Abstract
Recent evidence has identified substantial overlap between metabolic and oncogenic biochemical pathways, suggesting novel approaches to cancer intervention. For example, cholesterol lowering statins and the antidiabetes medication metformin both act as chemopreventive agents in prostate and other cancers. The natural compound resveratrol has similar properties: increasing insulin sensitivity, suppressing adipogenesis, and inducing apoptotic death of cancer cells in vitro. However, in vivo tumor xenografts acquire resistance to resveratrol by an unknown mechanism, while mouse models of metabolic disorders respond more consistently to the compound. Here we demonstrate that castration-resistant human prostate cancer C4-2 cells are more sensitive to resveratrol-induced apoptosis than isogenic androgen-dependent LNCaP cells. The MEK inhibitor U0126 antagonized resveratrol-induced apoptosis in C4-2 cells, but this effect was not seen with other MEK inhibitors. U0126 was found to inhibit mitochondrial function and shift cells to aerobic glycolysis independently of MEK. Mitochondrial activity of U0126 arose through decomposition, producing both mitochondrial fluorescence and cyanide, a known inhibitor of complex IV. Applying U0126 mitochondrial inhibition to C4-2 cell apoptosis, we tested the possibility that glutamine supplementation of citric acid cycle intermediate α-ketoglutarate may be involved. Suppression of the conversion of glutamate to α-ketoglutarate antagonized resveratrol-induced death in C4-2 cells. A similar effect was also seen by reducing extracellular glutamine concentration in the culture medium, suggesting that resveratrol-induced death is dependent on glutamine metabolism, a process frequently dysregulated in cancer. Further work on resveratrol and metabolism in cancer is warranted to ascertain if the glutamine dependence has clinical implications.
Introduction
Altered metabolic pathways in cancer are well documentedCitation1-Citation3 and are potential targets for therapeutic intervention.Citation4 Excess body weight alone is associated with an increase of cancer incidence,Citation5 implying the metabolic state of the patient can lead to cancer development. Recent observational studies have provided evidence that medical therapies that affect cellular metabolism, such as cholesterol lowering (e.g., statins) and antidiabetic agents (e.g., metformin), reduce the risk of some cancers and/or aggressive cancer.Citation6-Citation9 These findings suggest that metabolism-based chemopreventive and chemotherapeutic strategies could substantially decrease cancer incidence and prolong survival in some patients.
Anabolic and catabolic metabolism intersects with multiple oncogenic signal transduction nodes in tumor cells.Citation10-Citation13 This complex web of interactions starts from two major metabolic precursors: glucose and glutamine. In normal cells, glucose is the major energy source and carbon backbone for biosynthesis. Activation of the phosphoinositide 3′-kinase/AKT pathway, a common feature of human cancers, can result in increased glucose import and consumption,Citation14 necessary in cancer cells to fuel growth and proliferation. In contrast, glutamine is the most abundant amino acid in plasma and a necessary precursor for amino acid and nucleotide synthesis.Citation15 In the process of glutaminolysis, glutamine is successively converted into glutamate, followed by conversion to α-ketoglutarate, which can be supplemented into the citric acid cycle to drive production of citrate for lipogenesis.Citation15 In cancer, overexpression of MYC can increase the rate of glutaminolysis, leading to glutamine addiction.Citation11,Citation16 The interconnection between glucose and glutamine supports a strategy where both metabolic pathways, glycolysis and glutaminolysis, are targeted simultaneously.Citation17
Current options for metabolic therapy for cancer are limited. With malignant transformation, glucose metabolism is characteristically shifted away from mitochondrial ATP production to increased lactic acid production by aerobic glycolysis. The glucose analog, 2-deoxyglucose, has been used to inhibit and selectively kill cancer cells,Citation18 showing some clinical efficacy.Citation19 Targeting glutamine metabolism with non-metabolizable analogs like 6-diazo-5-oxo-L-norleucine has been effective in mouse models,Citation20 but side effects in humans limit clinical translation of these strategies.Citation21
One approach to metabolic targeting in cancer is evaluating natural compounds that display cancer-specific cytotoxicities. Dietary natural compounds are potentially advantageous clinically because they are well tolerated and may function as long-term chemopreventives.Citation22 Resveratrol is an example of a natural product that is selectively toxic to cancer and not normal cells,Citation23 though the mechanism of action is unknown. Resveratrol has been shown to act as an antioxidant,Citation24 inhibit COX2,Citation25 activate SIRT1Citation26 and AMPK,Citation27 elicit a DNA damage response,Citation28 and arrest the cell cycle.Citation29 Resveratrol can also alleviate a variety of metabolic disorders in mice including obesity, insulin resistance,Citation26 and liver dysfunction,Citation30 however the compound has generally elicited a poor response in tumor xenograft models.Citation31,Citation32 Nevertheless, the ability of resveratrol to target a variety of oncogenic mechanisms, along with its in vivo efficacy in non-cancer conditions, suggests the possibility that resveratrol is potentially a viable anti-cancer agent. The cancer toxicity and the metabolic effects from resveratrol indicate that this compound may be a powerful tool to learn about cancer cell vulnerabilities that arise from metabolic derangements that arise with neoplasia.
In this study, we investigated the actions of resveratrol in an isogenic pair of prostate cancer cell lines, LNCaP and C4-2, representing early and late stage disease, respectively.Citation39,Citation40 Our objective was to elucidate vulnerabilities targeted by resveratrol for therapeutic development. While exploring a novel function of the MEK inhibitor U0126, we discovered that resveratrol-induced apoptosis is dependent on glutamine metabolism.
Results
U0126 inhibits resveratrol induced apoptosis
Resveratrol is an established cytotoxic agent in prostate,Citation23 breast,Citation33 ovarian,Citation34 colon,Citation35 lung,Citation36 heptatic, and brain cancer cellsCitation37 and is a proposed chemopreventive agent.Citation22 As a component of grape skins, resveratrol is already in the diet,Citation38 implying it may be well tolerated if used in long-term clinical dosing. To determine if resveratrol might be a relevant chemopreventive compound to prevent prostate cancer progression, we exposed the LNCaP and C4-2 prostate cancer disease progression model to resveratrol. C4-2 cells are a subline of LNCaP derived by serial passage through miceCitation39 and display a castration resistant growth phenotype in vivo similar to advanced prostate cancer.Citation40 We found that resveratrol elicited a robust apoptotic response in C4-2 cells and less of a response in LNCaP cells under serum-depleted conditions. This differential response between the two cell lines was highly reproducible in over 20 independent trials. Cell death was measured by monitoring PARP cleavage by protein gel blot analysis () and quantifying DNA fragmentation by nucleosome ELISA after 12 h exposure to resveratrol (). We found extensive cell death at later time points in both cell lines by counting viable cells (data not shown). The increased toxicity in the more aggressive C4-2 cells implies that resveratrol may be more effective against aggressive prostate cancer cells and therefore has chemopreventive potential.
Figure 1. Resveratrol-induced apoptosis is inhibited by U0126. (A) Cell lysate from LNCaP and C4–2 cells, treated with the indicated doses of resveratrol for 12 h in serum-free conditions, were blotted with the indicated antibodies. The two cell lines exhibited differential cleavage of PARP. (B) Nucleosome ELISA data confirm differential apoptosis in LNCaP and C4–2 cells at 2 μM resveratrol after 12 h exposure. (C) Cell lysates from C4–2 cells treated with the indicated MEK inhibitors, at the indicated doses of resveratrol, were subjected to protein gel blot. PARP cleavage is selectively inhibited by 10 μM U0126. (D) U0126 inhibition of apoptosis by resveratrol is confirmed by nucleosome ELISA at 12 h. (E) Cell lysates from C4–2 cells treated with the indicated MEK inhibitors after EGF stimulation in serum-free conditions.
