Abstract
HDAC inhibitors (HDACi) suppress the growth of tumor cells due to induction of cell cycle arrest, senescence or apoptosis. Recent data demonstrate that HDACi can interfere with DNA Damage Response (DDR) thereby sensitizing the cells to DNA damaging agents. Here, we show that HDACi sodium butyrate (NaBut) potentiates the formation of γH2AX foci predominantly in S-phase E1A+Ras cells. Accumulation of γH2AX foci sensitizes the cells toward such DNA damaging agents as irradiation (IR) and adriamycin. In fact, NaBut potentiates the persistence of γH2AX foci induced by genotoxic agents. The synergizing effects depend on DNA damaging factors and on the order of NaBut treatment. Indeed, NaBut treatment for 24 h leads to an accumulation of G1-phase cells and a lack of S-phase cells, therefore, adriamycin, a powerful S-phase-specific inhibitor, when added to NaBut-treated cells, is unable to substantially add γH2AX foci. In contrast, IR produces both single- and double-strand DNA breaks at any stage of the cell cycle and was shown to increase γH2AX foci in NaBut-treated cells. Further, a lifetime of IR-induced γH2AX foci depends on the subsequent presence of HDACi. Correspondingly, NaBut withdrawal leads to the extinction of IR-induced γH2AX foci. This necessitates HDACi to hold the IR-induced γH2AX foci unrepaired. However, the IR-induced γH2AX foci persist after long-term NaBut treatment (72 h) even after washing the drug. Thus, although signaling pathways regulating H2AX phosphorylation in NaBut-treated cells remain to be investigated, the obtained results show that NaBut potentiates effects of DNA damaging agents by facilitating formation and persistence of γH2AX foci.
Introduction
Post-translational modifications of histones and non-histone proteins play an important role in the regulation of various cellular processes, such as cell proliferation and differentiation as well as cell transformation.Citation1,Citation2 Reversible DNA methylation/demethylation and histone acetylation/deacetylation are the main epigenetic events regulating gene expression.Citation3 Nucleosomes in transcriptionally active chromatin regions have acetylated histones, whereas histones are underacetylated in repressed genes.Citation4 It has been shown that transformed cells have a number of tumor-suppressor genes in a repressed state, whereas many proliferation-related genes are overexpressed. Consistently, DNA chip-hybridization data showed that the expression profile of 4–10% of genes is changed in HDAC inhibitor-treated tumor cells.Citation5 Therefore, histone deacetylases are considered as promising molecular targets for tumor therapy. By changing the expression of different sets of genes, HDAC inhibitors are capable of suppressing cell growth and inducing cell differentiation, senescence or apoptosis.Citation5–Citation6 There is data that indicates that HDACi-induced modulation of chromatin due to acetylation of histones and non-histone proteins can affect the pathways involved in DNA repair.Citation8 In addition to cell growth arrest, HDAC inhibitors augment the effects of cytotoxic agents traditionally used for tumor therapy.Citation9,Citation10 This sensitizing effect is likely due to the inhibition of the various components involved in DNA repair.Citation8,Citation9,Citation11 Due to their ability to augment the effect of conventional antitumor therapy, HDAC inhibitors are considered a new class of promising drugs.
E1A is capable of recruiting histone deacetylase activity for the promoters of target genes thereby modulating their expression and promoting tumor growth. Various HDAC inhibitors, including sodium butyrate (NaBut) and Trichostatin A (TSA), cause growth suppression of E1A+Ras transformed cells and induce cellular senescence.Citation12,Citation13 Senescent E1A+Ras cells display activation of DNA damage response signaling (DDR) as evidenced by the accumulation of γH2AX foci.Citation13 In this work we used cytometric bivariate distribution (DNA content vs γH2AX) to see the time-course of γH2AX foci appearance, preferable cell cycle phases for their induction and a lifetime of NaBut-induced γH2AX foci. We studied NaBut-induced sensitization of E1A+Ras transformed cells to irradiation and adriamycin treatment. It has been shown that the prior treatment of cells with NaBut enforced irradiation-induced accumulation of γH2AX foci. Adriamycin treatment had a lesser effect due to the lack of S-phase cells in NaBut-treated cells. By means of two-parametric FACS analysis, we showed that NaBut alone was able to promote the formation of γH2AX foci in S-phase E1A+Ras cells. The fate of the NaBut-induced γH2AX foci depends on the duration of treatment. In cells treated with NaBut for 24 h, γH2AX foci have been found to disappear after washing the drug. However after a long-term treatment for 72 h and longer, the foci do not disappear even in the absence of HDACi, implying an irreversible effect. Thus, HDAC inhibitors can interfere with DNA repair machinery and sensitize cells to DNA damaging agents by promoting the formation of γH2AX foci.
