKfyve mediates epidermal growth factor receptor trafficking to the nucleus. Cancer Res 2007; 67:9229 - 37
- Xia WY, Wei YK, Du Y, Liu JS, Chang B, Yu YL, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinogen 2009; 48:610 - 617
- Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801 - 3813
- Lin SY, Makino K, Xia WY, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3:802 - 808
- Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801 - 3813
- Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem 1998; 273:1568 - 1573
- Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther 2009; 8:696 - 703
- Nevo J, Mattila E, Pellinen T, Yamamoto DL, Sara H, Iljin K, et al. Mammary-derived growth inhibitor alters traffic of EGFR and induces a novel form of cetuximab resistance. Clin Cancer Res 2009; 15:6570 - 6581
- Huynh HT, Larsson C, Narod S, Pollak M. Tumor suppressor activity of the gene encoding mammary-derived growth inhibitor. Cancer Res 1995; 55:2225 - 2231
- Huynh H, Alpert L, Pollak M. Silencing of the mammary-derived growth inhibitor (MDGI) gene in breast neoplasms is associated with epigenetic changes. Cancer Res 1996; 56:4865 - 4870
- Specht B, Bartetzko N, Hohoff C, Kuhl H, Franke R, Borchers T, et al. Mammary derived growth inhibitor is not a distinct protein but a mix of heart-type and adipocyte-type fatty acid-binding protein. J Biol Chem 1996; 271:19943 - 19949
- Spener F, Unterberg C, Borchers T, Grosse R. Characteristics of fatty acid-binding proteins and their relation to mammary-derived growth inhibitor. Mol Cell Biochem 1990; 98:57 - 68
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17:548 - 58
- Thompson EW, Newgreen DF. Carcinoma invasion and metastasis: A role for epithelial-mesenchymal transition?. Cancer Res 2005; 65:5991 - 5995
- Basu D, Nguyen TTK, Montone KT, Zhang G, Wang LP, Diehl JA, et al. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene 2010; 29:4170 - 4182
- Strauss R, Sova P, Liu Y, Li ZY, Tuve S, Pritchard D, et al. Epithelial phenotype confers resistance of ovarian cancer cells to oncolytic denoviruses. Cancer Res 2009; 69:5115 - 5125
- Troussard AA, Mawji NM, Ong C, Mui A, St Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem 2003; 278:22374 - 22378
- Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res 2008; 68:2391 - 2399
- Kim SM, Kim JS, Kim JH, Yun CO, Kim EM, Kim HK, et al. Acquired resistance to cetuximab is mediated by increased PTEN instability and leads cross-resistance to gefitinib in HCC827 NSCLC cells. Cancer Lett 2010; 296:150 - 159
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol 2009; 6:519 - 527
- De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 2008; 19:508 - 15
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010; 28:1254 - 1261
- Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, et al. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2010; In press
- Hatakeyama H, Cheng HX, Wirth P, Counsell A, Marcrom SR, Wood CB, et al. Regulation of heparin-binding EGF-like growth factor by miR-212 and acquired cetuximab-resistance in head and neck squamous cell carcinoma. PLoS One 2010; 5:e12702