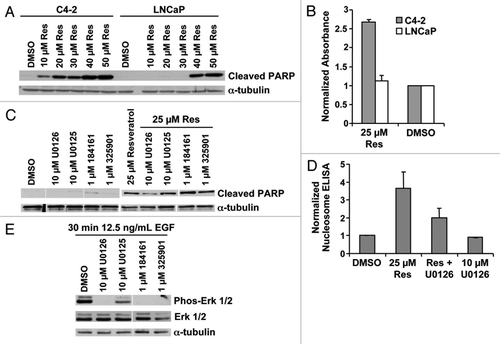
To probe the mechanism behind the higher sensitivity to resveratrol of the C4-2 cells, we investigated the signaling events reported to be linked with resveratrol’s biological effects. The MEK/ERK pathway has been shown to be a critical node in cell survival following resveratrol treatmentCitation41 and this pathway is downstream from growth factor receptors implicated in prostate cancer.Citation42 Intriguingly, inhibition of MEK with the selective MEK-1/2 inhibitor U0126 reduced the extent of resveratrol-induced apoptosis, as measured by PARP cleavage and DNA fragmentation (). However, other MEK inhibitors of similar potencies and specificities did not cause a similar reversion of the apoptotic effect (), suggesting that inhibition of resveratrol-induced apoptosis by U0126 was independent of MEK and was operating through an alternative mechanism.
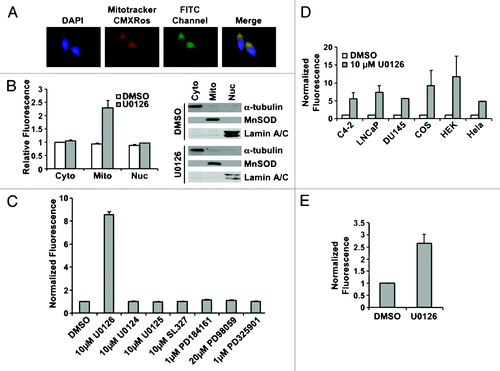
In performing these experiments, we noted that treatment of LNCaP or C4-2 cells with U0126 produced a perinuclear fluorescence signal detectable through the FITC channel in the absence of an FITC fluorochrome. The fluorescence colocalized with a mitochondrial-specific stain, MitoTracker CMXRos (). The mitochondrial localization was confirmed by protein gel blot of cytoplasmic, mitochondrial, and nuclear fractions. Fluorescence was only detected in the mitochondrial fraction of prostate cancer cells treated with U0126 (). The fluorescence signal had excitation and emission peaks of 365 ± 5 nm and 420 ± 5 nm, respectively, in an aqueous solvent, and was quenched in organic solvents including methanol, chloroform, and acetone. The excitation emission characteristics of the fluorescence signal implies it may originate from a conjugated Schiff base, but not a lipid peroxidation product, which would fluoresce in organic solvents.Citation43 Mitochondrial localization of the U0126-dependent fluorescence suggests that U0126 may react in the mitochondria or perturb mitochondrial function. Other MEK inhibitors (PD98059, PD325901 or PD184161) and analogs of U0126 (U0124, U0125 or SL327) did not produce fluorescence (). U0126 caused fluorescence in a variety of mammalian cell lines (HeLa, COS, DU145, or HEK293T) and in the BY4741 yeast strain (). To evaluate the dependence of appearance of the fluorescent product on MEK, a yeast knockout strain was used that lacks the MEK homolog STE7. The STE7 knockout strain produced comparable levels of fluorescence when exposed to U0126 (). We concluded that the specific structure of U0126 produces or induces fluorescence by a mechanism common to eukaryotic cells and independently of MEK.
Figure 2. U0126 induces mitochondrial fluorescence. (A) Fluorescence imaging of LNCaP cells treated with U0126 for 12 h and stained with the mitochondrial dye Mitotracker CMXRos. Note the FITC perinuclear signal that colocalizes with the mitochondrial stain. (B) Fluorescence localization in the mitochondria is confirmed by cell fractionation and fractionation purity is assayed by protein gel blotting of compartment specific markers: α-tubulin for cytoplasm (cyto), MnSOD for mitochondria (mito), and lamin A/C for nucleus (nuc). (C) Multiple MEK inhibitors did not produce a fluorescent signal when incubated in C4–2 cells for 12 h. (D) U0126 produced fluorescence in multiple mammalian cells after 12 h incubation and (E) in BY4741 yeast.
U0126 alters metabolism and inhibits mitochondrial function
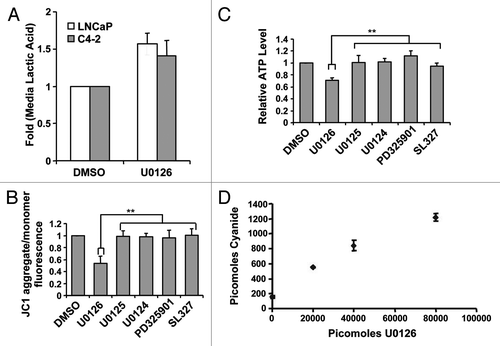
Given the generation of fluorescence by U0126 and its ability to suppress resveratrol-induced apoptosis, we hypothesized that U0126 was shifting the cellular state at the time resveratrol initiated apoptosis. Consistent with this, U0126 treatment of C4-2 cells altered cell morphology, producing a flattened cell shape (), and acidified the culture media after 12–24 h. Acidification of the media suggested that U0126-treated cells excrete more lactic acid, the final product of aerobic glycolysis. An absorbance-based assay confirmed increased lactic acid content in the media of U0126-treated cells at 24 h compared with DMSO treated control cells (). Considering the increased lactic acid excretion and the mitochondrial fluorescence, we postulated that U0126 may be a mitochondrial inhibitor. Consistent with this hypothesis, U0126 decreased mitochondrial membrane potential and ATP levels in the presence of 2-deoxyglucose, while other MEK inhibitors did not (). To understand how U0126 may inhibit mitochondria, we inspected its chemical structure and hypothesized that decomposition might release the nitrile groups as cyanide. U0126 dose-dependently generated higher levels of cyanide from C4-2 cells when assayed 24 h after treatment (). Cyanide levels detected were substantially lower than the theoretical limit and may be low estimates of total cyanide released due to cyanide outgassing from culture media.Citation44
Figure 4. Biological consequences of mitochondrial inhibition by U0126. (A) Cell morphology of C4–2 cells 48 h after drug treatment. U0126, U0124 were dosed at 10uM and PD325901(PD32) was dosed at 1 μM. (B) Crystal violet staining of C4–2 and T24 cells at 48 h after treatment with DMSO (D) or U0126 (U) and varying concentrations of sodium oxamate (Ox). (C) Viable cell counting with trypan blue of C4–2 cells 48 h after treatment with the indicated drugs. (D) Lactate dehydrogenase (LDH) enzyme activity from cell lysates incubated with varying concentrations of sodium oxamate to verify LDH inhibition.