Results
Sodium butyrate induces formation of γH2AX foci
One of the early markers of double-strand DNA breaks (DSBs) is the formation of so-called γH2AX foci, which represent clusters of histone H2AX molecules phosphorylated on Ser139 residue.Citation16 Phosphorylation of H2AX is a necessary signal to recruit a number of proteins also involved in DNA repair, such as ATM/ATR, p53, 53BP1 and others.Citation17 The modification is performed by protein kinases of a PI3K-related PIKK family: ATM, ATR and/or DNA-PK.Citation17,Citation18 Process phosphorylation starts from the ends of DNA breaks and spreads across several thousand nucleotides. One DSB is sufficient to induce the formation of a unique γH2AX focus. However, recent data indicate that the formation of γH2AX foci takes place even in the absence of genotoxic agents, in particular, on chromatin stretches with anomalies (irregularities) caused by endogenous reasons occurring during DNA replication and recombination. Thus, γH2AX foci are observed in fast-proliferating mouse embryonic stem cells,Citation19 in case of a stable association between single repair factors chromatinCitation20 and also in response to such stress factors such as heat-shock.Citation21,Citation22 Thus, the appearance of γH2AX foci is a very sensitive tool to visualize both early events of DNA damage and various modulations of chromatin structure.Citation14 Here we used immunofluorescent γH2AX staining in combination with confocal laser microscopy and two-parametric cytometry to demonstrate NaBut-induced γH2AX foci. Also, we addressed the issue of whether the accumulated γH2AX foci do sensitize E1A+Ras transformed cells to genotoxic agents (ionizing irradiation and adriamycin). First, representative immunofluorescence images of intracellular distribution of γH2AX and pATM in E1A+Ras cells (Fig. S1) and immunobloting () are given at various time points. According to these data, NaBut induced phosphorylation of H2AX histone but the formation of γH2AX foci is not accompanied by nuclear accumulation of pATM (Fig. S1). Second, we checked which phase of cell cycle is preferable for induction of γH2AX by HDAC inhibitors (). Data presented in demonstrate that NaBut-induced γH2AX foci are already seen at 2 h and increased for 24 h of treatment. After 24 h of treatment, γH2AX staining is still increased (See quantitation data of γH2AX foci in Fig. S2 in ref. Citation13), and the clear foci concentrate in cells, which are arrested in phases G1 and G2 of the cell cycle with a relative gap in the region of S-phase cells. NaBut treatment for 48 and 72 h led to additional H2AX phosphorylation (, right panel), which was not followed by additional binding of γH2AX-specific antibody in FACS analysis (). Accumulation of γH2AX foci was not due to apoptotic cell death because NaBut treatment was shown to suppress apoptosis in E1A+Ras cells.Citation23
Figure 1. Sodium butyrate induces H2AX phosphorylation in E1A+Ras cells. (A) Western blotting of total cell proteins probed with antibodies to Ser139-H2AX and GAPDH (loading control) at various time points of NaBut treatment. (B) Analysis of SSB DNA breaks in E1A + Ras cells. E1A + Ras cells were subjected to single-cell gel electrophoresis under denaturing conditions (comet assay): (1) untreated cells, (2) NaBut-treated cells, (3) cells irradiated at dosage 6 Gy. (C and D) Bivariate distributions (DNA content vs γH2AX) of E1A + Ras cells treated with 4 mM NaBut for time intervals as indicated. Results are present as DNA content (X-axis) vs γH2AX levels (Y-axis). Fluorescence intensity was measured by flow cytometry. Based on difference in DNA content, subpopulations of cells in G1 vs S vs G2M phases of the cycle may be distinguished and gated as shown in the Ctrl sample. The mean γH2AX IF may then be calculated for each subpopulation. Cells were treated with 4 mM NaBut for 2, 4, 8 and 24 h (C) or for 24, 48 and 72 h (D).
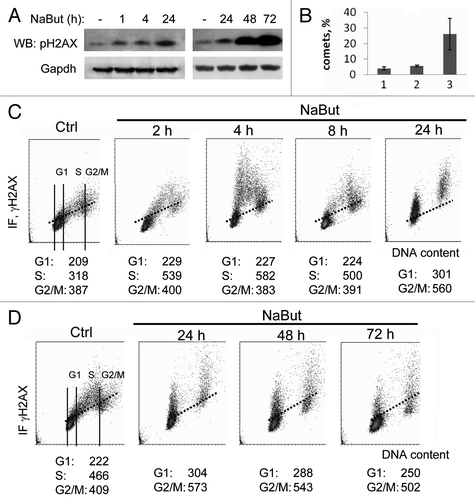
To see which phase of the cell cycle is preferable for γH2AX foci induction, we treated E1A+Ras cells with NaBut for 2, 4 and 8 h (). After short-term treatments, when there still a noticeable amount of cells in S-phase, one can see sharp clouds of γH2AX-positive cells, which then move to the G2 phase of the cell cycle. This implies that the accumulation of γH2AX foci in S-phase cells cannot induce S-phase block.
To assess whether γH2AX foci appear due to NaBut-induced single-strand DNA breaks (SSBs), we performed a single cell DNA comet assay (). According to the comet assay data, NaBut-induced accumulation of γH2AX foci is accompanied by only an insignificant increase of single-strand DNA breaks in cells treated with HDACi for 24 h. At that, there is no reliable increase of the ROS level in cells exposed to NaBut for 4 h (Fig. S2) even though γH2AX foci are accumulated (). NaBut-induced increase of the ROS levels is becoming visible only in cells treated for 24 h or 48 h. Hence it appears that the NaBut-mediated hyperacetylation of histones and the formation of chromatin stretches with altered structure stimulate H2AX histone phosphorylation. Sodium butyrate promotes γH2AX foci accumulation predominantly in S-phase cells (). The cells tolerate the accumulated γH2AX foci and progress over G2/M to G1 phase of the next cell cycle. The altered chromatin stretches still persist in these cells and serve as potential sites for DNA damaging agents such as cytostatic adriamycin and for binding IR-induced DNA repair proteins (see below). In any case, HDACi-induced γH2AX foci can be an important factor in enforcing the antitumor effect of such conventional DNA-damaging agents as irradiation and intercalating DNA cytostatics.
NaBut increases the lifetime of γH2AX foci induced by DNA damaging factors: ionizing irradiation and adriamycin
Taking into account data presented in that demonstrate the capability of HDAC inhibitors to promote the formation of γH2AX foci in E1A+Ras cells, we next studied the combined effect of NaBut with such known DNA damaging agents as ionizing irradiation and adriamycin (doxorubicin)—a powerful inhibitor of topoisomerase II activity. These two agents have an important difference: the primary effect of irradiation is the formation of single-strand DNA breaks occurring in any phase of the cell cycle, whereas adriamycin acts primarily in S-phase cells when DNA replication takes place, giving rise double-strand breaks. The kinetics of γH2AX foci formation in adriamycin-treated cells (40 min, then washing the drug) shows that maximal accumulation of γH2AX foci generally occurs 1–4 h after treatment, mostly in S-phase cells, then the amount of foci decrease, reaching the control values of 18–24 h (, Ctrl). In cells pre-treated with NaBut for 24 h, which have a very low portion of S-phase cells,Citation12 adriamycin still increases the amount of γH2AX foci in G2- and G1-arrested cells likely due to the fact that it works on “open” chromatin stretches, which are accessible by DNA damaging agents and persist after NaBut treatment (). This follows from two-parametric flow cytometry data indicating that adriamycin induces the accumulation of γH2AX in cells that are in phases G2 and G1(). The neutral comet assay presented on shows that NaBut-pretreated cells demonstrate a reduced proportion of adriamycin-induced DSBs. This also might reflect a decreased amount of target S-phase cells.
Figure 2. Induction of γH2AX foci in cells treated with adriamycin alone or in combination with NaBut. (A) Bivariate distributions (DNA content vs γH2AX) of E1A+Ras cells treated with NaBut for 24 h (NaBut) or left untreated (Ctrl) and exposed to adriamycin for 40 min (Adr) and then incubated in NaBut-free medium for 1, 4 or 18 h as indicated. (B) Immunoblotting of total proteins with antibodies to the γH2AX and GAPDH (loading control) from cells harvested 1 h or 4 h after adriamycin treatment alone or after NaBut-pretreatment for 24 h. (C) Cells were treated with adriamycin alone (light gray) or after NaBut pretreatment for 24 h (dark gray), harvested and subjected to single-cell gel electrophoresis under neutral conditions (neutral comet assay). Tail moment is defined as percentage of DNA in the tail multiplied by the distance between the means of the head and tail distributions.Citation15 Results are normalized to the tail moment in untreated cells. (D) Clonogenic survival after treatment with adriamycin alone, pretreatment with 4 mM NaBut for 24 h before adriamycin treatment, or adriamycin treatment followed by NaBut. (E) Caspase-3 activation assay. Cells were treated as indicated in previous legend. Twelve hours later, the cells were harvested, and caspase-3 activity was measured in triplicate. Results are normalized to the caspase-3 activity in untreated cells.
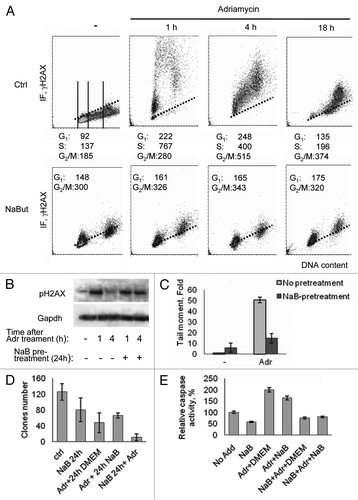
We have previously shown that HDAC inhibitors induced G1 arrest of E1A+Ras transformed cells and suppressed their proliferation.Citation12 To determine the cytotoxic effects of the combined action of the HDAC inhibitor and adriamycin, a clonogenic assay was performed to see a difference between prior- and post-NaBut treatment. E1A+Ras cells were exposed to sodium butyrate for 24 h and/or treated with adriamycin for 40 min, trypsinized into single cell suspensions, and plated into a NaBut-free medium for estimation of colony-forming efficiency as an indicator of proliferation capability. Alternatively, cells were exposed to adriamycin first and treated with sodium butyrate for 24 h. Our data () indicate that treatment of cells with adriamycin or NaBut alone decreased their clonability. However, the pretreatment of cells with NaBut markedly increased adriamycin killing efficiency. When the cells were treated with adriamycin first and then treated with NaBut, no significant difference was observed compared with adriamycin treatment alone (). However, a colorimetric caspase-3 activity assay demonstrates that sodium butyrate is able to decrease adriamycin-induced apoptosis in E1A+Ras cells only when sodium butyrate was added prior to adriamycin. Correspondingly, caspase-3 activity is markedly higher in cells treated with adriamycin and followed by NaBut, than vice versa ().