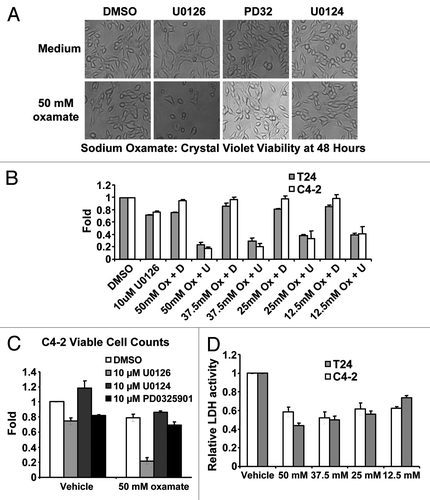
Figure 3. U0126 induces a metabolic shift in prostate cancer cells. (A) Lactic acid levels in the medium of LNCaP and C4–2 cells were assayed 24 h after treatment with U0126. DMSO was used as a vehicle. (B) Mitochondrial potential was assayed at 12 h after dosing various MEK inhibitors in C4–2 cells. (C) Cellular ATP levels were measured 9 h after treatment with U0126. Prior to assay, cells were preincubated with 10mM 2-deoxygluocse for 30 min to inhibit glycolytic ATP production, but not mitochondrial production. (D) Cyanide levels detected in cell lysates exposed to different concentrations of U0126 for 24 h. **p < 0.05, two-tailed Student’s t-test.
All mammalian cells require glycolysis to generate energy and carry out metabolic reactions. Glycolysis can be autoinhibited if the end product, pyruvate, is not consumed by the mitochondria or converted to lactate by lactate dehydrogenase (LDH), resulting in cell death.Citation45 If 10 μM U0126 substantially inhibits mitochondrial function, then dosing U0126 with an LDH inhibitor should result in synergistic cell death. To evaluate this hypothesis, we used U0126 in combination with sodium oxamate, an LDH inhibitor, and evaluated survival in C4-2 and T24 bladder cancer cell lines at 48 h by crystal violet staining and viable cell counting (). U0126 and sodium oxamate produced synergistic cell death in both cancer cell lines. In comparison, another MEK inhibitor, PD325901, or the U0126 analog U0124 were dosed as control compounds with sodium oxamate and did not induce cell death (). PD325901 is an important control in this experiment because it binds to the same site on MEKCitation46 with a ~100-fold lower IC50.Citation47 Taken together, these data indicate that U0126 inhibits mitochondrial function and stimulates aerobic glycolysis by a MEK-independent mechanism.
Role of glutamine metabolism in resveratrol-induced tumor cell death
The above results indicate that U0126 acts in the mitochondria to cause a shift in cellular metabolism. In cancer, mitochondrial function is altered toward the production of substrates for biosynthesis at the expense of energy production. Cancer cells use the TCA cycle substrate citrate for lipid synthesis, but must load α-ketoglutarate derived from glutamine into the cycle to maintain the mass flux. As a mitochondrial inhibitor, U0126 may therefore influence metabolite flux through the TCA cycle and reduce glutamine-dependent supplementation of α-ketoglutarate.
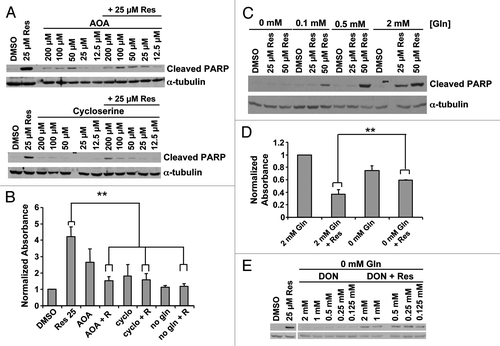
We tested the dependence of resveratrol-induced death on the supplementation of TCA cycle intermediates by inhibiting alanine and aspartate transaminases, which convert glutamate to α-ketoglutarate in the cytosol (aspartate transaminase also has a mitochondrial isoform) for import into the mitochondria. Two transaminase inhibitors, cycloserineCitation48 and amino oxyacetate (AOA), inhibited programmed cell death in response to resveratrol at 12 h, as measured by PARP cleavage () and nucleosome ELISA (). Transaminases produce α-ketoglutarate indirectly from imported glutamine and some cancer cells exhibit increased glutamine uptake.Citation1 Decreasing glutamine in the culture media produced a dose-dependent decrease in resveratrol-induced apoptosis at 12 h in C4-2 cells (). Glutamine depletion of the culture medium protected cells from resveratrol for up to 60 h (). Glutamine-free media did not induce morphological signs of cellular distress. A non-metabolizable analog, 6-diazo-5-oxo-L-norleucine (DON), failed to restore the resveratrol-induced PARP cleavage in glutamine-free media, suggesting that glutamine metabolism is likely to be required for the resveratrol-induced apoptosis (). Taken together, we conclude that resveratrol-induced programmed cell death is dependent on metabolism of glutamine, partially through a mitochondria-dependent mechanism.
Figure 5. Role of glutamine metabolism in resveratrol induced apoptosis. (A) Transaminase inhibitors AOA and cycloserine were used in serum-free conditions. Twelve hours later resveratrol was added and cell death was assayed at 24 h by cleaved PARP or (B) nucleosome ELISA. **p < 0.05, paired two tailed t-test. (C) Cell death induced by resveratrol was assayed at varying glutamine concentrations. (D) Cell viability by crystal violet staining 60 h after resveratrol treatment at varying glutamine concentrations. **p < 0.05, paired one tailed t-test (E) The non-metabolizable glutamine analog, 6-diazo-5-oxo-L-norleucine (DON), did not restore resveratrol-induced death in glutamine-free medium.
Discussion
In this study, we found that the MEK inhibitor U0126 is also a mitochondrial inhibitor capable of shifting cell metabolism toward aerobic glycolysis and sensitizing cells to glycolytic inhibitors. Attempts to understand the metabolic implications of our U0126 findings led us to discover that resveratrol-induced apoptosis is dependent on glutamine metabolism. As small molecule drugs, U0126 and resveratrol illuminate distinct metabolic vulnerabilities within cancer cell metabolic pathways. U0126 shifts prostate cancer cells toward increased glycolysis and sensitivity to lactic acid dehydrogenase inhibitors, while resveratrol targets glutamine addiction. The mechanisms proposed for both compounds may substantially influence the interpretation of the preceding U0126 literature, help to integrate the divergent conclusions from the resveratrol literature, and may suggest novel therapeutic strategies.
U0126 is a commonly used MEK1/2 inhibitor. Over 3,500 papers are retrievable from the NCBI Pubmed database using U0126 as a search term. The supposed specificity of U0126 for MEK1/2 is based on failure of the drug to inhibit a panel of 11 other kinases.Citation49 Other potential binding partners or cellular perturbations were not considered in this original report. Several subsequent studies have attributed other functions to U0126, including activation of AhRCitation50 and AMPK,Citation51,Citation52 inhibition of mitochondrial ATP productionCitation53 and interference with flow cytometry resulting from increased background fluorescence.Citation54 Our identification of U0126-induced fluorescence confirms the Blank et al.Citation54 findings on increased fluorescence associated with U0126 treatment of cells, and the mitochondrial ATP inhibition documented previously by Yung et al.Citation53 Furthermore, our findings demonstrated, using other MEK inhibitors and the MEK homolog STE7 knockout yeast strain, that the fluorescent signal is independent of MEK. In addition, we have shown that U0126 treatment results in release of cyanide, which likely results in a decrease in mitochondrial membrane potential. We also documented a metabolic shift toward aerobic glycolysis following U0126 treatment for 12–24 h. The excitation and emission peaks of the U0126-induced fluorescence correspond with the properties of a conjugated Schiff base,Citation43 but the molecular identity of the fluorescent species could not be ascertained. We suggest that the fluorescence signal is a direct product of chemical modification of U0126 or its reaction with other compounds in the oxidative environment of the mitochondria, but further work needs to be done to validate this hypothesis.