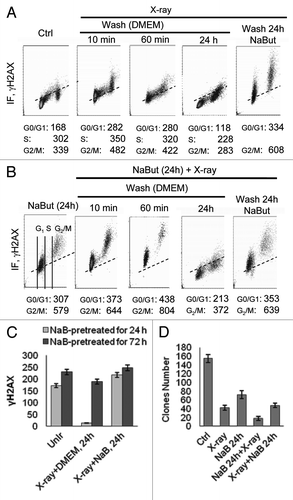
We have also checked the effect of irradiation, which induces single-strand DNA breaks independently the cell cycle phase, in cells pretreated with sodium butyrate (). In control cells, there is no preference for γH2AX accumulation in relation to the phase of the cell cycle (). In cells pre-treated with NaBut for 24 h, when S-phase cells are almost absent, irradiation induces accumulation of γH2AX in G1- and G2-arrested cells to a greater extent than adriamycin (). However, the amount of γH2AX-positive cells slows down over time, implying that repair of the IR-induced foci takes place when cells washed free of the drug (). We performed a functional clonogenic assay to check whether an order of treatment with HDACi and IR also has a functional meaning and found that maximal suppression of clone formation was observed in that case, when NaBut-arrested cells have been further irradiated (). The same combination was more effective in the case of NaBut + adriamycin treatment. Thus, there is a synergistic effect of IR and HDAC inhibitors on the accumulation of γH2AX foci in E1A+Ras transformed cells. For adriamycin, however, a synergistic effect looks less significant.
Figure 3. NaBut sensitizes E1A+Ras cells to irradiation. (A and B) Bivariate distributions (DNA content vs γH2AX) of E1A+Ras cells. (A) Cells were X-ray irradiated and incubated after irradiation for 10 min, 60 min or 24 h with or without NaBut as indicated. (B) Alternatively, cells were NaBut-pretreated for 24 h, irradiated and incubated after irradiation for 10 min, 60 min or 24 h with or without NaBut as indicated. (C) Histogram presented the dependence between duration of HDACi pretreatment and IR-induced γH2AX persistence. Phosphorylated histone H2AX was visualized by flow cytometry. To obtain the IR-induced increase in γH2AX IF (ΔH2AX IF), the means for the untreated cells were subtracted from respective means of the treated cells. Cells were pretreated with 4 mM NaBut for 24 h (light gray) or for 72 h (dark gray), irradiated and incubated after irradiation for 24 h with or without NaBut as indicated. (D) Colony formation assay after X-ray irradiation alone, pretreatment with 4 mM NaBut for 24 h before irradiation, or irradiation followed by NaBut in E1A+Ras cells. After treatment cells were trypsinized and plated as single cells in NaBut-free medium for determination of colony-forming efficiency. In each case, colonies were determined 10–14 d afterwards. Values represent the mean from three independent experiments.
Prolonged treatment with HDAC inhibitor leads to sustained γH2AX foci
As we described recently, NaBut induced p21-dependent irreversible cell cycle arrest and cellular senescence in E1A+Ras-transformed rodent cells.Citation13,Citation24 NaBut-induced cellular senescence is accompanied by irreversible G1 arrest, permanent loss of proliferative potential (cells did not resume proliferation, even when NaBut was removed), a large and flat morphology of senescent cells and development of a specific marker SA-β-Gal staining. In contrast, cells after short-term NaBut treatment show a reversible cell cycle arrest that is abrogated after washing free of the drug, implying a qualitative change in cells over prolonged treatment. Therefore, we addressed the issue of whether HDAC inhibitors must be present in the medium in order to achieve a synergistic effect on lifetime of γH2AX foci induced by genotoxic agents. Besides, we were interested to see whether there is a relationship between the duration of NaBut treatment and the necessity of its permanent presence together with DNA damaging factors. Results presented in demonstrate that if cells were pretreated with NaBut for 24 h before IR, then irradiated and plated without HDACi for another 24 h they lose the IR-induced γH2AX foci. However, the IR-induced γH2AX foci persist for a longer time in the presence of NaBut (). In turn, γH2AX foci created after NaBut treatment for 72 h followed by irradiation became more long-lasting and still persist on a high level up to 24 h irrespectively of the presence of NaBut (). Thus, the effect of sodium butyrate on DNA damage response (IR) depends on the duration of HDACi treatment. As NaBut suppresses apoptosis in E1A+Ras cells due to activation of the NF-kB pathway,Citation23 the persistence of γH2AX foci is not a consequence of apoptosis-mediated DNA fragmentation. Instead, there is a correlation between accumulation of γH2AX foci and NaBut-induced senescence.Citation13 Therefore, formation of γH2AX foci likely contributes to both cellular senescence and activation of DDR signaling. In summary, the obtained data indicate that a short treatment with an HDAC inhibitor gives a reversible effect on DDR, while a long-term exposure makes the changes less reversible.
Discussion
The antitumor effect of HDAC inhibitors is based on the activation of tumor-suppressor and pro-apoptotic genes, the expression of which is capable of inducing either irreversible cell cycle arrest or apoptotic cell death.Citation6,Citation7 According to a number of data, HDAC inhibitors activate transcription in at least 4–10% of the genome.Citation5 It is unlikely that all these activated genes belong to tumor-suppressors and pro-apoptotic genes. There should be alternative pathways, which are additionally involved in the suppression of cell growth. Here, by two-parametric cytometry and immunofluorescence (Fig. S1) we showed that HDAC inhibitor sodium butyrate induces both G1 cell cycle arrest and accumulation of γH2AX foci in E1A+Ras transformed fibroblasts. Thus, this indicates that the antiproliferative effect of HDAC inhibitors can be somehow based on the modulation of various components of the DNA damage response (DDR) pathway. It is well known that the Ser139-phosphorylated H2AX histone is a marker of double-strand DNA breaks (DSBs).Citation14 However, there is accumulating data evidencing that the phosphorylation of H2AX can take place in the absence of noticeable DSBs, for example, during mitosis independent from DNA damage,Citation25,Citation28 in fast-proliferating mouse embryonic stem cells,Citation19 in case of stable association of single repair factor with chromatinCitation20 and also in response to such stress factors such as heat-shock.Citation21,Citation22 The mechanism of DSB-independent H2AX phosphorylation is still unclear. Recently we suggested that histone acetylation, apart from local relaxation of chromatin structure, might promote H2AX phosphorylation even in the absence of DNA breaks.Citation13 In line with these data, the enforced loosening of chromatin structure by histone acetylation induced either by HDAC inhibitors or by HAT p300 had similarly augmented γH2AX accumulation and sensitized cells to irradiation or topoisomerase inhibitorsCitation29-Citation32 thereby increasing the efficiency of anticancer drugs targeting DNA. This may be advantageous for treating tumors intrinsically resistant to these drugs.
It is worthwhile to note that NaBut-induced accumulation of γH2AX is not accompanied by nuclear accumulation of a censor kinase-ATM and formation of 53BP1 foci, which are both involved in typical DDR signalingCitation13 (Fig. S1). HDACi valproic acid and panobinostat do not induce DNA damages according to TUNEL and comet assays data and do not alter gene expression of some DDR components (H2AX and ATM).Citation33 In our case, accumulation of γH2AX foci in cells treated with NaBut is not connected with apoptosis.Citation23 If ATM is not involved in phosphorylation of H2AX in NaBut-treated cells, what is the kinase phosphorylates H2AX? Recently we showed that rapamycin, an inhibitor of mTOR kinase activity, which was shown to suppress HDACi- and p21 overexpression-induced cellular senescence, also decreased NaBut-induced H2AX foci formation.Citation13 This may mean that phosphorylation of H2AX in HDACi-treated cells can be dependent on a kinase(s) of mTOR signaling pathway thereby making a contribution to cellular senescence. To this end, DNA-PK was shown to be involved in phosphorylation of prior-acetylated H2AX histones.Citation18 In contrast, hyperthermia (heat shock) induces both phosphorylation of ATM and accumulation of γH2AX foci, which, however, does not take place due to DNA double-strand breaks.Citation21,Citation22 It is of interest that, although predominant NaBut-sensitive cells are in S-phase of the cell cycle (), the cells with γH2AX foci seem to not undergo S-phase arrest and progress through G2/M phases to the next G1 phase, where they are blocked at the G1/S boundary. This may imply that (1) HDACi-induced H2AX phosphorylation marks not merely DNA breaks but rather somehow “open” (relaxed) acetylated chromatin stretches; (2) NaBut does not interfere with ongoing DNA replication but affects the G1 > S transition of the next cell cycle. This is a very important notion allowing to predict to some extent whether the effect of the HDAC inhibitor will be additive in case of combined treatment with other genotoxic agents or not.
An important outcome from the above considerations is that HDAC inhibitors create conditions for activation of some components of the DNA damage response, including an accumulation of γH2AX foci that promotes sensitizing cells to various DNA damaging factors. In addition, there are data showing that HDAC inhibitors modulate the activity of DNA repair machinery. Particularly, HDAC inhibitor SAHA (vorinostat) suppressed the expression of DNA repair proteins, e.g., Rad50 and MRE11 in cancer (but not in normal) cells that promoted cancer cell death.Citation34 Besides, sodium butyrate and trichostatin A were shown to downregulate on transcriptional levels the expression of DNA repair proteins Ku70, Ku80 and Ku86 as well as Rad50.Citation34,Citation35 Also, sodium butyrate increases acetylation of Ku70 that leads to a decrease in its binding affinity to DNA.Citation36 Interestingly, according to protein-protein interaction analysis, valproic acid caused downregulation of several DNA repair genes related to BRCA1 in prostate cancer cell lines.Citation37 Many of these genes are involved in the homologous recombination DNA repair pathway mediated by BRCA1 and executed by the RecA homolog Rad51 DNA recombinase, a key protein in the HR pathway. However, genes of the NHEJ DNA repair pathway remained unaffected.Citation38 Altogether, this suggests that HDACi-mediated inhibition of proteins and enzymes involved in DNA repair may be a condition of γH2AX accumulation.
There are data showing that HDAC inhibitors can be involved in the accumulation of γH2AX by interfering in the association of HDACs with enzymes that regulate H2AX phosphorylation. So HDACs can functionally interact with cellular proteins that may not necessarily be substrates for acetylation/deacetylation. The interaction of HDACs with other proteins may affect HDAC activity and/or the activity of the other partner protein. For example, in response to IR, HDAC1 interacts with ATM,Citation39 and HDAC2 interacts with ATR.Citation40 HDAC1 and DNA topoisomerase II isoforms physically interact both in vivo and in vitro, and the inhibition of HDAC suppresses DNA decatenation activity of DNA topoisomerase II.Citation41 Interaction between HDACs and protein phosphatases (PP1, PP2A, PP4), which participate in dephosphorylation of γH2AX, has been demonstrated.Citation42 HDACi treatment disrupted the HDAC-phosphatase interactions resulting in attenuated dephosphorylation of target proteins.Citation43,Citation44 By modulation of the serine-threonine phosphatase activity, HDACi can also affect Wip1 phosphatase, which was shown to dephosphorylate γH2AX.Citation45 Thus, the treatment of cells with HDACi not only results in hyperacetylation of HDAC targets but also induces altered phosphorylation of a range of target proteins which are potential effector proteins for the HDACi-mediated biological response.