The finding that U0126 is a mitochondrial inhibitor may be relevant to published studies that have employed this compound. For example, mitochondrial inhibition may be responsible for the decrease in infarct size attributed to MEK in brain ischemia reperfusion experiments performed with U0126.Citation55–Citation57 Other brain ischemia reperfusion experiments with the mitochondrial uncoupler 2,4-dinitrophenol have resulted in similar infarct reductions.Citation58 As a multifunctional drug, U0126 may be a useful therapeutic for targeting cancer cells like other “dirty drugs” documented in the literature such as the Abl inhibitor Imatinib (Gleevec).Citation59 MEK inhibition is clinically relevant in multiple tumor systems where MEK is hyperactivatedCitation60 or overexpressed, such as in prostate cancer.Citation61 As a therapeutic strategy in cancer, our results support pairing U0126 with a glycolysis inhibitor such as 2-deoxyglucose to target cells with high metabolic rates. In this scenario, U0126 could target both survival stimuli from oncogenic pathways, such as ERBB/HER receptor tyrosine kinases, and metabolic shifts characteristic of cancer cells.
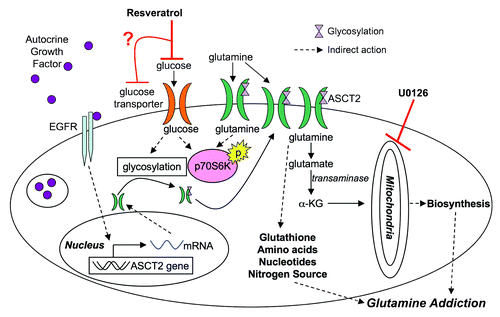
Based on our results, we propose that resveratrol is selectively toxic to cells with increased glutaminolysis. Building from what is known about this process, we postulate that glutamine metabolism may be inhibited through the scheme shown in . Resveratrol has been shown to inhibit glucose uptakeCitation62,Citation63 leading to loss of p70S6K phosphorylation.Citation64,Citation65 As glucose levels in the cell decline, glycosylation is inhibited and plasma membrane levels of growth factor receptors and transporters are reduced,Citation66 including the glutamine importer ASCT2. Glutamine importers are upregulated in a number of cancer cells linesCitation67,Citation68 possibly through autocrine growth factor signaling,Citation69 leading to a glutamine “addiction” phenotype.Citation11 Loss of glutamine influx may inhibit glutathione, nucleotide, and amino acid synthesis resulting in the induction of autophagy or apoptosis.Citation70 Normal cells may be resistant to the effects of resveratrol because the compound activates stress response networks that efficiently downregulate metabolic pathways. These networks may be lost or disabled with malignant transformation.
Figure 6. Model for resveratrol action in cancer metabolism. Based on the results presented, resveratrol may inhibit glucose uptake leading to a cascade of downstream effectors. Glutamine metabolism may be adversely targeted by downregulation of ASCT2 from the surface of the cell, depleting glutamine levels in the cell, which ultimately induce apoptosis in cancer cells (α-KG, α ketoglutarate).
Despite the ability of resveratrol to repress metabolic syndrome in mice,Citation71 it has shown minimal therapeutic efficacy in xenograft models of cancer (26, 30.). We suggest that this difference in potency in vivo may be attributable to lower glutamine concentrations in tumor cells and the tumor microenvironment compared with organs such as the liver, with intracellular glutamine concentrations between 4–8 mM.Citation72 Intratumor glutamine levels have been reported to be between 300–600 µM,Citation73-Citation75 which may be insufficient to efficiently drive resveratrol-dependent death. In addition, some tumors obtained from human subjects show a net efflux of glutamine.Citation74 Therapeutically, resveratrol may be more effective if tumor glutamine concentration and metabolic conversion could be increased. However, increasing glutamine availability in the tumor may also drive tumor cell growth and disease progression concurrently.
The proposed requirement for glutamine metabolism as a cofactor for resveratrol-induced cell death may relate to research reports in areas other than cancer, specifically to the ability of resveratrol to inhibit glutamate toxicityCitation76 and adipogenesis.Citation77 Glutamate toxicity in neurons results from elevated glutamate concentrations in the synaptic cleft and is modulated through interactions with astrocytes.Citation76 Inhibiting glucose or glutamine uptake may alter the downstream flux of glutamate within this dynamic system. Glutamine uptake is also critical in adipocytes, which need oxaloacetate replenished in the TCA cycle to drive citrate release for production of fatty acids and triglyceride storage.Citation78 Resveratrol inhibits adipogenesis in culture modelsCitation77 and modulation of glucose/glutamine metabolism could mechanistically account for this phenomenon.
In summary, this study found that the selective MEK inhibitor U0126 is a MEK-independent mitochondrial inhibitor. We employed this new information to explore several features of cancer cell metabolism. Our findings suggest one clinical strategy where U0126, or structurally similar analogs, would be used to selectively upregulate aerobic glycolysis in cancer cells under conditions where lactic acid dehydrogenase is simultaneously inhibited, thereby starving cancer cells of energy and inducing tumor regression. In this scenario, suppression of the prosurvival kinase MEK, an important oncogenic signaling node in many cancers, would occur in concert with the metabolic perturbation. The glutamine-dependence for resveratrol-induced apoptosis we have described is consistent with the glutaminolytic phenotype seen in advanced cancer and warrants further study of resveratrol to ascertain whether this finding has clinical implications.
Materials and Methods
Reagents
Cell lines (LNCaP, HEK293T, HELA, DU145, COS) were acquired from American Type Culture Collection (ATCC), with the exception of the C4–2 cells, which were a gift from Dan Gioeli (University of Virginia). BY4741 yeast and Protein BCA kit were obtained from Thermo Fisher Scientific. Resveratrol, PD325901, PD184161, U0126, JC-1 Mitochondrial Membrane Potential Assay Kit, and the LDH cytotoxicitiy assay kit, were obtained from Cayman Chemicals. U0124 and U0125 were obtained from EMD Chemicals. SL327 was obtained from Tocris Bioscience. Mitotracker CMXRos, H2DCFDA, 200 mM L-glutamine, RPMI 1640, DMEM low glucose, and McCoy’s medium were obtained from Invtirogen. Recombinant human EGF was obtained from R&D Systems. Vectashield mounting medium for fluorescence with DAPI was obtained from Vector Laboratories. Sodium oxamate, FCCCP, Antimycin A, 2-deoxy-D-glucose, O-(carboxymethyl) hydroxylamine hemihydrochloride (AOA), L-cycloserine, 6-diazo-5-oxo-L-norleucine (DON), dimethyl 2-oxoglutarate, crystal violet, and trypan blue were obtained from Sigma. Oligomycin was obtained from Cell Signaling Technologies. CellTiter Glo Luminescent Cell Viability Assay was obtained from Promega. Anitbodies against cleaved PARP (9541), Lamin A/C (2032), p44/42 MAPK (ERK1/2) (4695), and phospho-p44/42 MAPK (ERK1/2) (9101), total EGFR (2232), ATM/ATR substrate motif (2851), ASCT2(V051) (5345), phos (T68)-Chk2 (2661), phos(T389) p70S6K (9205), phos(T172)-AMPK(2535), CHOP (2895), phos(S209) eiF4e (9741), anti-mouse IgG, HRP-linked (7076) and anti-rabbit IgG, HRP-linked (7074) were obtained from Cell Signaling Technologies. Antibody against α-tubulin (TU-02, sc-8035) was obtained from Santa Cruz Biotechnology. Antibodies against β-actin (AC-15) and MnSOD (2A1) were obtained from Abcam.