We showed a difference in the lifetime of NaBut-induced γH2AX foci in cells treated for 24 h and for a longer period (72 h). While the 24 h foci disappear soon after the removal of NaBut, the 72 h foci still persist for an additional 24 h period after washing the drug (Fig. Citation3C). It may mean that stability of the 72 h foci is provided by sustained inability of long-term NaBut-treated cells to dephosphorylate γH2AX. Correspondingly, the sustained γH2AX foci provide a better synergistic effect with irradiation, if cells were treated first with NaBut for 72 h then irradiated and left without HDACi, the γH2AX foci do not disappear. However, such a synergistic effect is clearly evident in case of a combined treatment NaBut and IR, but not with adriamycin. This can be accounted for by well-known differences in the mechanisms of IR and adriamycin action. IR induces mostly single-strand breaks (SSBs) in any phase of the cell cycle; therefore, IR can induce γH2AX foci in G1- and G2-arrested cells. Moreover, this effect is more long-lasting in the case of long-term HDACi treatment. In contrast, DNA-intercalating agents such as adriamycin are unable to induce the comparable amount of additional γH2AX foci in the population of cells arrested by NaBut in G1 and G2 phases and therefore lacking S-phase cells. Nevertheless, pretreatment of cells with HDACi is useful as it stimulates both the formation and persistence of γH2AX foci thereby sensitizing the cells to any type of DNA damaging agents.
Materials and Methods
Cell cultures and treatment
The E1A+Ras cells were established by Ca-phosphate transfection of E1A Ad5 and activated cHa-Ras oncogenes, as described earlier.Citation12 Cells grown in log-phase were treated with 4 mM NaBut (Sigma) for 1–72 h and harvested at the indicated time points. For combined treatments (irradiation or adriamycin), cells were left untreated or treated with NaBut for 24–72 h, then 0.2 μg/ml adriamycin was added for 40 min or the cells were irradiated for 3 min (6 Gy). The medium was replaced immediately after exposure to genotoxic agents and cells were harvested at the indicated time points.
Immunofluorescence microscopy
Cells were seeded on coverslips, washed in PBS, fixed in fresh 3.7% paraformaldehyde for 15 min at room temperature, washed three times in PBS for 10 min and in 0.15 M glycine for 15 min, then permeabilized with 0.2% Triton X-100 for 15 min, and blocked by incubation with 3% bovine serum albumin in PBS for 1.5 h. Primary and secondary antibodies were diluted in blocking solution. The coverslips were incubated with primary antibody to phosphorylated histone H2AX (Ser139) (Cell Signaling) overnight at +4°C and then incubated with secondary Alexa Fluor 488-conjugated goat anti-rabbit antibody (Invitrogene) and 1 mM Topro III (Invitrogene) for 1 h at room temperature in the dark. Cells were washed in 0.05% Tween-20/PBS and mounted. Slides were viewed with a Leica DMRE fluorescence microscope.
Cell extracts and immunoblots
The cell extracts for immunoblotting were obtained as described earlier.Citation12 Proteins were separated by electrophoresis in 12% polyacrylamide gel in the presence of 0.1% SDS, transferred onto a PVDF membrane (Immobilon P), and probed with the appropriate antibodies. Broad molecular mass colored markers were used as molecular weight standards (BioLabs). Rabbit antibodies to phosphorylated H2AX (Ser139) (Cell Signaling) and mouse antibodies to GAPDH were used. Anti-mouse and anti-rabbit antibodies conjugated with horseradish peroxidase (Sigma) were used as secondary antibodies. The visualization of membrane-bound proteins was performed by SuperSignal West Femto Chemiluminescent Substrate (Pierce).
FACS analysis of γH2AX foci
Analysis was done mostly as described.Citation14 Cells were fixed in a freshly prepared 2% paraformaldehyde at room temperature for 10 min, resuspended in 70% ethanol for at least 2 h at -20°C. The cells were then washed twice in PBS and suspended in 1% BSA (Sigma) in PBS with 0.2% Triton X-100 (Sigma) for 15 min to suppress nonspecific antibody binding. After centrifugation, the pellet was suspended in 100 μl of 1% BSA containing anti-H2AX (Ser-139) (Cell Signaling) and incubated overnight at +4°C. The cells were rinsed with 1% BSA in PBS with 0.2% Triton X-100 (300 g, 5 min) and, after centrifugation, the cell pellets were resuspended in 100 μl of 1% BSA containing AlexaFluor-488 conjugated mouse anti-rabbit F(ab')2 fragment (Molecular Probes) for 1.5 h at room temperature in the dark. After washing with 1% BSA, the cells were counterstained with 10 μg/ml propidium iodide (Molecular Probes) dissolved in PBS containing 100 μg/ml RNase A (Sigma) for 30 min at room temperature in the dark. The fluorescence of individual cells induced by excitation with a 488 nm argon ion laser was measured using a FACScan cytometer Bechman Coulter Epicks XL.
Caspase assay
Ac-DEVD-pNA (caspase-3 substrate) was purchased in Calbiochem. Cell lysates were obtained with a lysis buffer and (50 mM Tris-HCl, pH 7.5; 120 mM NaCl; 1 mM EDTA; 1% NP-40) supplemented with protease inhibitors. Caspase assays were performed by pipetting 40 µl of cell lysates to a 96-well dish, containing 160 µl reaction buffer (20% glycerol; 0.5 mM EDTA; 5 mM DTT; 100 mM HEPES, pH 7.5) with the colorimetric substrate Ac-DEVD-pNA (Calbiochem). The substrate cleavage readout was the p-nitroanilide release as detected at 405 nm in a microplate reader Multiscan-EX (Labsystems).
Clonogenic assay
To evaluate modulation of cellular sensitivity to genotoxic agents by NaBut, HDACi-pretreated or untreated cells were irradiated (6 Gy) or treated with adriamycin for 40 min. Immediately after irradiation, cultures were trypsinized into a single cell suspension, and were equally reseeded at 200 cells/30-mm plate. After 10–14 d, the cells were fixed in 100% methanol (30 min at room temperature) and stained for 1 h with 0.1% crystal violet (Sigma). The stained colonies were counted and compared with an untreated control.
Measurement of DNA single-strand and double-strand breaks using denaturing and neutral comet assays
Comet assay of DNA breaks in a single cell by neutral and denaturing electrophoresis was done as described with some modifications.Citation13,Citation15
Additional material
Download Zip (1.1 MB)Acknowledgments
Authors thank Lyudmila Supanko for technical help in the process of performing the work and Xenia Strunnikova for critical reading the manuscript. The work was supported by Russian Foundation for Basic Research (RFBR): No. 09-04-00466 (VAP) and No. 10-04-01152 (TVP), Russian Academy of Sciences Program “Molecular and Cell Biology” (VAP), and grant from the St.Petersburg State University (VAP), Contract No. 1.37.122.2011, grant the committee on Science and Higher Education of the Government of St. Petersburg (MAV).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental material can be found at: www.landesbioscience.com/journals/cbt/article/18365/.
References
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997; 389:349 - 52; http://dx.doi.org/10.1038/38664; PMID: 9311776
- Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev 1999; 9:40 - 8; http://dx.doi.org/10.1016/S0959-437X(99)80006-9; PMID: 10072350
- Villar-Garea A, Esteller M. Histone deacetylase inhibitors: Understanding a new wave of anticancer agents. Int J Cancer 2004; 112:171 - 8; http://dx.doi.org/10.1002/ijc.20372; PMID: 15352027
- Wade PA, Wolffe AP. Histone acetyltransferases in control. Curr Biol 1997; 7:R82 - 4; http://dx.doi.org/10.1016/S0960-9822(06)00042-X; PMID: 9081669
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 1996; 5:245 - 53; PMID: 8723390
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 2000; 92:1210 - 6; http://dx.doi.org/10.1093/jnci/92.15.1210; PMID: 10922406
- Johnstone RW. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002; 1:287 - 99; http://dx.doi.org/10.1038/nrd772; PMID: 12120280
- Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst) 2011; 10:970 - 7; http://dx.doi.org/10.1016/j.dnarep.2011.07.003; PMID: 21824827
- Karagiannis TC, El-Osta A. Modulation of cellular radiation responses by histone deacetylase inhibitors. Oncogene 2006; 25:3885 - 93; http://dx.doi.org/10.1038/sj.onc.1209417; PMID: 16462761
- Lindemann RK, Gabrielli B, Johnstone RW. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle 2004; 3:779 - 88; http://dx.doi.org/10.4161/cc.3.6.927; PMID: 15153801
- Zhang Y, Adachi M, Zou H, Hareyama M, Imai K, Shinomura Y. Histone deacetylase inhibitors enhance phosphorylation of histone H2AX after ionizing radiation. Int J Radiat Oncol Biol Phys 2006; 65:859 - 66; http://dx.doi.org/10.1016/j.ijrobp.2006.03.019; PMID: 16751067
- Abramova MV, Pospelova TV, Nikulenkov FP, Hollander CM, Fornace AJ Jr., Pospelov VA. G1/S arrest induced by histone deacetylase inhibitor sodium butyrate in E1A+Ras-transformed cells is mediated through downregulation of E2F activity and stabilization of beta-catenin. J Biol Chem 2006; 2:21040 - 51; http://dx.doi.org/10.1074/jbc.M511059200
- Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle 2009; 8:1 - 7; http://dx.doi.org/10.4161/cc.8.24.10215; PMID: 19182532
- Tanaka T, Halicka D, Traganos F, Darzynkiewicz Z. Cytometric analysis of DNA damage: phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). Methods Mol Biol 2009; 523:161 - 8; http://dx.doi.org/10.1007/978-1-59745-190-1_11; PMID: 19381940
- Olive PL, Banath JP. The comet assay: A method to measure DNA damage in individual cells. Nat Protoc 2006; 1:23 - 9; http://dx.doi.org/10.1038/nprot.2006.5; PMID: 17406208
- Pehrson JR, Fuji RN. Evolutionary conservation of histone macroH2A subtypes and domains. Nucleic Acids Res 1998; 26:2837 - 42; http://dx.doi.org/10.1093/nar/26.12.2837; PMID: 9611225
- Riches LC, Lynch A, Gooderham NJ. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 2008; 23:331 - 9; http://dx.doi.org/10.1093/mutage/gen039; PMID: 18644834
- Park E-J, Chan DW, Park J-H, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res 2003; 31:6819 - 27; http://dx.doi.org/10.1093/nar/gkg921; PMID: 14627815
- Chuykin IA, Lianguzova MS, Pospelova TV, Pospelov VA. Activation of DNA damage response signaling in mouse embryonic stem cells. Cell Cycle 2008; 7:2922 - 8; http://dx.doi.org/10.4161/cc.7.18.6699; PMID: 18787397
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008; 320:1507 - 10; http://dx.doi.org/10.1126/science.1159051; PMID: 18483401