Cell culture
LNCaP, and C4–2 cells were grown in RPMI 160 medium with 2 mM L-glutamine 10% heat inactivated fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin. DU145, HeLa, COS, HEK293T cells were grown in low glucose DMEM with 10% heat inactivated fetal bovine serum and 100 units/mL penicillin and 100 µg/mL streptomycin. T24 cells were grown in McCoy’s 5A medium with 10% heat inactivated fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin. All cells were maintained in a 37°C humidified incubator with 5% CO2. For experiments, cells were plated at 2.5 × 104 cells/cm2 and allowed to attach to plate for 48 h in full growth media. Cells were then placed in serum-free medium for 12 h prior to drug exposure for an additional 9–60 h.
Protein gel blotting
Cells were lysed in lysis buffer (50 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 30 mM sodium fluoride, 50 mM Tris, pH 7.5, 1% Triton X-100, 150 mM NaCl, 1 mM benzamidine, 2 mM EGTA, 100 mM sodium orthovanadate with Complete Mini protease inhibitors from Roche). Samples were spun to pellet insoluble components and protein was quantified with Protein BCA kit from Thermo. Cleared lysates were run on ReadyGel format from Biorad. Proteins were transferred to PVDF membranes and block in 5% milk or 5% BSA in accordance with manufacturers recommendations. Primary antibodies were diluted in blocking solution at 1:2000 with the exception of α-tubulin (1:10,000) and β-actin (1:20,000) and incubated at 4°C overnight. Secondary antibodies were diluted at 1:2,500 in PBS-0.1% Tween and incubated for 1 h at room temperature. Blots were visualized on film with Western Lightning Chemiluminescence Reagent (Perkin Elmer).
Nucleosome ELISA
Nucleosome Cell Death ELISA kit was purchased from Roche Applied Sciences and used according to the instructions. Cells were lysed after 12 h of resveratrol treatment with incubation buffer supplied in the kit. Lysed cells were pelleted with a 12,000 rpm centrifugation for 20 min. Supernatant was diluted 1:10 in incubation buffer and 100 μl of dilutes samples was incubated in ELISA wells. ELISA was quantitated with a FLUOstar Omega plate reader by absorbance at 415 nm.
Cell counting
Media was removed and reserved. Trypsin was added to the plate and once cells rounded, trypsin was neutralized with serum containing media and added to the reversed media. Cells were pelleted for 5 min at 1,000 rpm and resuspended in PBS. Cells were then diluted 1:2 in trypan blue and counted with a hemocytometer.
Measurement of lactic acid in medium
Cells were incubated under desired conditions for 24 h. Media was collected from the plates and spun at 12,000 g to pellet any floating cells or debris. Media was diluted 1:4 for use in the assay. Assay contains 100 µL of cleared/diluted media in 280 mM hydrazine, 467 mM glycine, 2.6 mM EDTA, 2.5 mM β-nicotinamide adenine dinucleotide and 250 units of L-lactic dehydrogenase. Absorbance was monitored at 340 nm continuously in a FLUOstar Omega plate reader every 30 sec for 1 h. Steady-state absorbance values for each condition were normalized to cell protein content.
Immunofluorescence imaging
For U0126 fluorescence analysis, cells were plated on glass coverslips at a density of 1 × 104 cells/cm2. For mitochondrial visualization, MitoTracker CMXRos was added to the medium at 100nM for 15 min. At the indicated time cells were fixed in 3% paraformadehyde for 5 min, rinsed in PBS and mounted on microscope slides with Vectorshield mounting media with DAPI. Slides were analyzed on a Zeiss Microscope.
Cell fractionation
A cell fractionation kit was acquired from MitoSciences and used as per company instructions. C4–2 cells were serum starved and then incubated with 10 μM U0126 for 12 h.
Fluorophore purification and characterization
Cells were scrapped off plates and lysed in phosphate buffered saline with 1% NP-40 with protease inhibitors for 20 min on ice. Fluorophore intensity was measured on a FLUOstar Omega plate reader with an excitation filter of 355 nm and an emission filter of 460 nm. For fluorophore characteristics, cells were lysed in PBS with 1% NP-40. Cleared supernatant was heated to 95°C for 1 min and then put on ice to precipitate proteins. Proteins were removed by 5 min centrifugation at 12,000 g. Supernatant was then combined with a 1/10 volume of saturated ammonium acetate solution to precipitate the fluorophore into a hydrophobic drop on the surface. The hydrophobic precipitate was captured and used for excitation/emission characterization on a Spectramax M2E Molecular Devices.
Yeast cultures and fluorescence measurements
Yeast were grown in standard medium containing: 1% yeast extract, 2% peptone and 2% dextrose or on plates containing: 1% yeast extract, 2% peptone, 2% dextrose and 2% agar. Yeast were streaked onto plates and allowed to grow into colonies for 48 h. Liquid cultures were inoculated with single colonies and grown at 30°C with 300 rpm for 24 h. For fluorescence generation experiments, yeast were grown 1 mL cultures in the presence of 0.001% DMSO or 10 μM U0126 for 24 h. Cultures were collected and cell concentration was measured by absorbance at 600 nm. Individual culture volumes were adjusted to standardize total cells assayed across all conditions. Cells were then pelleted and lysed in 500 μL 1% NP40 in PBS with mechanical disruption from glass beads (Sigma, G8772) by vortexing for three 1 min intervals. Cellular debris was pelleted by centrifugation at 12,000 g for 5 min and supernatant was assayed on a FLUOstar Omega plate reader with excitation and emission filters of 355/460 nm.
STE7 knockout (STE7 KO) BY4741 yeast strain was verified by polymerase chain reaction (PCR) with primers designed in the upstream and downstream sequence surrounding the STE7 wild type (WT) gene, and within the STE7 WT gene or the KanMX cassette. Primers were designed by Samuel Hasson (NIH) with Clone Manager Suite 7 and include: KanMXForward: ATGCGCCAGAGTTGTTTC; KanMXReverse: AACTCACCGAGGCAGTTC; UpstreamForward: GCCCATCTGATGGATTAG; DownstreamReverse: GGTGAAACACAGGCTAAC; STE7Forward: TGGCGTACTAAATGGCTTGG; and STE7Reverse: TGCCTTCACCACAGTTCC. Yeast DNA template for PCR was prepared from individual yeast colonies of WT or STE7 KO. Colonies were collected from YPD agar plates, resuspended in 10 μL of 0.02 M sodium hydroxide, and heated to 95°C for 10 min. PCR was performed with Platinum PCR SuperMix High Fidelity (Invitrogen), 3 μL of yeast DNA template, and primers at a final concentration of 200 nM. Amplified DNA from PCR reactions was electrophoresed on 0.7% agarous gels and visualized with SYBR Gold Nuclei Acid Gel Stain (Invitrogen) under UV light.