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 67:3010 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-06-4328; PMID: 17409407
- Laszlo A, Fleischer I. The heat-induced gamma-H2AX response does not play a role in hyperthermic cell killing. Int J Hyperthermia 2009; 25:199 - 209; http://dx.doi.org/10.1080/02656730802631775; PMID: 19437236
- Abramova MV, Zatulovskiy EA, Svetlikova SB, Pospelov VA. HDAC inhibitor-induced activation of NF-κB prevents apoptotic response of E1A+Ras-transformed cells to proapoptotic stimuli. Int J Biochem Cell Biol 2010; 42:1847 - 55; http://dx.doi.org/10.1016/j.biocel.2010.08.001; PMID: 20692358
- Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, et al. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010; 9:3945 - 55; http://dx.doi.org/10.4161/cc.9.19.13160; PMID: 20935470
- Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol 2009; 10:243 - 54; http://dx.doi.org/10.1038/nrm2651; PMID: 19277046
- Ichijima Y, Sakasai R, Okita N, Asahina K, Mizutani S, Teraoka H. Phosphorylation of histone H2AX at M phase in human cells without DNA damage response. Biochem Biophys Res Commun 2005; 336:807 - 12; http://dx.doi.org/10.1016/j.bbrc.2005.08.164; PMID: 16153602
- McManus KJ, Hendzel MJ. ATM-dependent and DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol Biol Cell 2005; 16:5013 - 25; http://dx.doi.org/10.1091/mbc.E05-01-0065; PMID: 16030261
- Ziegler-Birling C, Helmrich A, Tora L, Torres-Padilla ME. Distribution of p53 binding protein 1 (53BP1) and phosphorylated H2A.X during mouse preimplantation development in the absence of DNA damage. Int J Dev Biol 2009; 53:1003 - 11; http://dx.doi.org/10.1387/ijdb.082707cz; PMID: 19598117
- Camphausen K, Burgan W, Cerra M, Oswald KA, Trepel JB, Lee MJ, et al. Enhanced radiation-induced cell killing and prolongation of γH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res 2004; 64:316 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-03-2630; PMID: 14729640
- Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES, Camphausen K, et al. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res 2008; 14:5410 - 5; http://dx.doi.org/10.1158/1078-0432.CCR-08-0643; PMID: 18765532
- Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 2003; 63:7291 - 300; PMID: 14612526
- Kim M-K, Shin J-M, Eun HC, Chung JH. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS ONE 2009; 4:e4864; http://dx.doi.org/10.1371/journal.pone.0004864; PMID: 19287485
- Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 2011; 13:292 - 302; http://dx.doi.org/10.1038/ncb2170; PMID: 21336312
- Lee OP, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA 2010; 107:14639 - 44; http://dx.doi.org/10.1073/pnas.1008522107; PMID: 20679231
- Munshi A, Kurland JF, Nishikawa T, Tanaka T, Hobbs ML, Tucker SL, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res 2005; 11:4912 - 22; http://dx.doi.org/10.1158/1078-0432.CCR-04-2088; PMID: 16000590
- Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther 2006; 5:1967 - 74; http://dx.doi.org/10.1158/1535-7163.MCT-06-0022; PMID: 16928817
- Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res 2007; 67:5318 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-06-3996; PMID: 17545612
- Kachhap SK, Rosmus N, Collis SJ, Kortenhorst M, Wissing MD, Hedayati M, et al. Downregulation of Homologous Recombination DNA Repair Genes by HDAC Inhibition in Prostate Cancer Is Mediated through the E2F1 Transcription Factor. PLoS ONE 2010; 5:e11208; http://dx.doi.org/10.1371/journal.pone.0011208; PMID: 20585447
- Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem 1999; 274:31127 - 30; http://dx.doi.org/10.1074/jbc.274.44.31127; PMID: 10531300
- Schmidt DR, Schreiber SL. Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry 1999; 38:14711 - 7; http://dx.doi.org/10.1021/bi991614n; PMID: 10545197
- Johnson CA, Padget K, Austin CA, Turner BM. Deacetylase Activity Associates with Topoisomerase II and Is Necessary for Etoposide-induced Apoptosis. J Biol Chem 2001; 276:4539 - 42; http://dx.doi.org/10.1074/jbc.C000824200; PMID: 11136718
- Peng A, Maller JL. Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene 2010; 29:5977 - 88; http://dx.doi.org/10.1038/onc.2010.371; PMID: 20838380
- Brush MH, Guardiola A, Connor JH, Yao TP, Shenolikar S. Deacetylase inhibitors disrupt cellular complexes containing protein phosphatases and deacetylases. J Biol Chem 2004; 279:7685 - 91; http://dx.doi.org/10.1074/jbc.M310997200; PMID: 14670976
- Canettieri G, Morantte I, Guzm´n E, Asahara H, Herzig S, Anderson SD, et al. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol 2003; 10:175 - 81; http://dx.doi.org/10.1038/nsb895; PMID: 12567184
- Cha H, Lowe JM, Li H, Lee J, Belova G, Bulavin D, et al. Wip1 Directly Dephosphorylates γ-H2AX and Attenuates the DNA Damage Response. Cancer Res 2010; 70:1213 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-09-4244