Mitochondrial potential with JC1
At the 12 h after drug exposure, JC1 dye was added to culture media and incubated for 15 min in a 37°C humidified incubator. Media and dye were removed and cells were washed three times with JC-1 wash buffer. Fluorescence was measured with the 485/520 and 544/590 filter sets on an FLUOstar Omega plate reader. Control wells treated with drugs, but not JC1 were run in parallel and corresponding fluorescent readings were subtracted from stained wells. Mitochondrial potential was calculated as the ratio between (544/590–background) / (485/520–background).
Measurement of ATP levels
Cells were plated in white, opaque 96-well plates and treated with drugs for 9 h. Cell TiterGlo reagent was added directly to the wells and incubated for 2 min on an orbital rotator plate. Luminescence was measured over 10 min on an FLUOstar Omega plate reader. Steady-state values were compared after normalization with protein concentrations from adjacent wells under the same drug treatment conditions.
Lactic acid dehydrogenase (LDH) activity assay
Following the directions on the Cayman LDH cytotoxicity assay kit. Briefly, cells were plated and treated with sodium oxamate for 12 h at variable concentrations. Cells from 6-well plates were lysed in 200 uL in PBS containing 0.1% Triton X-100 and diluted 1:10 in PBS with appropriate sodium oxamate concentration to match culture conditions. Resulting cleared supernatant was combined with LDH reaction buffer provided in the kit and absorbance at 490 nm was monitored every 20 sec for 20 min on a FLUOstar Omega plate reader. LDH activity was calculated from the slope of the early linear kinetic phase and normalized to the vehicle treated control sample.
Crystal violet staining
Media was removed from the cells and stained with 0.5% crystal violet in 25% methanol for 30 min. Cells were washed extensively with water and plates were allowed to dry. Dye was solubilized from dried plates with 10% acetic acid for 20 min and absorbance was quantified at 595 nm on a FLUOstar Omega plate reader.
In vitro fluorophore generation
To generate fluorescence from U0126 in an Eppendorf tube, the following components were mixed and allowed in incubate at room temperature for 1 h. 50 µL of PBS, 1 µL of 10 mM U0126 dissolved in DMSO or 2 µL of 100U/µL superoxide dismutase (Sigma, S-2515) or 1 uL of 10 mM EUK134 and 2 μL of hydrogen peroxide, 30% by weight (Sigma, 216763). All permutations of reactions were run in parallel and after 1 h incubation samples were read on a fluorimeter with excitation of 320 nm and emission 510 nm.
Measurement of cyanide
Adapted from reference Citation2. Briefly, C4–2 cells were plated, serum starved and treated with 10, 20 or 40 µM U0126 for 24 h. Cells were scrapped off plates and pelleted. Cells were lysed with 120 µL of 1% NP-40 in PBS for 30 min and centrifuged to remove insoluble material. An aliquot of supernatant was removed for protein quantification by BCA. All samples had protein contents within 10%. Twenty microliters of supernatant was added to the following reaction mixture: 200 µL of 0.01 M potassium phosphate, pH 7.4, 200 µL pyridoxal phosphate (0.5 mg/mL) and 20 µL water. The reaction was incubated in a heat block at 80°C for 45 min and then allowed to cool to room temperature. Reactions were acidified with 200 µL of 10% trichloroacetic acid and centrifuged at 1,500 g for 5 min to pellet insoluble debris. Fifty-seven microliters of the acidified reaction was mixed with 143 µL 2M potassium acetate pH 3.8. Fluorescence was measured from the potassium acetate mixture by exciting at 320 nm and recording emission at 427 nm. Each set of samples was run with a standard curve of sodium cyanide samples and the standard curve was used to convert experimental sample fluorescence to cyanide concentration. Each condition was run in triplicate.
Acknowledgments
This study was supported by NIH R01CA143777, R01DK57691 and W81XWH-08-1-0150 from the US Army (M.R.F), NCI R00CA131472-04 (D.D.V), and New York Academy of Medicine; and Children’s Hospital Boston Faculty Development (J.K.); J.K. is an Eleanor and Miles Shore Scholar of Harvard Medical School.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104:19345 - 50; http://dx.doi.org/10.1073/pnas.0709747104; PMID: 18032601
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010; 329:1492 - 9; http://dx.doi.org/10.1126/science.1188015; PMID: 20847263
- Chhipa RR, Wu Y, Mohler JL, Ip C. Survival advantage of AMPK activation to androgen-independent prostate cancer cells during energy stress. Cell Signal 2010; 22:1554 - 61; http://dx.doi.org/10.1016/j.cellsig.2010.05.024; PMID: 20570728
- Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol 2011; 223:283 - 94; http://dx.doi.org/10.1002/path.2809; PMID: 21125681
- Cao Y, Ma J. Body-mass index, prostate cancer-specific mortality and biochemical recurrence:A systematic review and meta-analysis. Cancer Prev Res 2011; 4:486 - 501
- Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009; 52:1766 - 77; http://dx.doi.org/10.1007/s00125-009-1440-6; PMID: 19572116
- Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, Melani C, et al. Metformin and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care 2011; 34:129 - 31; http://dx.doi.org/10.2337/dc10-1287; PMID: 20980415
- Jacobs EJ, Newton CC, Thun MJ, Gapstur SM. Long-term use of cholesterol-lowering drugs and cancer incidence in a large united states cohort. Cancer Res 2011; 71:1763 - 71
- Breau RH, Karnes RJ, Jacobson DJ, McGree ME, Jacobsen SJ, Nehra A, et al. The association between statin use and the diagnosis of prostate cancer in a population based cohort. J Urol 2010; 184:494 - 9; http://dx.doi.org/10.1016/j.juro.2010.03.149; PMID: 20620405
- Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005; 24:6465 - 81; PMID: 16007182
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 2008; 105:18782 - 7; http://dx.doi.org/10.1073/pnas.0810199105; PMID: 19033189
- Palmada M, Speil A, Jeyaraj S, Bohmer C, Lang F. The serine/threonine kinases SGK1, 3 and PKB stimulate the amino acid transporter ASCT2. Biochem Biophys Res Commun 2005; 331:272 - 7; http://dx.doi.org/10.1016/j.bbrc.2005.03.159; PMID: 15845389
- Heemers H, Vanderhoydonc F, Roskams T, Shechter I, Heyns W, Verhoeven G, et al. Androgens stimulate coordinated lipogenic gene expression in normal target tissues in vivo. Mol Cell Endocrinol 2003; 205:21 - 31; http://dx.doi.org/10.1016/S0303-7207(03)00205-3; PMID: 12890564
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 2007; 18:1437 - 46; http://dx.doi.org/10.1091/mbc.E06-07-0593; PMID: 17301289
- Shanware NP, Mullen AR, Deberardinis RJ, Abraham RT. Glutamine: Pleiotropic roles in tumor growth and stress resistance. J Mol Med 2011; 89:229 - 36; http://dx.doi.org/10.1007/s00109-011-0731-9; PMID: 21301794
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762 - 5; http://dx.doi.org/10.1038/nature07823; PMID: 19219026
- Molavian HR, Kohandel M, Milosevic M, Sivaloganathan S. Fingerprint of cell metabolism in the experimentally observed interstitial pH and pO2 in solid tumors. Cancer Res 2009; 69:9141 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-09-2112; PMID: 19920192
- Qin JZ, Xin H, Nickoloff BJ. 2-deoxyglucose sensitizes melanoma cells to TRAIL-induced apoptosis which is reduced by mannose. Biochem Biophys Res Commun 2010; 401:293 - 9; http://dx.doi.org/10.1016/j.bbrc.2010.09.054; PMID: 20851102
- Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 2010; 70:1388 - 94; http://dx.doi.org/10.1002/pros.21172; PMID: 20687211
- Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int J Cancer 2010; 127:2478 - 85; http://dx.doi.org/10.1002/ijc.25431; PMID: 20473919
- Lynch G, Kemeny N, Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol 1982; 5:541 - 3; PMID: 7180833
- Ratan HL, Steward WP, Gescher AJ, Mellon JK. Resveratrol–a prostate cancer chemopreventive agent?. Urol Oncol 2002; 7:223 - 7; http://dx.doi.org/10.1016/S1078-1439(02)00194-1; PMID: 12504842
- Shih A, Zhang S, Cao HJ, Boswell S, Wu YH, Tang HY, et al. Inhibitory effect of epidermal growth factor on resveratrol-induced apoptosis in prostate cancer cells is mediated by protein kinase C-alpha. Mol Cancer Ther 2004; 3:1355 - 64; PMID: 15542774
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997; 275:218 - 20; http://dx.doi.org/10.1126/science.275.5297.218; PMID: 8985016
- Subbaramaiah K, Chung WJ, Michaluart P, Telang N, Tanabe T, Inoue H, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem 1998; 273:21875 - 82; http://dx.doi.org/10.1074/jbc.273.34.21875; PMID: 9705326
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006; 127:1109 - 22; http://dx.doi.org/10.1016/j.cell.2006.11.013; PMID: 17112576
- Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010; 59:554 - 63; http://dx.doi.org/10.2337/db09-0482; PMID: 19934007
- Gatz SA, Keimling M, Baumann C, Dork T, Debatin KM, Fulda S, et al. Resveratrol modulates DNA double-strand break repair pathways in an ATM/ATR-p53- and -Nbs1-dependent manner. Carcinogenesis 2008; 29:519 - 27; http://dx.doi.org/10.1093/carcin/bgm283; PMID: 18174244
- Ahmad N, Adhami VM, Afaq F, Feyes DK, Mukhtar H. Resveratrol causes WAF-1/p21-mediated G(1)-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin Cancer Res 2001; 7:1466 - 73; PMID: 11350919
- Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 2008; 295:G833 - 42; http://dx.doi.org/10.1152/ajpgi.90358.2008; PMID: 18755807
- Wang TT, Hudson TS, Wang TC, Remsberg CM, Davies NM, Takahashi Y, et al. Differential effects of resveratrol on androgen-responsive LNCaP human prostate cancer cells in vitro and in vivo. Carcinogenesis 2008; 29:2001 - 10; http://dx.doi.org/10.1093/carcin/bgn131; PMID: 18586690
- Niles RM, Cook CP, Meadows GG, Fu YM, McLaughlin JL, Rankin GO. Resveratrol is rapidly metabolized in athymic (nu/nu) mice and does not inhibit human melanoma xenograft tumor growth. J Nutr 2006; 136:2542 - 6; PMID: 16988123
- Sakamoto T, Horiguchi H, Oguma E, Kayama F. Effects of diverse dietary phytoestrogens on cell growth, cell cycle and apoptosis in estrogen-receptor-positive breast cancer cells. J Nutr Biochem 2010; 21:856 - 64; http://dx.doi.org/10.1016/j.jnutbio.2009.06.010; PMID: 19800779
- Opipari AW Jr., Tan L, Boitano AE, Sorenson DR, Aurora A, Liu JR. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res 2004; 64:696 - 703; http://dx.doi.org/10.1158/0008-5472.CAN-03-2404; PMID: 14744787
- Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem 2008; 56:4813 - 8; http://dx.doi.org/10.1021/jf800175a; PMID: 18522405
- Kim YA, Lee WH, Choi TH, Rhee SH, Park KY, Choi YH. Involvement of p21WAF1/CIP1, pRB, bax and NF-kappaB in induction of growth arrest and apoptosis by resveratrol in human lung carcinoma A549 cells. Int J Oncol 2003; 23:1143 - 9; PMID: 12963997
- Michels G, Watjen W, Weber N, Niering P, Chovolou Y, Kampkotter A, et al. Resveratrol induces apoptotic cell death in rat H4IIE hepatoma cells but necrosis in C6 glioma cells. Toxicology 2006; 225:173 - 82; http://dx.doi.org/10.1016/j.tox.2006.05.014; PMID: 16843582
- Simopoulos AP. The mediterranean diets: What is so special about the diet of greece? the scientific evidence. J Nutr 2001; 131:Suppl 3065S - 73S; PMID: 11694649
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int J Cancer 1994; 57:406 - 12; http://dx.doi.org/10.1002/ijc.2910570319; PMID: 8169003
- Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 1994; 54:2577 - 81; PMID: 8168083
- Kim AL, Zhu Y, Zhu H, Han L, Kopelovich L, Bickers DR, et al. Resveratrol inhibits proliferation of human epidermoid carcinoma A431 cells by modulating MEK1 and AP-1 signalling pathways. Exp Dermatol 2006; 15:538 - 46; http://dx.doi.org/10.1111/j.1600-0625.2006.00445.x; PMID: 16761963
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest 2008; 118:3051 - 64; PMID: 18725989
- Trombly R, Tappel A. Fractionation and analysis of fluorescent products of lipid peroxidation. Lipids 1975; 10:441 - 7; http://dx.doi.org/10.1007/BF02532426; PMID: 1160518
- Arun P, Moffett JR, Ives JA, Todorov TI, Centeno JA, Namboodiri MA, et al. Rapid sodium cyanide depletion in cell culture media: Outgassing of hydrogen cyanide at physiological pH. Anal Biochem 2005; 339:282 - 9; http://dx.doi.org/10.1016/j.ab.2005.01.015; PMID: 15797569
- Griffiths M, Keast D, Patrick G, Crawford M, Palmer TN. The role of glutamine and glucose analogues in metabolic inhibition of human myeloid leukaemia in vitro. Int J Biochem 1993; 25:1749 - 55; http://dx.doi.org/10.1016/0020-711X(88)90303-5; PMID: 8138012
- Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 2009; 48:2661 - 74; http://dx.doi.org/10.1021/bi801898e; PMID: 19161339
- Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett 2008; 18:6501 - 4; http://dx.doi.org/10.1016/j.bmcl.2008.10.054; PMID: 18952427
- Wong DT, Fuller RW, Molloy BB. Inhibition of amino acid transaminases by L-cycloserine. Adv Enzyme Regul 1973; 11:139 - 54; http://dx.doi.org/10.1016/0065-2571(73)90013-7; PMID: 4150977
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 1998; 273:18623 - 32; http://dx.doi.org/10.1074/jbc.273.29.18623; PMID: 9660836
- Bachleda P, Dvorak Z. Pharmacological inhibitors of JNK and ERK kinases SP600125 and U0126 are not appropriate tools for studies of drug metabolism because they activate aryl hydrocarbon receptor. Gen Physiol Biophys 2008; 27:143 - 5; PMID: 18645229
- Dokladda K, Green KA, Pan DA, Hardie DG. PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP:ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett 2005; 579:236 - 40; http://dx.doi.org/10.1016/j.febslet.2004.11.084; PMID: 15620719
- LaRosa C, Downs SM. MEK inhibitors block AICAR-induced maturation in mouse oocytes by a MAPK-independent mechanism. Mol Reprod Dev 2005; 70:235 - 45; http://dx.doi.org/10.1002/mrd.20200; PMID: 15570612
- Yung HW, Wyttenbach A, Tolkovsky AM. Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem Pharmacol 2004; 68:351 - 60; http://dx.doi.org/10.1016/j.bcp.2004.03.030; PMID: 15194007
- Blank N, Burger R, Duerr B, Bakker F, Wohlfarth A, Dumitriu I, et al. MEK inhibitor U0126 interferes with immunofluorescence analysis of apoptotic cell death. Cytometry 2002; 48:179 - 84; http://dx.doi.org/10.1002/cyto.10127; PMID: 12210141
- Wang ZQ, Chen XC, Yang GY, Zhou LF. U0126 prevents ERK pathway phosphorylation and interleukin-1beta mRNA production after cerebral ischemia. Chin Med Sci J 2004; 19:270 - 5; PMID: 15669185
- Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation 2010; 7:14; http://dx.doi.org/10.1186/1742-2094-7-14; PMID: 20187933
- Maddahi A, Edvinsson L. Enhanced expressions of microvascular smooth muscle receptors after focal cerebral ischemia occur via the MAPK MEK/ERK pathway. BMC Neurosci 2008; 9:85; http://dx.doi.org/10.1186/1471-2202-9-85; PMID: 18793415
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem 2005; 94:1676 - 84; http://dx.doi.org/10.1111/j.1471-4159.2005.03328.x; PMID: 16045446
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 2007; 25:1035 - 44; http://dx.doi.org/10.1038/nbt1328; PMID: 17721511
- Schubbert S, Shannon K, Bollag G. Hyperactive ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295 - 308; http://dx.doi.org/10.1038/nrc2109; PMID: 17384584
- Gioeli D, Mandell JW, Petroni GR, Frierson HF Jr., Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res 1999; 59:279 - 84; PMID: 9927031
- Kueck A, Opipari AW Jr., Griffith KA, Tan L, Choi M, Huang J, et al. Resveratrol inhibits glucose metabolism in human ovarian cancer cells. Gynecol Oncol 2007; 107:450 - 7; http://dx.doi.org/10.1016/j.ygyno.2007.07.065; PMID: 17825886
- Park JB. Inhibition of glucose and dehydroascorbic acid uptakes by resveratrol in human transformed myelocytic cells. J Nat Prod 2001; 64:381 - 4; http://dx.doi.org/10.1021/np000411t; PMID: 11277764
- Faber AC, Dufort FJ, Blair D, Wagner D, Roberts MF, Chiles TC. Inhibition of phosphatidylinositol 3-kinase-mediated glucose metabolism coincides with resveratrol-induced cell cycle arrest in human diffuse large B-cell lymphomas. Biochem Pharmacol 2006; 72:1246 - 56; http://dx.doi.org/10.1016/j.bcp.2006.08.009; PMID: 16979140
- Haider UG, Sorescu D, Griendling KK, Vollmar AM, Dirsch VM. Resveratrol suppresses angiotensin II-induced Akt/protein kinase B and p70 S6 kinase phosphorylation and subsequent hypertrophy in rat aortic smooth muscle cells. Mol Pharmacol 2002; 62:772 - 7; http://dx.doi.org/10.1124/mol.62.4.772; PMID: 12237323
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 2010; 24:2784 - 99; http://dx.doi.org/10.1101/gad.1985910; PMID: 21106670
- Sidoryk M, Matyja E, Dybel A, Zielinska M, Bogucki J, Jaskolski DJ, et al. Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. Neuroreport 2004; 15:575 - 8; http://dx.doi.org/10.1097/00001756-200403220-00001; PMID: 15094455
- Witte D, Ali N, Carlson N, Younes M. Overexpression of the neutral amino acid transporter ASCT2 in human colorectal adenocarcinoma. Anticancer Res 2002; 22:2555 - 7; PMID: 12529963
- Avissar NE, Sax HC, Toia L. In human entrocytes, GLN transport and ASCT2 surface expression induced by short-term EGF are MAPK, PI3K, and rho-dependent. Dig Dis Sci 2008; 53:2113 - 25; http://dx.doi.org/10.1007/s10620-007-0120-y; PMID: 18157695
- Hwang SO, Lee GM. Nutrient deprivation induces autophagy as well as apoptosis in chinese hamster ovary cell culture. Biotechnol Bioeng 2008; 99:678 - 85; http://dx.doi.org/10.1002/bit.21589; PMID: 17680685
- Lee MH, Choi BY, Kundu JK, Shin YK, Na HK, Surh YJ. Resveratrol suppresses growth of human ovarian cancer cells in culture and in a murine xenograft model: Eukaryotic elongation factor 1A2 as a potential target. Cancer Res 2009; 69:7449 - 58; http://dx.doi.org/10.1158/0008-5472.CAN-09-1266; PMID: 19738051
- Bode B, Tamarappoo BK, Mailliard M, Kilberg MS. Characteristics and regulation of hepatic glutamine transport. JPEN J Parenter Enteral Nutr 1990; 14:Suppl 51S - 5S; http://dx.doi.org/10.1177/014860719001400404; PMID: 2402055
- Wasa M, Bode BP, Abcouwer SF, Collins CL, Tanabe KK, Souba WW. Glutamine as a regulator of DNA and protein biosynthesis in human solid tumor cell lines. Ann Surg 1996; 224:189 - 97; http://dx.doi.org/10.1097/00000658-199608000-00012; PMID: 8757383
- Kallinowski F, Runkel S, Fortmeyer HP, Forster H, Vaupel P. L-glutamine: A major substrate for tumor cells in vivo?. J Cancer Res Clin Oncol 1987; 113:209 - 15; http://dx.doi.org/10.1007/BF00396375; PMID: 3584211
- Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: Comparison with normal adult brain. Brain Res 1976; 117:105 - 13; http://dx.doi.org/10.1016/0006-8993(76)90559-X; PMID: 186155
- de Almeida LM, Pineiro CC, Leite MC, Brolese G, Tramontina F, Feoli AM, et al. Resveratrol increases glutamate uptake, glutathione content, and S100B secretion in cortical astrocyte cultures. Cell Mol Neurobiol 2007; 27:661 - 8; http://dx.doi.org/10.1007/s10571-007-9152-2; PMID: 17554623
- Rayalam S, Yang JY, Ambati S, Della-Fera MA, Baile CA. Resveratrol induces apoptosis and inhibits adipogenesis in 3T3-L1 adipocytes. Phytother Res 2008; 22:1367 - 71; http://dx.doi.org/10.1002/ptr.2503; PMID: 18688788
- Ballard FJ, Hanson RW. The citrate cleavage pathway and lipogenesis in rat adipose tissue: Replenishment of oxaloacetate. J Lipid Res 1967; 8:73 - 9; PMID: 14564711